

Summary
Richter syndrome (RS) is mostly due to the direct transformation of the chronic lymphocytic leukaemia (CLL) clone, as documented by the same immunoglobulin heavy‐chain variable region (IGHV) rearrangement in both CLL and RS cells. In rare cases characterized by a better outcome, the RS clone harbours a different IGHV rearrangement compared to the CLL phase. We investigated the CLL phase of clonally unrelated RS to test whether the RS clone was already identifiable prior to clinicopathologic transformation, albeit undetectable by conventional approaches. CLL cells of eight patients with unrelated RS were subjected to an ultra‐deep next‐generation sequencing (NGS) approach with a sensitivity of 10−6. In 7/8 cases, the RS rearrangement was not identified in the CLL phase. In one case, the RS clone was identified at a very low frequency in the CLL phase, conceivably due to the concomitance of CLL sampling and RS diagnosis. Targeted resequencing revealed that clonally unrelated RS carries genetic lesions primarily affecting the TP53, MYC, ATM and NOTCH1 genes. Conversely, mutations frequently involved in de novo diffuse large B‐cell lymphoma (DLBCL) without a history of CLL were absent. These results suggest that clonally unrelated RS is a truly de novo lymphoma with a mutational profile reminiscent, at least in part, of clonally related RS.
Keywords: CAPP‐Seq, disease dissemination, NGS IGHV analysis, Richter syndrome
INTRODUCTION
Richter syndrome (RS) is defined by the occurrence of an aggressive lymphoma in patients with a previous or concomitant diagnosis of chronic lymphocytic leukaemia (CLL) or small lymphocytic lymphoma (SLL). 1 , 2 RS is a rare, but frequently lethal, disease with an incidence ranging from 2% to 10% among CLL patients. A recent report analysing patients enrolled in first‐line prospective clinical trials of the German CLL study group (GCCLSG) showed an incidence of RS of 3.6%. 3
Analysis of the immunoglobulin heavy‐chain gene (IGHV) rearrangement allows assessment of the clonal relationship between the RS and the CLL phase of the same patient and identifies two distinct RS types, namely clonally related RS (80% of the cases) and the less frequent clonally unrelated RS (20% of the cases). 1 , 4 , 5 From the biological standpoint, clonally related RS derives from the direct transformation of the CLL clone into an aggressive lymphoma due to the acquisition of detrimental genetic lesions. 6 , 7 Conversely, clonally unrelated RS is considered a de novo lymphoma arising from a different clone and conceivably favoured by the intrinsic higher incidence of secondary tumours in CLL patients. 8
From a clinical standpoint, patients with unrelated RS are characterized by a better prognosis than clonally related RS. 6 , 7 Consistently, patients with clonally related RS are treated with intensive chemotherapy and, if suitable, are candidate for allogeneic haematopoietic stem cell transplantation (HSCT), whereas clonally unrelated RS patients are managed as de novo lymphomas without the need of consolidation by transplant. 1 , 9 Whereas the molecular histogenesis and pathogenesis of clonally related RS has been investigated in detail, the biological knowledge regarding clonally unrelated RS is scant.
Till now, the clonal relationship of RS has been investigated by DNA Sanger sequencing that allows the identification of the major IGHV clone but, due to the sensitivity limits of the technique, is not able to identify smaller IGHV subclones. 10 Conversely, IGHV mutational analysis by next‐generation sequencing (NGS) is able to overcome this technical hurdle and is able to identify small IGHV subclones in up to 20%–25% of cases in CLL. 11 On these bases, we used an ultra‐deep NGS approach to investigate the CLL phase of patients who developed clonally unrelated RS to test whether the RS clone was already present, albeit undetectable by conventional DNA sequencing, at the time of CLL prior to clinicopathologic transformation. In parallel, to clarify the molecular pathogenesis of clonally unrelated RS, we used a targeted resequencing NGS approach to investigate the mutational profile of cancer‐related genes involved in this rare, but clinically relevant, RS variant.
PATIENTS AND METHODS
Patients and samples
This study included paired CLL and RS tumour samples from eight CLL patients who transformed into clonally unrelated RS and for whom biological material was longitudinally available. Four patients with clonally related RS were also included for comparative purposes. Diagnosis of RS was assessed by two experienced pathologists (Annalisa Andorno and Renzo Boldorini). Tumour genomic DNA (gDNA) from both the CLL and the RS clone was retrospectively analysed. Patients provided informed consent in accordance with institutional review board requirements and the Declaration of Helsinki. The study was approved by the Ethical Committee of the Ospedale Maggiore della Carità di Novara associated with the Università del Piemonte Orientale (study number CE 67/14 and CE 120/19).
Peripheral blood was collected during the CLL phase, mononuclear cells were isolated by centrifugation using the Ficoll density gradient and genomic DNA (gDNA) from the CLL clone was extracted using the ‘salting out’ protocol. 12 The diagnosis of RS was based on the histology of lymph node or extra‐nodal tissue excisional biopsies according to the World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues. 2 In RS samples, tumour gDNA was extracted from formalin‐fixed paraffin‐embedded (FFPE) tissue specimens using the NucleoSpin kit (Macherey‐Nagel) or from fresh tissues, using the ‘salting out’ protocol.
Immunoglobulin gene rearrangement analysis
To test the IGHV‐D‐J (IGH variable‐delta‐joining region) gene rearrangement harboured by CLL and RS samples and to investigate the clonal relationship between the two phases of the disease, the gDNA from the CLL phase and the gDNA from the RS sample were amplified by a polymerase chain reaction (PCR) with family‐specific primers that hybridize to sequences in the IGHV leader region in conjunction with the IGHJ primers. PCR products were then subjected to bi‐directional Sanger sequencing using the BigDye®Terminator v1.1 Cycle Sequencing kit (Applied Biosystems) and subjected to capillary electrophoresis with the automated ABI PRISM 3130XL Genetic Analyser (Applied Biosystems) sequencer. Following the European Research Initiative on CLL (ERIC) guidelines for IGHV mutational analysis, 10 the nucleotide sequences obtained were aligned using the IMGT/V‐QUEST tool and the ARResT/AssignSubsets tool. The clonal relationship between the CLL phase and the RS phase was established by comparing IGHV‐D‐J and VH CDR3 (complementarity‐determining region 3) sequences.
To test whether the RS clone was already present in the CLL phase at low frequency also in cases of clonally unrelated RS, 2 μg of gDNA of the CLL clone was subjected to ultra‐deep NGS analysis to obtain a sensitivity of 10−6. The IGHV rearrangement in the CLL phase was also tested in four patients with clonally related RS for comparative purposes. Libraries were prepared using the LymphoTrack® Dx IGH FR3Assay‐MiSeq® Kit (Invivoscribe). The amplicon libraries were quantified by using the Quant‐iT™ PicoGreen dsDNA Assay kit (ThermoFisher Scientific) and the Bioanalyzer 2100 (Agilent Technologies) was used to verify the size of the PCR products. The libraries were then pooled together to obtain a concentration of 4 nM and were subjected to paired‐end sequencing using a 300‐bp paired‐end cycle kit on the MiSeq sequencer (Illumina) platform. The FASTQ files obtained by this approach were then processed into fully analysed data and the immunogenetic sequence of the IGHV region was assessed using the LymphoTack® Dx Data Analysis‐MiSeq v2.0.1 Application. The total read counts for each IGHV rearrangement are reported in supplementary Table S1. The sequences were aligned with the IMGT/V‐QUEST tool to investigate the IGHV‐D‐J gene rearrangement and VH CDR3 sequences.
Mutational analysis by CAPP‐Seq
The LyV4.0 Cancer Personalized Profiling by deep Sequencing Assay 13 was used for the study; it comprised a panel of 124 genes known to be relevant for B‐cell malignancies. A background error‐suppressed approach was used for variant calling. The NGS libraries for gDNA were constructed using the KAPA Library Preparation Kit (Kapa Biosystems) and were sequenced using 300‐bp paired‐end runs on a MiSeq sequencer (Illumina). A robust and previously validated bioinformatics pipeline was used for variant calling. 14 , 15 , 16
RESULTS
Clinical features of patients with clonally unrelated RS
The present study was based on eight patients with clonally unrelated RS for whom DNA was available from both the CLL phase and the RS transformed phase. The median age at the time of RS diagnosis was 65 years, five patients were male and all eight patients presented with lactate dehydrogenase (LDH) above the upper limit of normal with a median LDH of 971 u/L (upper limit of normal 450 u/L). At the time of RS diagnosis, CLL scored as stage Binet A in three patients, as Binet B in two patients and as Binet C in three patients. Six patients received R‐CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone)‐like therapy and two patients were treated with an intensified schedule (Table S2).
Ultra‐deep NGS of IGHV genes reveals that clonally unrelated RS are truly de novo aggressive lymphomas
As a first step, the immunogenetic features of the CLL phase before transformation to RS were analysed. By applying the cut‐off value of 98% sequence identity to the germline counterpart that is conventionally used to distinguish IGHV‐mutated CLL from IGHV‐unmutated cases, the immunogenetic analysis of the CLL phase documented that six out of eight cases had a CLL carrying unmutated IGHV genes. In particular, three out of eight cases harboured the IGHV 1–69 rearrangement and one case (ID2) was assigned to the B‐cell receptor stereotyped subset #1.
We then proceeded to analyse the immunogenetic features of the RS phase of each of the eight patients. The IGHV gene rearrangements harboured by tumour cells, their similarity to the germline IGHV sequence and the CDR3 utilized by each case of clonally unrelated RS are reported in Table 1. Three patients harboured unmutated IGHV genes and five patients carried mutated IGHV rearrangements in the lymph node biopsy. Two RS cases utilized the IGHV 1–69 gene, but none of the eight cases of clonally unrelated RS was assigned to a specific stereotyped subset.
TABLE 1.
Immunoglobulin gene rearrangements identified in the CLL clone and in the RS clone of patients who developed clonally unrelated RS
| ID sample | CLL | RS | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IGHV | IGHD | IGHJ | Identity % | CDR3 | IGHV | IGHD | IGHJ | Identity % | CDR3 | |
| ID1 | 1–69*01 or 1–69*12 | 3–16*02 | 6*02 | 100 | CASKGVDDYIWGSYRYTDYYYYYGMDVW | 1–69*02 | 3–3*01 | 6*02 | 100 | CAREEGLTIFGVVGYYYYGMDVW |
| 3–3*01 | 6*02 | 100 | CAREEGLTIFGVVGYYYYGMDVW | |||||||
| ID2 | 1–3*01 | 6–19*01 | 4*02 | 100 | CAFEQWLMIPAFDYW | 1–69*01 or 1–69*12 | 3–3*01 | 6*02 | 100 | CASPTMYDFWSGYSYYWYGMDVW |
| ID3 | 4–31*03 or *04 | 3–3*01 | 6*03 | 100 | CARGVYYDFWSGWYKPYMYYMDVW | 1–8*01 | 4–17*01 | 4*03 | 95.83 | CTDELRRFDWW |
| ID4 | 1–69*01 | 1–7*01 | 6*02 | 99.65 | CAKTPPLWNSPPHYYYYYGMDVW | 3–30*03 or *18 or 3–30‐5*01 | 2–2*01 | 4*02 | 92 | CAKTSCDSINCYIPFDYW |
| ID5 | 1–02*02 or 1–02*05 | 3–9*01 | 4*02 | 92.36 | CARSSEPPRYYDSWSGHTAAW | 1–02*02 or 1–02*05 | 3–9*01 | 4*02 | 92.36 | CARSSEPPRYYDSWSGHTAAW |
| 3–21*01 | 3–22*01 | 3*02 | 87.15 | CTRGPLAYESDGFDMW | ||||||
| ID6 | 1–69*01 | 4–17*01 | 6*03 | 100 | CAGISKVGDLVDYGDRETYYYYMDVW | 4–59*01 | 6–6*01 | 4*02 | 92.98 | CARVRGRQLASDYW / CARVRGRHLASDYW |
| ID7 | 3–23*01 | 3–9*01 | 4*02 | 92.01 | CAKDLEVENKNWLLKLDYW | 3–23*01 | 6–19*01 | 4*02 | 98.96 | CAKDEASGWYDYFDYW |
| ID8 | 6–1*01 | – | 6*02 | 98.32 | CARDFYYGMDVW | 6–1*01 | – | 6*02 | 96.97 | CARDFYYGMDVW |
| 4–34*01 O *02 | 3–9*01 | 6*02 | 96.79 | CARHLKTLRGYPGRYYYYGMDVW | ||||||
Note: Italic font denotes cases in which the IGHV rearrangement was detected only by IGHV analysis by next‐generation sequencing.
Abbreviations: CDR3, complementarity‐determining region 3; CLL, chronic lymphocytic leukaemia; IGHD, immunoglobulin heavy‐chain delta region; IGHJ, immunoglobulin heavy‐chain joining region; IGHV, immunoglobulin heavy‐chain variable region; RS, Richter syndrome.
By conventional Sanger sequencing, the IGHV rearrangement carried by the cases of clonally unrelated RS was undetectable in the corresponding samples of the CLL phase. Because of the low sensitivity of Sanger sequencing, however, this observation could not rule out that traces of the RS clone might be already present before clinicopathologic transformation, albeit undetectable by conventional DNA sequencing.
To test whether traces of the RS clone were already present at the time of the CLL phase prior to clinicopathologic transformation, ultra‐deep NGS analysis with a sensitivity of 10−6 was applied to investigate the presence or absence of RS‐specific IGHV rearrangements in the corresponding CLL phase of each patient of clonally unrelated RS. In seven out of the eight patients, the RS clone was not identifiable in the CLL phase even with a deep sensitivity of 10−6. In only one case (ID1), the RS clone was identified at a very low frequency in the CLL phase, accounting for 0.9% of the total reads of the NGS output. Notably, as opposed to the other seven samples in which the CLL diagnosis and sampling were performed prior to RS diagnosis, in ID1 the diagnosis of RS was concomitant to the diagnosis of CLL.
In cases ID5 and ID8, the lymph node biopsy was affected by both CLL and RS (Figure 1A). As expected, in both these patients IGHV analysis in the lymph node biopsy revealed two IGHV clones, one related to the CLL phase and one related to the RS clone. However, as previously stated, in these cases the IGHV rearrangement specific to the RS phase was not identified in the peripheral blood, thus confirming the clonally unrelated origin of the RS phase. In ID8, who showed both CLL and RS in the same tissue biopsy, the V‐gene of the IGHV rearrangement of the CLL clone had acquired six point mutations and one in‐frame insertion that could be ascribed to the phenomenon of intraclonal diversification.
FIGURE 1.
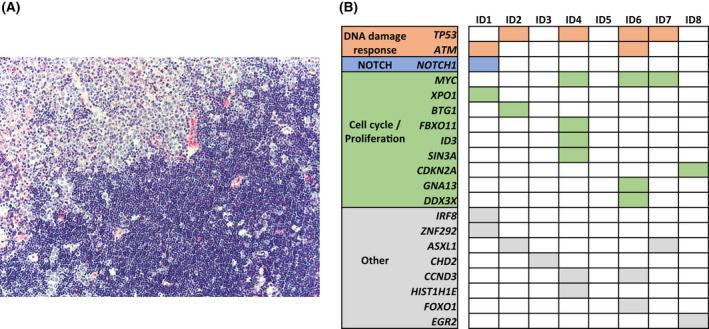
(A) This figure depicts the lymph node biopsy of case ID5 representative of clonally unrelated Richter syndrome (RS) and affected by both chronic lymphocytic leukaemia (CLL) and RS phases. In the top left part of the panel, the physiological lymph node structure is disrupted by the proliferation of large B cells with prominent nucleoli consistent with the RS diffuse large B‐cell lymphoma (DLBCL) variant. In the bottom right part of the panel, the lymph node biopsy is affected by the proliferation of small lymphocytes consistent with CLL infiltration. (B) The heatmap shows the mutational profile of the RS clone in the eight cases of clonally unrelated RS. Rows correspond to the indicated genes and columns represent samples. Genes are coloured‐coded and grouped for pathways.
Ultra‐deep NGS of IGHV genes corroborates the origin of clonally related RS from the CLL clone that is already predominant at the time of diagnosis
To evaluate the presence of additional clones in the peripheral blood of patients with clonally related RS, we analysed for comparative purposes the CLL phase of four clonally related RS patients. Ultra‐deep NGS analysis of the CLL phase did not detect any additional rearrangements in addition to the one related to the CLL clone that transformed into clonally related RS (Table S3). These data validated the notion that clonally related RS stems from the CLL clone that is already predominant at the time of diagnosis. 7
Analysis of the mutational profile of clonally unrelated RS reveals the involvement of genes that regulate DNA damage response and cell cycle
Ultra‐deep NGS analysis confirmed that clonally unrelated RS does not stem from a circulating clone already present at the time of the CLL phase. In order to analyse the mutation profile of this rare variant of RS, all eight lymph node biopsies of clonally unrelated RS were subjected to NGS by targeted resequencing using a panel of 124 cancer‐related genes that are known to be involved in mature B‐cell neoplasia. At least one somatic and non‐synonymous mutation in the genes included in our panel was found in seven out of eight patients (Table S4). Disrupting mutations of the TP53 tumour suppressor gene were present in four (50%) patients. The analysis of the CLL phase of these patients did not identify TP53 mutations, thus demonstrating that this mutation emerged in the tumour cell population of clonally unrelated RS at the time of clinicopathologic transformation. In addition to TP53 disruption, the other mutations identified in clonally unrelated RS affected genes that are known to be also involved in the pathogenesis of clonally related RS, including mutations of NOTCH1, ATM, MYC, CDKN2A and CCND3 (Figure 1B). 7 , 17 , 18 , 19
Notably, mutations of genes that are frequently involved in de novo diffuse large B‐cell lymphoma (DLBCL) without a previous history of CLL, including KMT2D, EZH2, CREEBP, BCL2, PIM1, were not found in this cohort of clonally unrelated RS. 20 , 21
DISCUSSION
In this study, we aimed at characterizing the molecular origin and the pathogenesis of a cohort of the rare, but clinically important, variant of clonally unrelated RS that is known to develop in a fraction of CLL patients, accounting for approximately 20% of all RS cases. Our comprehensive approach by ultra‐deep and highly sensitive NGS unequivocally documents that clonally unrelated RS represent cases of DLBCL that truly arise de novo and may display a different mutational profile compared to that of DLBCL in patients without CLL.
One unresolved question concerning the molecular origin of clonally unrelated RS is whether the DLBCL clone might be already detectable in the CLL phase before transformation and whether the DLBCL cells might derive from one of the subclones that are known to co‐occur in several cases of CLL. 11 Our analysis of IGHV gene rearrangements of clonally unrelated RS documents that, in seven out of eight cases, the RS clone was undetectable in the CLL phase explored at different time points even with the use of an ultra‐deep NGS approach reaching a sensitivity of 10−6. These data clearly define that the overwhelming majority of clonally unrelated RS arise as a de novo DLBCL in the context of a disease, i.e. CLL, that predisposes to the development of secondary tumours, including secondary lymphoid malignancies, because of the profound immune suppression of the host. 22 , 23
Only in one case of our cohort (ID1) the RS clone, that carried a different IGHV rearrangement, was present at very low frequency also in the leukaemic phase. This finding might be explained by two different scenarios. The first scenario is consistent with the inter‐ and intraclonal diversity of CLL, that may favour the selection of a CLL subclone undergoing clonal evolution and Richter transformation. 24 However, is important to note that, in this specific patient, the date of CLL sampling was one week after the lymph node biopsy. Therefore, a second possible scenario is that the surgical procedure may have led to the dissemination of the RS clone into the bloodstream and that the presence of the RS clone at a very low abundance in the CLL phase and detectable only by ultra‐deep NGS analysis may reflect the concomitance of CLL sampling with RS diagnosis, rather than the pre‐existence of a circulating RS clone. Blood samples of this patient before the time of the lymph node biopsy were not available for further characterization.
Since clonally unrelated RS represents only 20% of all RS cases and is a rare disease, the genetic landscape of these tumours has not yet been characterized. Our CAPP‐Seq (cancer personalized profiling by deep sequencing) analysis exploiting a well‐characterized panel of 124 genes involved in mature B‐cell neoplasia provides insights into the mutational profile of clonally unrelated RS. This mutational analysis revealed that genetic lesions primarily affected tumour suppressor genes that are involved in DNA damage response and in the regulation of cell cycle and cell proliferation such as TP53, ATM, NOTCH1, ID3 and MYC. Mutations of genes specific to B‐cell signalling pathways and other mutations that frequently occur in DLBCL patients without a history of CLL, such as alterations of KMT2D, CREBBP, EP300 and TNFAIP3, were not identified in any of these patients. Overall, the mutational profile of the cases of clonally unrelated RS included in this study appears to be reminiscent, at least in part, of that previously described in cases of clonally related RS. 17 The observation that clonally unrelated RS shares several genetic lesions with clonally related RS deserves further investigations and might be potentially ascribed to commonalities in the lymph node microenvironment and degree of immune suppression in both conditions, and/or to previous exposure to anti‐leukaemic therapy that might have favoured the accumulation specific genetic lesions.
The stringent requirements of the ultra‐deep NGS investigations performed in this study, coupled with the rarity of clonally unrelated RS that accounts for only 20% of all RS developing in CLL patients, posed limitations to the number of cases that could be tested. Future investigations on multicentric series of RS are needed to assess whether the CLL microenvironment favours the accumulation and/or selection of some specific genetic lesions that are common to both clonally unrelated and clonally related RS.
The treatment algorithm of clonally unrelated RS differs from that of clonally related RS, especially in young and transplant‐eligible patients. 1 In view of precision medicine, this fact mandates the need for a better understanding of the molecular origin and pathogenesis of clonally unrelated RS, that until now has not been extensively investigated with next‐generation approaches, also due to the disease rarity. This study provides evidence that clonally unrelated RS is represented by a truly de novo lymphoma and that it does not derive from one of the minor subclones detectable in the CLL phase. 11 Besides documenting the truly de novo origin of clonally unrelated RS, our data show a certain degree of commonality between the pathogenetic lesions of clonally unrelated and clonally related RS, including a high frequency of TP53 disruption. Importantly, TP53 disruption predisposes to failure of R‐CHOP‐like chemoimmunotherapy regimens 25 in de novo DLBCL without a history of CLL. 25 The molecular pattern of clonally unrelated RS identified in this study might deserve consideration when designing future treatment algorithms of RS. For clonally related RS, expert opinions propose transplant consolidation after the achievement of first remission in transplant‐eligible patients. 1 , 6 , 19 Conversely, transplant consolidation is not currently indicated for clonally unrelated RS. 1 On these grounds, the high prevalence of TP53 disruption in clonally unrelated RS prompts the need for large collaborative studies for devising a precision medicine approach for this neoplasm.
AUTHOR CONTRIBUTIONS
Riccardo Moia and Gianluca Gaidano designed the study, interpreted data and wrote the manuscript; Chiara Favini and Donatella Talotta, contributed to data interpretation and manuscript preparation; Mohammad Almasri, Silvia Rasi, Ramesh Adhinaveni, Sreekar Kogila, Bassel Awikeh, Mattia Schipani, Paola Boggione, Samir Mouhssine, Joseph Ghanej, Wael Al Essa, Abdurraouf Mokhtar Mahmoud, Riccardo Dondolin and Nariman Alessa performed molecular studies and contributed to manuscript revision; Annalisa Andorno, Gloria Margiotta Casaluci, Valter Gattei and Renzo Boldorini provided study material and contributed to manuscript revision; Annalisa Andorno and Renzo Boldorini performed pathological revision.
FUNDING INFORMATION
Molecular bases of disease dissemination in lymphoid malignancies to optimize curative therapeutic strategies, (5 × 1000 No. 21198), Associazione Italiana per la Ricerca sul Cancro Foundation Milan, Italy; Progetti di Rilevante Interesse Nazionale (PRIN; 2015ZMRFEA), Rome, Italy; the AGING Project – Department of Excellence — DIMET, Università del Piemonte Orientale, Novara, Italy; and Ricerca Finalizzata 2018 (project RF‐2018‐12365790), MoH, Rome, Italy; and Digital Morphology Program, Novara‐AIL Onlus, Novara, Italy.
Supporting information
Tables S1–S4
ACKNOWLEDGEMENT
Open Access Funding provided by Universita degli Studi del Piemonte Orientale Amedeo Avogadro within the CRUI‐CARE Agreement.
Favini C, Talotta D, Almasri M, Andorno A, Rasi S, Adhinaveni R, et al. Clonally unrelated Richter syndrome are truly de novo diffuse large B‐cell lymphomas with a mutational profile reminiscent of clonally related Richter syndrome. Br J Haematol. 2022;198:1016–1022. 10.1111/bjh.18352
Chiara Favini and Donatella Talotta contributed equally to this work.
Gianluca Gaidano and Riccardo Moia contributed equally.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761–72. [DOI] [PubMed] [Google Scholar]
- 2. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Al‐Sawaf O, Robrecht S, Bahlo J, Fink AM, Cramer P, V Tresckow J, et al. Richter transformation in chronic lymphocytic leukemia (CLL)‐a pooled analysis of German CLL study group (GCLLSG) front line treatment trials. Leukemia. 2021;35(1):169–76. [DOI] [PubMed] [Google Scholar]
- 4. Mao Z, Quintanilla‐Martinez L, Raffeld M, Richter M, Krugmann J, Burek C, et al. IgVH mutational status and clonality analysis of Richter's transformation: diffuse large B‐cell lymphoma and Hodgkin lymphoma in association with B‐cell chronic lymphocytic leukemia (B‐CLL) represent 2 different pathways of disease evolution. Am J Surg Pathol. 2007;31(10):1605–14. [DOI] [PubMed] [Google Scholar]
- 5. Moia R, Patriarca A, Deambrogi C, Rasi S, Favini C, Kodipad AA, et al. An update on: molecular genetics of high‐risk chronic lymphocytic leukemia. Expert Rev Hematol. 2020;13(2):109–16. [DOI] [PubMed] [Google Scholar]
- 6. Parikh SA, Rabe KG, Call TG, Zent CS, Habermann TM, Ding W, et al. Diffuse large B‐cell lymphoma (Richter syndrome) in patients with chronic lymphocytic leukaemia (CLL): a cohort study of newly diagnosed patients. Br J Haematol. 2013;162(6):774–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rossi D, Spina V, Deambrogi C, Rasi S, Laurenti L, Stamatopoulos K, et al. The genetics of Richter syndrome reveals disease heterogeneity and predicts survival after transformation. Blood. 2011;117(12):3391–401. [DOI] [PubMed] [Google Scholar]
- 8. Kumar V, Ailawadhi S, Bojanini L, Mehta A, Biswas S, Sher T, et al. Trends in the risk of second primary malignancies among survivors of chronic lymphocytic leukemia. Blood Cancer J. 2019;9(10):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cwynarski K, van Biezen A, de Wreede L, Stilgenbauer S, Bunjes D, Metzner B, et al. Autologous and allogeneic stem‐cell transplantation for transformed chronic lymphocytic leukemia (Richter's syndrome): a retrospective analysis from the chronic lymphocytic leukemia subcommittee of the chronic leukemia working party and lymphoma working party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2012;30(18):2211–7. [DOI] [PubMed] [Google Scholar]
- 10. Rosenquist R, Ghia P, Hadzidimitriou A, Sutton LA, Agathangelidis A, Baliakas P, et al. Immunoglobulin gene sequence analysis in chronic lymphocytic leukemia: updated ERIC recommendations. Leukemia. 2017;31(7):1477–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stamatopoulos K, Agathangelidis A, Rosenquist R, Ghia P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia. 2017;31(2):282–91. [DOI] [PubMed] [Google Scholar]
- 12. Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moia R, Favini C, Ferri V, Forestieri G, Terzi Di Bergamo L, Schipani M, et al. Multiregional sequencing and circulating tumour DNA analysis provide complementary approaches for comprehensive disease profiling of small lymphocytic lymphoma. Br J Haematol. 2021;195(1):108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Diop F, Moia R, Favini C, Spaccarotella E, De Paoli L, Bruscaggin A, et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica. 2020;105(2):448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferrero S, Rossi D, Rinaldi A, Bruscaggin A, Spina V, Eskelund CW, et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high‐dose therapy: a FIL study. Haematologica. 2020;105(6):1604–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fabbri G, Khiabanian H, Holmes AB, Wang J, Messina M, Mullighan CG, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210(11):2273–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chigrinova E, Rinaldi A, Kwee I, Rossi D, Rancoita PM, Strefford JC, et al. Two main genetic pathways lead to the transformation of chronic lymphocytic leukemia to Richter syndrome. Blood. 2013;122(15):2673–82. [DOI] [PubMed] [Google Scholar]
- 19. Condoluci A, Rossi D. Biology and treatment of Richter transformation. Front Oncol. 2022;12:829983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morin RD, Mendez‐Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone‐modifying genes in non‐Hodgkin lymphoma. Nature. 2011;476(7360):298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B‐cell lymphoma. Nat Genet. 2011;43(9):830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Royle JA, Baade PD, Joske D, Girschik J, Fritschi L. Second cancer incidence and cancer mortality among chronic lymphocytic leukaemia patients: a population‐based study. Br J Cancer. 2011;105(7):1076–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schöllkopf C, Rosendahl D, Rostgaard K, Pipper C, Hjalgrim H. Risk of second cancer after chronic lymphocytic leukemia. Int J Cancer. 2007;121(1):151–6. [DOI] [PubMed] [Google Scholar]
- 24. Stamatopoulos B, Timbs A, Bruce D, Smith T, Clifford R, Robbe P, et al. Targeted deep sequencing reveals clinically relevant subclonal IgHV rearrangements in chronic lymphocytic leukemia. Leukemia. 2017;31(4):837–45. [DOI] [PubMed] [Google Scholar]
- 25. Chiappella A, Diop F, Agostinelli C, Novo M, Nassi L, Evangelista A, et al. Prognostic impact of TP53 mutation in newly diagnosed diffuse large B‐cell lymphoma patients treated in the FIL‐DLCL04 trial. Br J Haematol. 2022;196(5):1184–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S4
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.