

Abstract
Nonketotic hyperglycinemia (NKH) is caused by deficient glycine cleavage enzyme activity and characterized by elevated brain glycine. Metabolism of glycine is connected enzymatically to serine through serine hydroxymethyltransferase and shares transporters with serine and threonine. We aimed to evaluate changes in serine and threonine in NKH patients, and relate this to clinical outcome severity. Age‐related reference values were developed for cerebrospinal fluid (CSF) serine and threonine from 274 controls, and in a cross‐sectional study compared to 61 genetically proven NKH patients, categorized according to outcome. CSF d‐serine and l‐serine levels were stereoselectively determined in seven NKH patients and compared to 29 age‐matched controls. In addition to elevated CSF glycine, NKH patients had significantly decreased levels of CSF serine and increased levels of CSF threonine, even after age‐adjustment. The CSF serine/threonine ratio discriminated between NKH patients and controls. The CSF glycine/serine aided in discrimination between severe and attenuated neonates with NKH. Over all ages, the CSF glycine, serine and threonine had moderate to fair correlation with outcome classes. After age‐adjustment, only the CSF glycine level provided good discrimination between outcome classes. In untreated patients, d‐serine was more reduced than l‐serine, with a decreased d/l‐serine ratio, indicating a specific impact on d‐serine metabolism. We conclude that in NKH the elevation of glycine is accompanied by changes in l‐serine, d‐serine and threonine, likely reflecting a perturbation of the serine shuttle and metabolism, and of one‐carbon metabolism. This provides additional guidance on diagnosis and prognosis, and opens new therapeutic avenues to be explored.
Keywords: cerebrospinal fluid, d‐serine, l‐serine, nonketotic hyperglycinemia, threonine
1. INTRODUCTION
Nonketotic hyperglycinemia (NKH; OMIM# 605899) is an ultrarare neurogenetic condition characterized by deficient activity of the glycine cleavage enzyme (GCE), with an estimated incidence of 1:76 000 births. 1 , 2 It is caused by pathogenic variants in the genes encoding the protein components of the GCE: P‐protein encoded by GLDC gene (OMIM# 238300), and the T‐protein encoded by the AMT gene (OMIM# 238310). 2 Clinically, patients with the severe phenotype of NKH have developmental delay evolving into profound cognitive dysfunction, intractable epilepsy, spasticity, and cortical blindness. 3 , 4 Patients with an attenuated form of NKH have a better outcome with variable cognitive dysfunction, treatable or no epilepsy, and do not develop spasticity, but develop chorea and sometimes ataxia. Based on the developmental outcome, attenuated NKH is subdivided into attenuated poor (developmental quotient [DQ] <20), attenuated intermediate (DQ 20–50), and attenuated good (DQ >50). 4 The GCE breaks down glycine into carbon dioxide and ammonia, and the remaining one‐carbon unit is transferred to tetrahydrofolate (THF) as 5,10‐methylene‐tetrahydrofolate. 5 Deficient activity of the GCE results in accumulation of glycine in plasma and tissues, including in brain tissue, reflected in elevation of cerebrospinal fluid (CSF) glycine levels and an increase of the CSF/plasma glycine ratio. 6 The CSF glycine relates to outcome with higher CSF glycine levels correlating with lower DQ values. 4 The pathophysiology of NKH is not well‐known, and current therapy, focused exclusively at mitigating the effects of increased glycine has only limited impact. 7 , 8 , 9 To develop new therapeutic approaches to NKH, a more in‐depth evaluation of the biochemistry is indicated. Mouse models of NKH using a gene‐trap in the GLDC gene have been described. 10 , 11 , 12 All affected mice had elevated glycine levels. A metabolomics analysis showed increased glycine conjugates. 13 Also, in these mouse models deficient amounts of 5,10‐methylene‐THF and 5‐methyl‐THF have been documented in affected fetuses. 10 , 11
The biochemistry and transport of glycine is interconnected with that of serine and threonine (Figure 1). The serine hydroxymethyltransferases (SHMT) are key enzymes in this pathway, catalyzing the reversible equilibrium reaction of glycine with 5,10‐methylene‐THF to serine and THF. While a majority of serine is produced via a de novo synthesis pathway, 14 a substantial amount of serine is formed from glycine by the action of the SHMT enzymes. 15 , 16 The mitochondrial SHMT (SHMT2) is a major donor of one‐carbon units via 5,10‐methylene‐THF, whereas the cytoplasmic enzyme SHMT1 is only a minor donor. 17 On a whole‐body level, both GCE and SHMT are the major one carbon donors to the folate pool with serine being the main one‐carbon donor. 16 , 17 , 18 l‐serine, produced by the de novo synthesis pathway and from glycine, can be converted to d‐serine, which has an important physiological role in the developing brain. 19 , 20 This reaction is catalyzed by a serine racemase enzyme present in glutamatergic and GABA‐ergic neurons. 21 An increase in glycine in patients with NKH may be expected to result in increased l‐serine levels via the reversible reaction catalyzed by the SHMT enzymes (Figure 1, blue bold arrows), and in turn increase the production of d‐serine. However, in three patients with NKH decreased levels of d‐serine were reported in brain tissue, suggesting a disruption of d‐serine synthesis in NKH. 22
FIGURE 1.
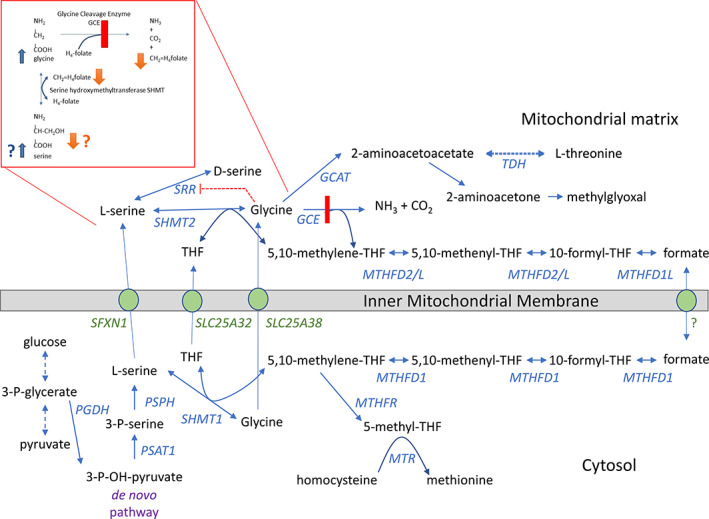
Biochemistry of brain one‐carbon metabolism including glycine in relation to serine, threonine, and folate metabolism. Glycine is in equilibrium through the serine hydroxymethyltransferase enzymes (SHMT1 and SHMT2) with serine, using 5,10‐methylene‐tetrahydrofolate. In nonketotic hyperglycinemia there is deficient activity of the glycine cleavage enzyme (GCE), which breaks glycine down into carbon dioxide, ammonia, and generates 5,10‐methylene‐tetrahydrofolate. In the presence of excess glycine due to deficiency in the glycine cleavage system, one would expect an increase in serine through the SHMT enzymes (blue bold arrows). If 5,10‐methylene‐tetrahydrofolate concentrations drop due to deficient generation by dysfunction of the glycine cleavage enzyme, one would expect a decrease in the generation of serine (orange arrows). Furthermore, high glycine levels are purported to inhibit the serine racemase enzyme (orange interrupted line). Gene symbols and reactions in blue, transporters across the inner mitochondrial membrane in green. The threonine dehydrogenase (TDH) is active in mice but inactive in humans. GCAT, glycine acetyltransferase; GCE, glycine cleavage enzyme; MTHFD, methylene‐tetrahydrofolate dehydrogenase; MTHFR, methylene‐tetrahydrofolate reductase; MTR, methionine synthase; SHMT, serine hydroxymethyltransferase; PGDH, 3‐phosphoglycerate dehydrogenase; PSAT, phosphoserine aminotransferase; PSPH, phosphoserine phosphatase; SRR, serine racemase; THF, tetrahydrofolate
Threonine is another small neutral amino acid related to glycine and serine, through biochemistry and membrane transporters. In contrast to other animals, a biochemical link between threonine and glycine does not appear functional in humans. Humans do not have the Escherichia coli bacterial enzyme threonine aldolase, also known as l‐serine/l‐threonine ammonia lyase, which converts threonine directly into glycine. In mice, glycine C‐acetyltransferase catalyzes the conversion of glycine to l‐2‐amino‐acetoacetate, which then can be converted to l‐threonine by threonine dehydrogenase. 23 , 24 In humans, the latter enzyme is an expressed pseudogene without activity. The small neutral amino acids glycine, serine, and threonine share several transporters, which can influence their levels. For instance, the neutral amino acid transporter 1 ASCT1 (gene SLC1A4) transports serine, threonine and glycine, in addition to other neutral amino acids, whereas ASCT2 (gene SLC1A5) transports alanine, serine, glutamine. Dysregulation of ASCT1, but not ASCT2, affects brain levels of d‐serine, l‐serine, l‐alanine, l‐threonine, and l‐glycine. 25 The Asc1 transporter (gene SLC7A10) carries these amino acids over the neuronal membrane. Other transporters for small neutral amino acids (including ATB0+ [encoded SLC1A5], SNAT1 [encoded SLC38A1], SNAT2 [encoded SLC38A2], SNAT5, and LAT2) display broad amino acid specificity and have differential contributions to glial glycine transport. 26 The transporters SNAT1 (SLC38A1) and SNAT2 (SLC38A2) carriers also share transport for serine, threonine, and glycine. Transporters across the mitochondrial membrane SFXN1 and SFXN3 carry both racemic forms of serine and to a lesser extent alanine, cysteine, and glycine, and impact one‐carbon metabolism. 27 The transporter SLC25A38 is the main transporter for glycine into mitochondria, and also transports alanine and serine, but not threonine. 28
In this cross‐sectional study, we evaluated the impact of NKH on CSF levels of glycine, l‐serine, d‐serine, and threonine, in relation to age‐matched controls, and related the findings to clinical severity.
2. METHODS
2.1. Human studies
The study was performed under IRB‐approved protocols (Colorado COMIRB# 05‐0790, Pittsburgh IRB# STUDY19090211) for NKH patients and COMIRB# 17‐1830 for controls. Informed consent was obtained from all living subjects. All procedures were done in accordance with the ethical standards of the ethics committee in accordance with the Helsinki Declaration of 1975 and as revised in 2000.
2.2. Control values
Deidentified data for the calculation of age‐specific reference ranges were obtained from CSF amino acid analyses with apparently normal results from two clinical laboratories: Duke University Health System (N = 224; ion‐exchange method on a Hitachi L‐8800 amino acid analyzer) and Children's Hospital Colorado (N = 50; ion‐exchange method on a Biochrom 30+ Amino Acid analyzer). Gender and age were recorded. A well‐known decline in the early years for serine and threonine concentration was noted to evolve to a stable value in older children and adults, with the correlation between age and CSF amino acids values no longer significant after age 10 years.
To account for this age‐related decline in concentrations of serine and threonine, we sought to calculate age‐adjusted z‐scores for NKH patients in our study. To create the normal average values at each age represented, we modeled the trend of serine and threonine by age in the normal control population. As proposed by DeVore et al., 36 potential models that use various functions and fractional polynomials were considered. 29 In addition, exponential declines to an established asymptote in biology are best modeled as a Gompertz curve with formula , so this was also considered. The best fitting model from the 37 options was chosen based on the highest R 2 value, separately for serine and threonine. It was evident that the normal standard deviation of serine and threonine also varied by age, so a normalized age‐dependent standard deviation was also derived. The same series of models described above were fitted, but with the absolute scaled residuals from the mean model as outcome and with age as predictor. The best model for the standardized residuals was chosen based on highest R 2 value. For each patient, the Z‐score was calculated using the formula: Z = (measured value − predicted mean value)/predicted SD, where predicted mean is the predicted average value using the best fitting mean model, and predicted SD is the predicted residual from the best fitting standard deviation model. Visualization was done by scatterplots of the analyte versus age with the modeled mean and SD (Figure 2). This allows for a comparison of age‐adjusted patient values as described before. 30
FIGURE 2.
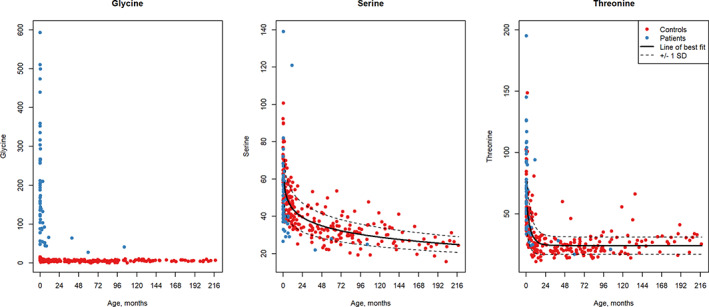
Modeling of CSF serine and threonine values in normal controls. Values of CSF glycine (A), CSF serine (B), and CSF threonine (C) are shown in relation to age in months with controls in red and nonketotic hyperglycinemia patients shown in blue. The line of best fit developed by modeling control data is provided and the 1 SD curve in a dotted line. The CSF serine values of patients are in the lower half of the control values (except for two patients), and the CSF threonine tend to cluster above the average particularly during early months. CSF, cerebrospinal fluid
2.3. Patient values
Inclusion criteria for this cross‐sectional study included a certain diagnosis of NKH based on biochemical value, clinical presentation, and molecular results, 1 and the availability of results for CSF amino acids glycine, serine and threonine. Patient values were extracted from the clinical records or via a questionnaire collected between 2005 and January 2022 for the results of CSF glycine, serine and threonine, and for plasma levels of glycine and serine. Levels of CSF protein and other amino acids were reviewed where available to exclude blood contamination. Patients were clinically characterized into severe, attenuated poor, attenuated intermediate, or attenuated good classes according to previously published criteria. 4 The molecular diagnosis of NKH was recorded from the medical record as the pathogenic variants identified in GLDC and AMT. 2
2.4. Stereoselective levels of serine
Stereoselective amino acids l‐Ser, d‐Ser, Gly, Thr, and Met were measured after chiral derivatization with N‐isobutyryl‐l‐cysteine and OPA by reverse phase HPLC on an XBridge C18 column with fluorescence detection, with details in Supporting Information Materials. 31
2.5. Statistical analysis
Normality of distribution was evaluated by Wilk–Shapiro and Kolmogorov–Smirnov tests. An initial analysis was done for neonates age ≤1 months of age, where control values were not substantively changed by age. When analyzing all subjects (neonates and older) to correct for age, the age‐adjusted Z scores for serine and threonine were computed as described above, and used for comparison, whereas for glycine raw values were compared, as the influence of age was limited. An initial analysis of the raw values was also done.
Comparison between controls and patients and between severe and attenuated NKH patients was done by two‐sided independent samples Student t‐test or by Mann–Whitney U (MWU) test if normally distributed or not, respectively. The strength of the difference was evaluated by Cohen's d. Comparison for all parameters in neonates was done by both MWU and t‐test using discriminator sex or discriminator protein type. Comparison of amino acid levels between controls, severe and attenuated NKH patients or between different classes of NKH was done by one‐way analysis of variance (ANOVA), and if significant, Tukey's post hoc adjustments were used to report pairwise comparisons. For nonnormal distributions, Kruskal–Wallis and Dunn tests were used to compare across class. A p < 0.05 was considered significant. For relationships analysis, a Pearson or Spearman correlation statistic was computed depending on normality of distribution. Statistical comparisons were done using IBM SPSS version 28, or by using R version 4.0.2.
3. RESULTS
3.1. Patient and controls characteristics
We collected data on 61 patients with proven NKH, 29 female (48%) and 32 male (52%) (Table S1). There were pathogenic variants documented in AMT in 10 patients (16%) and in GLDC in 51 patients (84%), of which for 1 patient a pathogenic variant in only one allele had been identified. All patients were characterized to disease severity: 32 patients with severe NKH (55%) and 26 patients with attenuated NKH, of which 6 attenuated poor (10%), 7 attenuated intermediate (12%), 7 attenuated good, and 6 attenuated where the subset was not established (attenuated not otherwise specified), except for three patients who were too young to assign severity class. Data of the CSF values were collected at an age ≤7 days in 32 patients (52%), >7 days but ≤31 days (first month) in 11 patients (18%), >1 month and <1 year in 14 patients (23%), and at age >1 year in 4 patients (7%).
We also collected data from 274 controls (139 females and 132 males), of which 224 were from Duke University Health System and 50 from Children's Hospital Colorado, with the Duke group being older (median: 3.7 years) than the Colorado group (median: 0.47 years, MWU p < 0.001). There were no statistical differences between both centers for CSF glycine, serine and threonine. There were 34 neonates (age ≤1 month), and 105 infants (age >1 month and <1 year), which allowed to map the age‐related trends.
3.2. Control values for CSF glycine, serine, and threonine
The distribution of control values for glycine, serine and threonine followed a decline in early life, attaining an apparently stable value. The decline was most rapid in the first 6 months for glycine, the first year for threonine, and the first 5 years for serine (Figure 2). After age 10 years a significant correlation with age was not observed. The age‐related changes in control CSF glycine were minor compared with the marked elevations of glycine observed in CSF from NKH patients. The CSF serine values did not fit well with a Gompertz curve, and the best fit was described with a fractional polynomial: showing an R 2 = 0.69 (Figure 2B). The model of best fit for CSF threonine was the Gompertz curve as with a = 24.16, b = −0.96, and c = 0.15, with R 2 = 0.47 (Figure 2C). The age‐adjusted mean, SD, and percentiles are listed in Table S2.
3.3. Patient values for CSF glycine, serine, and threonine
Of the 61 patients, plasma glycine levels of five patients were not recorded or recorded in an imprecise way (i.e., >1000 μM). Plasma serine was available for 22 patients. CSF glycine and serine were available on all 61 patients, and CSF threonine in 60 patients.
3.4. Neonates
We first examined neonates (age <1 month), where most patients are diagnosed and the age‐related decline in CSF amino acids is insignificant (Table 1). In patients with NKH, CSF amino acids were significantly different from the normal distribution, but plasma glycine was not. As expected, CSF glycine levels were elevated compared to controls (all patients 233 ± 127 μM [mean ± SD] vs. controls 9.9 ± 3.4 μM). CSF threonine levels were significantly increased in patients compared to controls (patients 80.6 ± 32.7 μM vs. controls 58.8 ± 19.9 μM) and CSF serine levels were significantly decreased compared to controls (patients 52.6 ± 17.9 μM vs. controls 65.4 ± 14.1 μM). The CSF glycine/serine ratio was elevated in patients compared to controls (patients 4.62 ± 2.36 vs. controls 0.155 ± 0.057) and the CSF serine/threonine ratio was significantly decreased (patient 0.066 ± 0.041 vs. control 1.18 ± 0.29), both with a larger Cohen's d effect size than CSF glycine alone. In this age group, in controls there was a moderate positive correlation between CSF threonine and serine (r = 0.636, p < 0.001) and fair correlation of CSF threonine with CSF glycine (r = 0.355) but not between glycine and serine. In patients, there was a fair positive correlation between CSF glycine and threonine (Spearman ρ = 0.467, p < 0.001), and inverse with CSF serine (Spearman ρ = −0.308, p = 0.006), but not between CSF serine and CSF threonine. There was no significant difference by sex or by affected protein (P or T) for any parameter.
TABLE 1.
Comparisons for neonates age <31 days
| Parameter | Controls mean ± SD (N) | All patients mean ± SD (N) | Patients severe mean ± SD (N) | Patients attenuated mean ± SD (N) | Contr. vs. patients p value a | Pats. vs. control effect size Cohen's d | Severe vs. Att. p value a | Severe vs. Att. effect size Cohen's d |
|---|---|---|---|---|---|---|---|---|
| CSF glycine b | 9.9 ± 3.4 (35) | 233 ± 127 (43) | 276.6 ± 125.3 (30) | 127.3 ± 54.4 (11) | <0.001 | 2.37 (1.78–2.95) | <0.001 | 1.34 (0.58–2.08) |
| CSF serine b | 65.4 ± 14.1 (35) | 52.6 ± 17.9 (43) | 52.6 ± 20.2 (30) | 54.2 ± 12.0 (11) | <0.001 | 0.78 (0.32–1.24) | 0.424 | −0.07 (−0.78–0.61) |
| CSF threonine b | 58.8 ± 19.9 (35) | 80.6 ± 32.7 (42) | 83.3 ± 34.4 (30) | 76.2 ± 29.1 (10) | 0.001 | 0.79 (0.32–1.25) | 0.724 | 0.21 (−0.51–0.93) |
| Plasma glycine | NA | 1174 ± 521 (37) | 1276 ± 577 (26) | 926 ± 240 (10) | NA | NA | 0.015 | 0.69 (−0.06–1.43) |
| Plasma serine b | NA | 208 ± 126 (15) | 199 ± 129 (10) | 254 ± 132 (4) | NA | NA | 0.304 | −0.42 (−1.59–0.76) |
| CSF/plasma Gly b | NA | 0.21 ± 0.10 (37) | 0.239 ± 0.107 (26) | 0.146 ± 0.069 (10) | NA | NA | 0.012 | 0.95 (0.18–1.70) |
| CSF/plasma Ser | NA | 0.28 ± 0.12 (15) | 0.293 ± 0.126 (10) | 0.244 ± 0.103 (4) | NA | NA | 0.506 | 0.41 (−0.77–1.57) |
| CSF Gly/Ser | 0.155 ± 0.057 (35) | 4.62 ± 2.36 (43) | 5.48 ± 2.22 (30) | 2.41 ± 1.11 (11) | <0.001 | 2.54 (1.94–3.14) | <0.001 | 1.54 (0.76–2.30) |
| CSF Ser/Thr b | 1.18 ± 0.29 (35) | 0.066 ± 0.041 (42) | 0.075 ± 0.043 (30) | 0.037 ± 0.018 (10) | <0.001 | 5.65 (4.64–6.65) | 0.001 | 1.00 (0.24–1.74) |
Note: Significant test results are highlighted by bold text.
Abbreviations: Att., attenuated; Contr., controls; CSF, cerebrospinal fluid; Gly, glycine; N, count; Pats., patients; SD, standard deviation; Ser, serine; Thr, threonine.
Comparisons are done by two‐sided Student t‐test if normally distributed or by the Mann–Whitney U test for those not normally distributed.
Statistically significantly different from the normal distribution.
We next compared severe NKH patients (n = 30) to attenuated NKH patients (n = 11) (Table S2). Severe NKH patients compared to attenuated patients had significantly higher plasma glycine (severe 1276 ± 577 μM vs. attenuated 926 ± 240 μM), higher CSF glycine levels (severe 276 ± 125 μM vs. attenuated 127 ± 54 μM) and higher CSF/plasma glycine ratio (severe 0.239 ± 0.107 vs. attenuated 0.146 ± 0.069). There was no significant difference in CSF threonine, CSF serine or plasma serine levels between severe and attenuated NKH. However, the CSF glycine/serine ratio discriminated between severe 5.48 ± 2.22 and attenuated NKH 2.41 ± 1.11, p < 0.001, and the effect size using Cohen's d statistic was larger for CSF glycine/serine ratio than for CSF glycine alone, although the confidence intervals overlapped.
Controls, attenuated, and severe NKH neonates were combined in an ANOVA analysis (Figure 3). CSF glycine showed significant differences (p < 0.001, F = 86.4) with post hoc analysis showing significant differences between all three classes (p < 0.001 for each comparison). The CSF serine showed significant difference (p = 0.007, F = 5.3) with post hoc analysis showing a significant difference between controls and severe (p = 0.007), but not with the attenuated patients (p = 0.13). Similarly, CSF threonine showed significant differences (p = 0.003, F = 6.5) with post hoc analysis showing significant difference between controls and severe (p = 0.002), but not with the attenuated patients (p = 0.20). Similarly, significant differences were present for the CSF glycine/serine ratio (p < 0.001, F = 107.7), with significant differences between all three classes (p < 0.001 for each comparison). The CSF serine/threonine ratio showed significant differences (p < 0.001, F = 281.3) with post hoc analysis showing significant differences between controls and both severe and attenuated NKH (p < 0.001 for each comparison), but not between severe and attenuated classes (p = 0.86). Thus, the strongest distinction, according to the largest F value, between severe and attenuated NKH patients was by the CSF glycine/serine ratio, whereas the CSF serine/threonine ratio showed a strong distinction between controls and affected patients but was in effective at discriminating between severe and attenuated patients (Figure 3).
FIGURE 3.
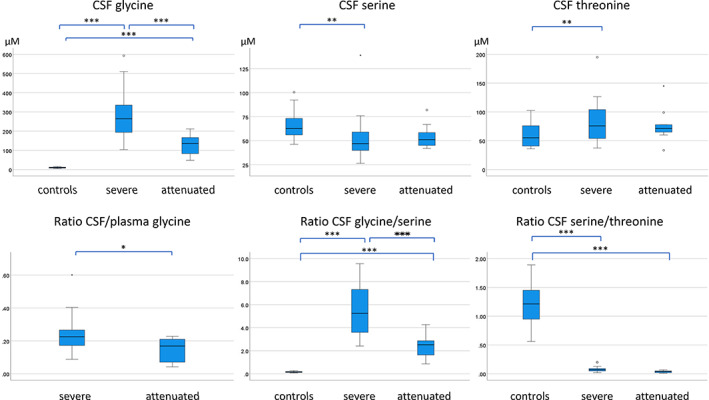
Difference between controls, severe and attenuated patients with nonketotic hyperglycinemia in the neonatal period. In the neonatal period (age < 31 days), the difference between controls, severe nonketotic hyperglycinemia patients, and attenuated nonketotic hyperglycinemia patients is shown for several parameters with differences evaluated by ANOVA analysis. CSF levels of glycine, serine and threonine (μM) are shown, and the ratios of CSF glycine/CSF serine and of CSF serine/CSF threonine. The ratio of CSF/plasma glycine is shown for nonketotic hyperglycinemia patients only. Pairwise comparisons on post hoc Tukey analysis: ***p < 0.001, **p < 0.01, *p < 0.05. ANOVA, analysis of variance; CSF, cerebrospinal fluid
3.5. All ages
In the neonatal period, there were insufficient numbers of each of the attenuated outcome subclasses to allow a comparison by severity, and this analysis required the full age range. An ANOVA analysis showed significant differences between the outcome classes for CSF glycine, CSF/plasma glycine ratio, CSF serine, CSF threonine, and particularly for CSF glycine/serine ratio (Supporting Information Results, Table S3 and Figure S1). However, there was also an age‐related difference (p < 0.001 with attenuated good having a significantly older age). Furthermore, moderate correlations were found between NKH class and: CSF glycine (Spearman ρ = −0.781), the CSF glycine/serine ratio (ρ = −0.724), and the CSF/plasma glycine ratio (ρ = −0.682) (each p < 0.001). Fair correlations were found between NKH class: and CSF threonine (ρ = −0.546, p < 0.001) and CSF serine (ρ = −0.321, p = 0.02).
Because of the impact of age on NKH class, and the age‐related decline in CSF serine and threonine, these values were transformed into Z‐values based on values derived from modeling of the control CSF values. Comparing all controls (n = 257) with all patients, the CSF serine z‐score was significantly lower in patients z = −0.98 ± 2.0 (n = 52) compared to controls z = 0.009 ± 1.02 (p < 0.001). The CSF threonine z‐score was higher in patients z = 0.62 ± 1.34 (n = 51) compared to controls z = 0.004 ± 1.11 (p = 0.004). Comparing severe with attenuated NKH patients, the serine z‐score and the threonine z‐score were not statistically different (p = 0.15 and p = 0.44 respectively), nor was the serine z‐score/threonine z‐score (p = 0.91) (Table S4). Only the CSF glycine/serine z‐score remained significant (p < 0.001). On a comparison across all classes, the CSF glycine (and particularly after log transformation) provided the best discrimination between the various outcome classes (Table S5), whereas after using the age‐corrected z‐score, neither serine or threonine showed significant differences. Finally, the difference between attenuated poor and the combination of attenuated intermediate and attenuated good is particularly pertinent to families. Attenuated intermediate/good group had a later CSF sampling, and was best predicted by lower CSF glycine levels (even more significant after log transformation) (Table S6).
3.6. Clinical applications
CSF glycine has been used as the primary biochemical diagnostic marker for NKH, and indeed over all ages, the values in patients (range 27–593 μM) did not overlap with those in controls (range 0.30–17.30 μM). Strikingly, the CSF serine/threonine ratio also provided diagnostic discrimination between patients (range 0.01–0.2) and controls (range 0.46–3.0). The value of this new criterion was exemplified by differentiation of transient NKH patients from classic NKH (see Supporting Information Results, Table S7). For prognosis, the CSF glycine and the CSF glycine/serine can be used with a correct prediction in 62% and 58% of cases, respectively (Supplemental Information F).
3.7. Serine stereoisomers
In brain tissue, serine consists of both stereoisomers l‐serine and d‐serine, which were not separated by the methods used in clinical laboratory amino acid analyses. We evaluated these two isomers using a stereospecific analysis in CSF samples from 29 controls in the first year of life, five neonates with NKH (Patients 648, 679, 680, 681, 683), one 6‐month‐old infant with attenuated poor NKH (Patient 674), and one 5.5‐month‐old infant (Patient 682) treated with benzoate with concomitant relatively low CSF glycine level of 40.8 μM and plasma glycine 213 μM. Patient 648 had an elevation of all CSF amino acids suggesting a likely serum contamination, which can increase the level of l‐serine but should not affect the level of d‐serine, which is absent in serum. All values were normally distributed. In controls, d‐serine showed a strong age‐related decline (Pearson correlation with age r = −0.8, p < 0.001), which for l‐serine was minimal (r = −0.4, p = 0.034). l‐serine was decreased in untreated NKH patients (33.6 ± 5.0 μM mean ± SEM) compared to age‐matched controls (45.1 ± 1.4 μM, two‐sample t‐test p = 0.007) (Figure 4A). Young infants age ≤2 months with untreated NKH had reduced d‐serine (7.9 ± 1.5 μM, n = 5) versus controls (18.9 ± 1.1 μM, n = 8, p < 0.001) (Figure 4B). Furthermore, the ratio of d‐serine/l‐serine and of d‐serine/total serine was reduced in all untreated patients (age ≤2 months: 0.17 ± 0.02, 14 ± 1%, respectively) compared to age‐matched controls (0.37 ± 0.02, 27 ± 1%, respectively, p < 0.001 for both), indicating an additional lowering effect on d‐serine (Figure 4C,D). Interestingly, the one patient treated with benzoate showed a low normal d‐serine and d‐serine/l‐serine ratio (Figure 4, red diamond).
FIGURE 4.
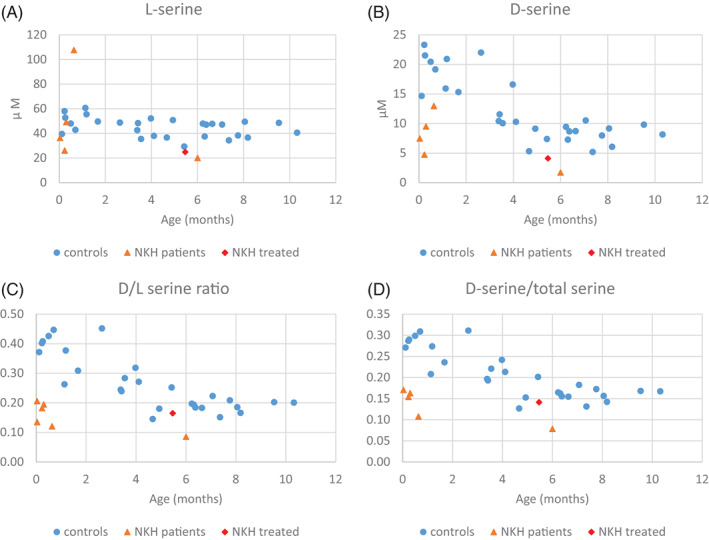
Serine enantiomers comparing patients with nonketotic hyperglycinemia with controls in the first year of life. The levels of l‐serine (A) and d‐serine (B) as a function of age are shown for controls as blue dots and for patients with untreated nonketotic hyperglycinemia as orange triangles and the benzoate‐treated patient as a red diamond. This shows a decrease of both l‐serine and d‐serine in nonketotic hyperglycinemia patients. The ratio of d‐serine/l‐serine (C) and of d‐serine/total serine are shown for controls and nonketotic hyperglycinemia patients. Nonketotic hyperglycinemia patients show a greater decrease in d‐serine than in l‐serine
The SHMT enzymes use 5,10‐methylene‐THF as a cofactor, which is only present in tissue, whereas CSF only contains 5‐methyl‐THF. Retrieval from the clinical charts of CSF 5,10‐methyl‐THF levels measured in a clinical laboratory (Medical Neurogenetics) showed in three cases normal levels of 73, 209, and 65 nM compared to the normal range 40–240 nM.
4. DISCUSSION
Confirming previous observations, NKH patients show elevated levels of glycine in plasma and CSF with an increased CSF/plasma glycine ratio. These measures were more increased in severe NKH than in attenuated NKH patients. 4 , 6 This reflects accumulation of the substrate glycine due to the deficient GCE activity. This was accompanied by significant changes in two other neutral amino acids threonine and serine.
CSF threonine was also elevated in NKH patients and had moderate correlation with CSF glycine. It provided a good distinction between affected patients and controls, but not between severe and attenuated NKH. The reason for this increase is not clear. In humans, no metabolic pathway connects threonine metabolism to glycine, and the threonine dehydrogenase enzyme is an inactive pseudogene, 23 , 24 although the GCE and the threonine dehydratase enzyme are both pyridoxal‐phosphate‐containing enzymes. Whereas the transport into mitochondria does not appear to be shared amongst these amino acids, multiple small neutral amino acid carriers such as ATB0+, SNAT1, SNAT2, SNAT5, and LAT2 provide shared transport over the plasma membrane between threonine and glycine. These carriers have broad substrate specificity over small neutral amino acids and contribute to glial glycine transport. 26 Interestingly, the ASCT1 transporter (gene SLC1A4) is primarily involved in the transfer of small neutral amino acids from astrocytes to neurons and transports both serine and threonine as well as glycine.
Since these small neutral amino acid carriers also transport serine, an increase in serine would also be expected. Furthermore, an increase in the substrate glycine for the equilibrium reaction catalyzed by SHMT enzymes would be expected to increase the formation of serine. Surprisingly, this is not what was observed. Rather, a significant decrease in CSF serine levels was documented in NKH patients. Accurate recognition of this deficiency required taking the substantial age‐related decline in CSF serine values into account. 32 , 33 When the normal range is broadened to the first 6 months of life rather than the month‐by‐month values, low values in the first month of life (where normal serine is 65.8 ± 14 μM) would then be inaccurately reported as middle of the normal range in the sixth month of life (where normal serine is 47.9 ± 8.6 μM). The decrease in serine impacted both l‐serine and d‐serine, which is synthesized from l‐serine via serine racemase. The decrease in d‐serine (25%–69% of average control) was more pronounced than the decrease in l‐serine as reflected in the decreased d‐serine/l‐serine ratio, implying an additional effect on d‐serine beyond l‐serine synthesis. Fair correlations were found between CSF serine or CSF threonine and outcome severity classes, but the best distinction by severity classes was by the CSF glycine level.
Several lines of evidence have previously documented disturbances in serine metabolism in NKH. First, a glycine load in control subjects increases serine levels through the action of SHMT; however, this rise is absent in NKH patients. 34 , 35 , 36 , 37 , 38 In contrast, glycine levels rise in NKH patients following a serine load, similar to controls. 34 , 37 , 38 Second, the rate of conversion of 2‐14C‐glycine into serine was reduced. 38 , 39 , 40 , 41 This indicates that the SHMT reactions are reversible in controls, but in NKH patients only serine can be converted into glycine and not the reverse. It was reported that the serine level in the brain of an NKH patient was 56% of controls. 42 Low levels of d‐serine (38% of controls) in brain cortex were previously documented in three patients with NKH, whereas in that study l‐serine was normal. 22
The biochemistry of glycine and serine is closely related. Mitochondrial complexome analysis shows the coexistence of both the GLDC and the SHMT2 proteins as part of a 175 kDa complex. 43 The GCE provides a substantial contribution to serine synthesis, even in comparison with the well‐known de novo serine biosynthetic pathway starting from 3‐phospho‐glycerate. 14 , 15 , 16 The decrease in serine levels reflects that this synthesis from glycine was not sufficiently effective in NKH. The rapid increase of glycine upon loading with serine indicates that the SHMT protein is present and active, and the inability of the reverse reaction thus implicates insufficient cofactor for serine synthesis, namely, mitochondrial 5,10‐methylene‐THF, which should have been provided by the GCE. Both SHMT2 and GCE are main contributors to mitochondrial one‐carbon charging of folate. 17 Indeed, such a deficiency of one‐carbon charged folates was shown in the NKH mouse. 10 In brain tissue the major sources of 5,10‐methylene‐THF are serine through SHMT2, formate via formyl‐THF synthase and formyl‐THF dehydrogenase, and the GCE (Figure 1). 14 Intramitochondrial 5,10‐methylene‐THF is critically important for several reactions: (1) synthesizing thymidylate from 5,10‐methylene‐THF, 44 (2) using formyl‐THF by methionyl‐tRNA transformylase to make formyl‐methionine‐tRNA which is required for mitochondrial protein synthesis initiation, 17 , 45 , 46 and (3) using 5,10‐methylene‐THF to generate the taurinomethyl‐modification of the wobble uridine base of certain mitochondrial tRNAs such as tRNALeu(UUR), in which absence mitochondrial protein translation stalls. 47 Insufficient mitochondrial one‐carbon charged folates causes severe mitochondrial dysfunction. 45 , 48 Furthermore, decreased methylation of folate vitamers impairs brain growth. The decreased serine levels reflects that this synthesis from glycine with GCE‐derived 5,10‐methylene‐THF is ineffective in NKH patients, and may even reflect additional serine catabolism to glycine via SHMT2 to provide sufficient levels of intramitochondrial 5,10‐methylene‐THF. Cytosolic one‐carbon folates donate to the homocysteine‐methionine cycle, which through S‐adenosylmethionine contributes to multiple methylation reactions and in the nucleus supports purine and pyrimidine synthesis. 17 , 49 Only a mild increase in CSF homocysteine was previously documented in NKH patients, 50 and CSF 5‐methyl‐THF was normal. In mouse studies, decreased levels of both 5,10‐methylene‐THF and 5‐methyl‐THF were described in brain tissue of NKH mice. 11 , 13 Further studies in brain tissue of human patients with NKH would be required to provide direct evidence of the impact on folate vitamers in this condition.
Serine biosynthesis and the GCE are primarily located in the astrocyte, and donate l‐serine to the neuron, called the serine shuttle. 51 , 52 In the neuron, serine racemase catalyzes synthesis of d‐serine. The decrease in the d‐serine/total serine ratio indicates that d‐serine synthesis is affected beyond the decrease in its precursor l‐serine. This could be due to a dysfunction of serine racemase or to a dysfunction in the serine shuttle. Glycine has been shown to competitively inhibit serine racemase in mouse tissue, but the equivalence in humans and applicable concentrations will require further study. 53 , 54 It is tempting to consider that the less reduction in d‐serine in patient 682 related to his glycine lowering treatment with benzoate. Glycine elevation also resulted in release of d‐serine from neurons through the Asc1 transporter. 54 Deficiency of the ASCT1 transporter (SLC1A4), which is the main transporter of d‐serine, as well as for the shuttle of l‐serine to neurons, resulted in decreased d‐serine and l‐serine in mouse brain, and increased threonine, glycine and l‐alanine, a finding surprisingly similar to what we observed in CSF of human NKH patients. 25 Moreover, human patients with pathogenic variants in SCL1A4 (OMIM# 616657) have a phenotype of progressive severe spastic quadriplegia, thin corpus callosum, and developmental delays, which are also core symptoms of severe NKH, not explained by previous hypotheses on pathophysiology. 55 , 56 , 57 , 58 , 59 The exact mechanism for these amino acid changes will require further exploration in human brain samples and in cell experiments.
Both l‐serine and d‐serine have important functions in brain. 14 , 19 , 20 , 60 , 61 d‐serine is, like glycine, a major modulator of NMDA‐receptors with the roles differing by development and brain region. 62 , 63 Glycine is a preferential activator of GluN1/GluN2B receptors and d‐serine a preferential activator of GluN1/GluN2A NMDA receptors. 62 d‐serine is also essential for long term potentiation. 63 In mouse brain, d‐serine increases with age as glycine is reduced probably due to activation of the GCE, and d‐serine is more important as an NMDA coagonist in older mice. 54 , 62 However, we confirm previous findings that in human CSF d‐serine levels decrease with age in the first year, 32 illustrating the need for caution when extrapolating from mouse data to human.
Our study has several clinical implications. Diagnosis of NKH can be made by elevated glycine, but the ratio of CSF serine/threonine also provides diagnostic evidence even without involvement of glycine. This can be used to discriminate classic NKH from transient NKH, where CSF glycine and CSF/plasma glycine ratio are elevated, but we show that the CSF serine/threonine ratio is normal. The CSF glycine level provides the best biochemical discriminator between severity outcome classes, which allows prognostic prediction in up to 60% of patients. These new insights also provide opportunities for new therapeutic avenues beyond glycine reduction, currently employed with benzoate therapy. Addressing one‐carbon charging of folates in mouse models with either formate or methionine has improved fetal brain growth in NKH mouse models. 11 , 13 l‐serine supplementation has been provided in serine deficiency syndromes, and d‐serine has been tried in neurological disorders. 64 , 65 , 66 , 67 , 68 These strategies should be carefully explored as untoward effects are possible. For instance, serine treatment at 200 and 400 mg/kg/day accompanied by an increase in sodium benzoate to accommodate increased glycine formation resulted in reduction of CSF glycine from 122 to 56 μM, and clinically with disappearance of stereotypic movements, convulsions, and myoclonic jerks. 69 However, the patient developed drowsiness evolving into severe apathy, hypotonia, and cessation of spontaneous drinking, followed by episodes of restlessness, crying and tonic convulsions necessitating treatment discontinuation. 64 The unfortunate evolution in this single case should not be a deterrent to novel therapeutic trials, but rather provide a warning that, after thorough studies in animals, clinical trials should be conducted with proper planning and careful management to control for adverse effects. Whether reduction of brain glycine will be sufficient to restore brain serine and threonine levels will be important to be evaluated.
Our study has several limitations. Due to the ultrarare nature of the condition, only a cross‐sectional study was possible, and required collection over a period of more than 15 years. This does not allow a systematic control over all variables of collection and analysis in the same way as a controlled research study, even though we systematically collected measures of blood contamination such as CSF protein levels. It does, however, reflect the data found in clinical practice. We were only able to collect a limited number of CSF samples for the measurements of serine stereoisomers since sampling for research only was not ethically justified and most laboratories do not routinely keep left‐over materials, and, in addition, one sample had apparent serum contamination. Since every sample had the same profile, we believe that the findings can be generalized, but this will benefit from future confirmatory studies.
We conclude that in NKH the elevation of glycine is accompanied by changes in l‐serine, d‐serine, and threonine, indicating a perturbation of the serine shuttle and metabolism and of one‐carbon metabolism. This provides additional guidance on diagnosis and prognosis and opens new therapeutic avenues to be explored.
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
AUTHOR CONTRIBUTIONS
Study concept and design: Johan L. K. Van Hove. Patient care data: Johan L. K. Van Hove, Lina Ghaloul‐Gonzalez, and Juanita Neira‐Fresneda. Control data: Johan L. K. Van Hove, Lisa Schlichting, Cheryl Peck, Linda Gabel, and Marisa W. Friederich. Laboratory studies: Michael A. Swanson. Statistical analysis: Kristen Miller, Suhong Tong, and Johan L. K. Van Hove. First draft writing: Johan L. K. Van Hove, Michael A. Swanson, and Kristen Miller. Critical rewriting: Johan L. K. Van Hove, Kristen Miller, Michael A. Swanson, Sarah P. Young, and Marisa W. Friederich Lina Ghaloul‐Gonzalez. Leadership and funding: Johan L. K. Van Hove. Final responsibility, guarantor, and communicating author: Johan L. K. Van Hove.
ETHICS STATEMENT
The study was performed under IRB‐approved protocols (Colorado COMIRB# 05‐0790, Pittsburgh IRB# STUDY19090211) for nonketotic hyperglycinemia patients and COMIRB# 17‐1830 for controls. Informed consent was obtained from all living subjects. All procedures were done in accordance with the ethical standards of the ethics committee in accordance with the Helsinki Declaration of 1975 and as revised in 2000.
Supporting information
Appendix S1 Supporting Information
Table S2 Age adjusted values for serine and threonine
Legend: Based on the best fit modeling for CSF serine and CSF threonine in control patients as described in methods and results, for each month, the mean, standard deviation (SD) and the 2nd, 5th, 10th, 90th, 95th, and 98th percentile are given. These can be used by clinical laboratories to identify patients with low CSF serine levels or elevated CSF threonine levels for age.
ACKNOWLEDGMENTS
The authors thank the many colleagues who provided clinical information and could not be individually recognized for their contribution. The authors thank Drs S. Wortmann and H. Björneson who provided information on their patients with transient nonketotic hyperglycinemia or ischemic brain damage with increased CSF glycine levels. The authors would like to acknowledge the philanthropic support of the patient organizations that funded this study: NKH Crusaders, Hope for NKH Foundation, Brodyn's Friends, Nora Jane Almany Foundation, the Dickens Family Foundation, Les Petits Bourdons, the Lucas John Foundation, the Jacqueline Kirby Foundation, Exelon Corporation, and the University of Colorado Foundation NKH research fund. Funding sources had no role in the design or execution of the study, the interpretation of data, or the writing of the study.
Swanson MA, Miller K, Young SP, et al. Cerebrospinal fluid amino acids glycine, serine, and threonine in nonketotic hyperglycinemia. J Inherit Metab Dis. 2022;45(4):734‐747. doi: 10.1002/jimd.12500
Communicating Editor: Nenad Blau
Funding information Brodyn's Friends; Dickens Family Foundation; Exelon Corporation; Hope for NKH Foundation; Jacqueline Kirby Foundation; Les Petits Bourdons; Lucas John Foundation; NKH Crusaders; Nora Jane Almany Foundation; University of Colorado Foundation, Grant/Award Number: NKH research fund
DATA AVAILABILITY STATEMENT
My manuscript has data included as electronic supplementary material.
REFERENCES
- 1. Van Hove J, Coughlin CR II, Swanson M, Hennermann JB. Nonketotic hyperglycinemia. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. University of Washington, Seattle; 2002. [PubMed] [Google Scholar]
- 2. Coughlin CR II, Swanson MA, Kronquist K, et al. The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT . Genet Med. 2017;19:104‐111. [DOI] [PubMed] [Google Scholar]
- 3. Hennermann JB, Berger J‐M, Grieben U, Scharer G, Van Hove LJK. Prediction of long‐term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis. 2012;35(2):253‐261. [DOI] [PubMed] [Google Scholar]
- 4. Swanson MA, Coughlin CR II, Scharer GH, et al. Biochemical and molecular predictors of prognosis in nonketotic hyperglycinemia. Ann Neurol. 2015;78:606‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kikuchi G, Motokawa Y, Yoshida T, Hiraga K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc Jpn Acad Ser B Phys Biol Sci. 2008;84:246‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perry TL, Urquhart N, MacLean J, et al. Nonketotic hyperglycinemia—glycine accumulation due to absence of glycine cleavage in the brain. N Engl J Med. 1975;292:1269‐1273. [DOI] [PubMed] [Google Scholar]
- 7. Tada K, Kure S. Non‐ketotic hyperglycinaemia: molecular lesion, diagnosis and pathophysiology. J Inherit Metab Dis. 1993;16:691‐703. [DOI] [PubMed] [Google Scholar]
- 8. Korman SH, Wexler ID, Gutman A, Rolland M‐O, Kanno J, Kure S. Treatment from birth of nonketotic hyperglycinemia due to a novel GLDC mutation. Ann Neurol. 2006;59:411‐415. [DOI] [PubMed] [Google Scholar]
- 9. Bjoraker KJ, Swanson MA, Coughlin CR II, et al. Neurodevelopmental outcome and treatment efficacy of benzoate and dextromethorphan in siblings with attenuated nonketotic hyperglycinemia. J Pediatr. 2016;170:234‐239. [DOI] [PubMed] [Google Scholar]
- 10. Pai YJ, Leung KY, Savery D, et al. Glycine decarboxylase deficiency causes neural tube defects and features of non‐ketotic hyperglycinemia in mice. Nat Comm. 2015;6:6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Leung KY, Pai YJ, Chen Q, et al. Partitioning of one‐carbon units in folate and methionine metabolism is essential for neural tube closure. Cell Rep. 2017;21(7):1795‐1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farris J, Alam MS, Rajashekara AM, Haldar K. Genomic analyses of glycine decarboxylase neurogenic mutations yield a large‐scale prediction model for prenatal disease. PLoS Genet. 2021;17(2):e1009307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leung K‐Y, De Castro SCP, Santos C, et al. Regulation of glycine metabolism by the glycine cleavage system and conjugation pathway in mouse models of non‐ketotic hyperglycinemia. J Inherit Metab Dis. 2020;43(6):1186‐1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalhan SC, Hanson RW. Resurgence of serine: an often neglected but indispensable amino acid. J Biol Chem. 2012;287:19786‐19791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thureen PJ, Narkewicz MR, Battaglya FC, Tjoa S, Fennessey PV. Pathways of serine and glycine metabolism in primary culture of ovine fetal hepatocytes. Pediatr Res. 1995;38:775‐782. [DOI] [PubMed] [Google Scholar]
- 16. Lamers Y, Williamson J, Golbet LR, Stacpoole PW, Gregory JF III. Glycine turnover and decarboxylation rate quantified in healthy men and women using primed constant infusions of [1,2‐13C2]glycine and [2H3]leucine. J Nutr. 2007;137:2647‐2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tibbetts AS, Appling DR. Compartmentalization of mammalian folate‐mediated one‐carbon metabolism. Annu Rev Nutr. 2010;30:57‐81. [DOI] [PubMed] [Google Scholar]
- 18. Davis SR, Stacpoole PW, Williamson J, et al. Tracer‐derived total and folate‐dependent homocysteine remethylation and synthesis rates in humans indicate that serine is the main one‐carbon donor. Am J Physiol Endocrinol Metab. 2004;286:E272‐E279. [DOI] [PubMed] [Google Scholar]
- 19. Fuchs SA, Berger R, de Koning TJ. D‐serine: the right or wrong isoform? Brain Res. 2011;1401:104‐117. [DOI] [PubMed] [Google Scholar]
- 20. Fuchs SA, Dorlans L, de Sain‐van der Velden MG, et al. D‐serine in the developing human central nervous system. Ann Neurol. 2006;60:476‐480. [DOI] [PubMed] [Google Scholar]
- 21. Wolosker H, Mori H. Serine racemase: an unconventional enzyme for an unconventional transmitter. Amino Acids. 2012;43(5):1895‐1904. [DOI] [PubMed] [Google Scholar]
- 22. Iwama H, Takahashi K, Kure S, et al. Depletion of D‐serine in non‐ketotic hyperglycinemia: possible involvement of glycine cleavage system in control of endogenous D‐serine. Biochem Biophys Res Commun. 1997;231:793‐796. [DOI] [PubMed] [Google Scholar]
- 23. Kim D, Fiske BP, Birsov K, et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520:363‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Edgar AJ. The human L‐threonine 3‐dehydrogenase gene is an expressed peudogene. BMC Genet. 2002;3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaplan E, Zubedat S, Radzishevsky I, et al. ASCT1 (Slc1a4) transporter is a physiologic regulator of brain D‐serine and neurodevelopment. Proc Natl Acad Sci U S A. 2018;115:9628‐9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. López‐Corcuera B, Benito‐Muñoz C, Aragón C. Glycine transporters in glia cells: structural studies. Adv Neurobiol. 2017;16:13‐32. [DOI] [PubMed] [Google Scholar]
- 27. Kory N, Wyant GA, Prakash G, et al. SFXN1 is a mitochondrial serine transporter required for one‐carbon metabolism. Science. 2018;362(6416):eaat9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lunetti P, Damiano F, De Benedetto G, et al. Characterization of human and yeast mitochondrial glycine carriers with implications for heme biosynthesis and anemia. J Biol Chem. 2016;291:19746‐19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. DeVore GR. Computing the Z‐score and centiles for cross‐sectional analysis: a practical approach. J Ultrasound Med. 2017;36:459‐473. [DOI] [PubMed] [Google Scholar]
- 30. Stence NV, Fenton LZ, Levek C, et al. Brain imaging in classic nonketotic hyperglycinemia: quantitative analysis and relation to phenotype. J Inherit Metab Dis. 2019;42:438‐450. [DOI] [PubMed] [Google Scholar]
- 31. Grant SL, Shulman Y, Tibbo P, Hampson DR, Baker GB. Determination of D‐serine and related neuroactive amino acids in human plasma by high‐performance liquid chromatography with fluorescence detection. J Chromatogr B. 2006;844:278‐282. [DOI] [PubMed] [Google Scholar]
- 32. Moat S, Carling R, Nix A, et al. Multicentre age‐related reference intervals for cerebrospinal fluid serine concentrations: implications for the diagnosis and follow‐up of serine biosynthesis disorder. Mol Genet Metab. 2010;101:149‐152. [DOI] [PubMed] [Google Scholar]
- 33. Fuchs SA, de Sain‐van der Velden MGM, de Barse MMJ, et al. Two mass‐spectrometric techniques for quantifying serine enantiomers and glycine in cerebrospinal fluid: potential confounders and age‐dependent ranges. Clin Chem. 2008;54:1443‐1450. [DOI] [PubMed] [Google Scholar]
- 34. Steinman GS, Yudkof M, Berman PH, Blazer‐Yost B, Segal S. Late‐onset nonketotic hyperglycinemia and spinocerebellar degeneration. J Pediatr. 1979;94:907‐911. [DOI] [PubMed] [Google Scholar]
- 35. Cole DE, Meek DC. Juvenile non‐ketotic hyperglycinaemia in three siblings. J Inherit Metab Dis. 1985;8(suppl 2):123‐124. [DOI] [PubMed] [Google Scholar]
- 36. Palmer T, Oberholzer VG. Amino acid loading tests in a patient with non‐ketotic hyperglycinaemia. J Inherit Metab Dis. 1985;8(suppl 2):125‐126. [DOI] [PubMed] [Google Scholar]
- 37. Trauner DA, Page T, Greco C, Sweetman L, Kulovich S, Nyhan WL. Progressive neurodegenerative disorder in a patient with nonketotic hyperglycinemia. J Pediatr. 1981;98:272‐275. [DOI] [PubMed] [Google Scholar]
- 38. Baumgartner R, Ando T, Nyhan WL. Nonketotic hyperglycinemia. J Pediatr. 1969;75:1022‐1030. [DOI] [PubMed] [Google Scholar]
- 39. Ando T, Nyhan WL, Gerritsen T, Gong L, Heiner DC, Bray PF. Metabolism of glycine in the nonketotic form of hyperglycinemia. Pediatr Res. 1968;2:254‐263. [DOI] [PubMed] [Google Scholar]
- 40. Ando T, Nyhyan WL, Bicknell J, Harris R, Stern J. Non‐ketotic hyperglycinaemia in a family with an unusual phenotype. J Inherit Metab Dis. 1978;1:79‐83. [DOI] [PubMed] [Google Scholar]
- 41. Tada K, Corbeel L, Eeckels R, Eggermont E. A block in glycine cleavage reaction as a common mechanism in ketotic and nonketotic hyperglycinemia. Pediatr Res. 1974;8:721‐723. [DOI] [PubMed] [Google Scholar]
- 42. Bachmann C, Mihatsch MJ, Baumgartner RE, et al. Solifluktionsdecken im Schweizerischen Nationalpark und ihre Beziehungen zur postglazialen Landschaftsentwicklung. Helv Paediatr Acta. 1971;26:228‐243. [PubMed] [Google Scholar]
- 43. Wessels HJCT, Vogel RO, Lightowlers RO, et al. Analysis of 953 human proteins from a mitochondrial HEK293 fraction by complexome profiling. PLoS One. 2013;8:e68340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Anderson DD, Quintero CM, Stover PJ. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011;108:15163‐15168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tani H, Ohnisihi S, Shitara H, et al. Mice deficient in the Shmt2 gene have mitochondrial respiration defects and are embryonic lethal. Sci Rep. 2018;8:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Minton DR, Nam M, McLaughlin DJ, et al. Serine catabolism by SHMT2 is required for proper mitochondrial translation initiation and maintenance of formylmethionyl‐tRNAs. Mol Cell. 2018;69:610‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morscher RJ, Ducker GS, Li SH‐J, et al. Mitochondrial translation requires folate‐dependent tRNA methylation. Nature. 2018;554:128‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lucas S, Chen G, Aras S, Wang J. Serine catabolism is essential to maintain mitochondrial respiration in mammalian cells. Life Sci Alliance. 2018;1:e201800036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ducker GS, Rabinowitz JD. One‐carbon metabolism in health and disease. Cell Metab. 2017;25:27‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Van Hove JLK, Lazeyras F, Zeisel SH, et al. One‐methyl group metabolism in non‐ketotic hyperglycinaemia: mildly elevated cerebrospinal fluid homocysteine levels. J Inherit Metab Dis. 1998;21:799‐811. [DOI] [PubMed] [Google Scholar]
- 51. Wolosker H, Radzishevsky I. The serine shuttle between glia and neurons: implications for neurotransmission and neurodegeneration. Biochem Soc Trans. 2013;41:1546‐1550. [DOI] [PubMed] [Google Scholar]
- 52. Wolosker H. Serine racemase and the serine shuttle between neurons an astrocytes. Biochim Biophys Acta. 2011;1814:1558‐1566. [DOI] [PubMed] [Google Scholar]
- 53. Dunlop DS, Neidle A. Regulation of serine racemase activity by amino acids. Mol Brain Res. 2005;113:208‐214. [DOI] [PubMed] [Google Scholar]
- 54. Neame S, Safory H, Radzishevsky I, et al. The NMDA receptor activation by D‐serine and glycine is controlled by an astrocytic Phgdh‐dependent serine shuttle. Proc Natl Acad Sci U S A. 2019;116:20736‐20742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Heimer G, Marek‐Yagel D, Eyal E, et al. SLC1A4 mutations cause a novel disorder of intellectual disability, progressive microcephaly, spasticity and thin corpus callosum. Clin Genet. 2015;88:327‐335. [DOI] [PubMed] [Google Scholar]
- 56. Conroy J, Allen NM, Gorman K, et al. Novel European SLC1A4 variant: infantile spasms and population ancestry analysis. J Hum Genet. 2016;61:761‐764. [DOI] [PubMed] [Google Scholar]
- 57. Srour M, Hamdan FF, Gan‐Or Z, et al. A homozygous mutation in SLC1A4 in siblings with severe intellectual disability. Clin Genet. 2015;88:e1‐e4. [DOI] [PubMed] [Google Scholar]
- 58. Pironti E, Salpietro V, Cucinotta F, et al. A novel SLC1A4 homozygous mutation causing congenital microcephaly, epileptic encephalopathy and spastic tetraparesis: a video‐EEG and tractography—case study. J Neurogenet. 2018;32:316‐321. [DOI] [PubMed] [Google Scholar]
- 59. Abdelrahman HA, Al‐Shamsi A, John A, Ali BR, Al‐Gazali L. A novel SLC1A4 mutation (p.Y191*) causes spastic tetraplegia, think corpus callosum, and progressive microcephaly (SPATCCM) with seizure disorder. Child Neurol Open. 2019;6:2329048X19880647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. De Koning TJ, Snell K, Duran M, Berger R, Poll‐The B‐T, Surtees R. L‐serine in disease and development. Biochem J. 2003;371(Pt 3):653‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hirabayashi Y, Furuya S. Roles of L‐serine and sphingolipid synthesis in brain development and neuronal survival. Prog Lipid Res. 2008;47:188‐203. [DOI] [PubMed] [Google Scholar]
- 62. Le Bail M, Martineau M, Sacchi S, et al. Identity of the NMDA receptor coagonist is synapse specific and developmentally regulated in the hippocampus. Proc Natl Acad Sci U S A. 2014;112:E204‐E213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Henneberger C, Papouin T, Oliet SHR, Rusakov DA. Long term potentiation depends on release of D‐serine from astrocytes. Nature. 2010;463:232‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. De Koning TJ, Duran M, Van Maldergem L, et al. Congenital microcephaly and seizures due to 3‐phosphoglycerate dehydrogenase deficiency: outcome of treatment with amino acids. J Inherit Metab Dis. 2002;25:119‐125. [DOI] [PubMed] [Google Scholar]
- 65. El‐Hattab AW. Serine biosynthesis and transport defects. Mol Genet Metab. 2016;118:153‐159. [DOI] [PubMed] [Google Scholar]
- 66. Tsai G, Yang P, Chung LC, Lange J, Coyle JT. D‐serine added to antipsychotics for the treatment of schizophrenia. Biol Psychiatry. 1998;44:1081‐1089. [DOI] [PubMed] [Google Scholar]
- 67. Kantrowitz JT, Epstein ML, Lee M, et al. Improvement in mismatch negativity generation during d‐serine treatment in schizophrenia: correlation with symptoms. Schizophr Res. 2018;191:70‐79. [DOI] [PubMed] [Google Scholar]
- 68. Guercio GD, Panizzutti R. Potential and challenges for the clinical use of d‐serine as a cognitive enhancer. Front Psych. 2018;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wijburg FA, de Groot CJ, Schutgens RBH, Barth PG, Tada K. Clinical effects of serine medication in non‐ketotic hyperglycinaemia due to deficiency of P‐protein of the glycine cleavage complex. J Inherit Metab Dis. 1988;11(suppl 2):218‐220. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supporting Information
Table S2 Age adjusted values for serine and threonine
Legend: Based on the best fit modeling for CSF serine and CSF threonine in control patients as described in methods and results, for each month, the mean, standard deviation (SD) and the 2nd, 5th, 10th, 90th, 95th, and 98th percentile are given. These can be used by clinical laboratories to identify patients with low CSF serine levels or elevated CSF threonine levels for age.
Data Availability Statement
My manuscript has data included as electronic supplementary material.