

Abstract
Guidelines for variant interpretation include criteria for incorporating phenotype evidence, but this evidence is inconsistently applied. Systematic approaches to using phenotype evidence are needed. We developed a method for curating disease phenotypes as highly or moderately predictive of variant pathogenicity based on the frequency of their association with disease‐causing variants. To evaluate this method's accuracy, we retrospectively reviewed variants with clinical classifications that had evolved from uncertain to definitive in genes associated with curated predictive phenotypes. To demonstrate the clinical validity and utility of this approach, we compared variant classifications determined with and without predictive phenotype evidence. The curation method was accurate for 93%–98% of eligible variants. Among variants interpreted using highly predictive phenotype evidence, the percentage classified as pathogenic or likely pathogenic was 80%, compared with 46%–54% had the evidence not been used. Positive results among individuals harboring variants with highly predictive phenotype‐guided interpretations would have been missed in 25%–37% of diagnostic tests and 39%–50% of carrier screens had other approaches to phenotype evidence been used. In summary, predictive phenotype evidence associated with specific curated genes can be systematically incorporated into variant interpretation to reduce uncertainty and increase the clinical utility of genetic testing.
Keywords: curated gene‐disease relationships, diagnostic yield, genetic testing, phenotype, variant interpretation, variants of uncertain significance
1. INTRODUCTION
When evaluating the pathogenicity of DNA variants, clinical information from ordering clinicians can be critical. As clinical genetic testing increasingly involves multi‐gene panels, exomes, and genomes, a great number of variants can be detected. In clinically affected individuals, phenotype data can be instrumental in differentiating causative variants from benign or likely benign (B/LB) ones.
Although the 2015 guidelines for sequence variant interpretation issued by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) clearly delineate the major categories and relative strengths of evidence to be considered when classifying variants, discordance in variant interpretation across clinical genetic testing laboratories has been reported (Amendola et al., 2016; Richards et al., 2015) In particular, the most inconsistently applied criteria category for classifying pathogenic variants (i.e., PP4) involves phenotype evidence: “Patient's phenotype or family history is highly specific for a disease with a single genetic etiology.” (Richards et al., 2015) The inconsistent application of this category may be due partly to the subjective nature of identifying components of a phenotype that are “highly specific” for a disease.
To improve concordance in clinical variant classification among labs, the National Institutes of Health‐funded Clinical Genome Resource (ClinGen) group has convened Variant Curation Expert Panels that rigorously curate gene‐disease relationships and phenotype variability in relation to the ACMG/AMP guidelines (Rivera‐Muñoz et al., 2018). In addition, ClinGen has tasked its Sequence Variant Interpretation Working Group with systematically reviewing ACMG/AMP criteria to evaluate how those criteria can be quantitatively assessed and how gene‐phenotype relationships should be integrated into that assessment. These efforts have been applied to a small subset of genes; but, there is not yet a unifying framework for incorporating the PP4 criterion across all disease genes.
With the goal of improving the consistency and scalability of variant interpretation, the clinical genetic testing lab Invitae iteratively refined the ACMG/AMP criteria by adding greater granularity around each evidence criterion. The result was the previously described ACMG/AMP‐guided variant interpretation framework, Sherloc (Nykamp et al., 2017). To date, Sherloc has undergone more than a dozen revisions and has been used to classify nearly 1 million variants from more than 15,000 genes (National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, 2022). Despite this expansive use, incorporating phenotype information into Sherloc remains challenging because that information can be difficult to accurately quantify and consistently apply. For example, although independent variant classification by two Invitae geneticists showed >95% concordance across 177 variants (internal data), the few discordances between them were almost all attributable to disagreements in the interpretation of phenotype information and its relation to the observed genotypes. In addition, ultra‐rare variants in individuals with well‐defined syndromes and highly specific clinical signs were sometimes classified as variant(s) of uncertain significance (VUS), despite their high likelihood of being pathogenic. This is not a new observation in clinical genetic testing labs, and clinicians have also voiced concerns about the inability of current variant interpretation approaches to systematically account for prior probability of pathogenicity based on phenotype (Cederbaum, 2015).
To increase the consistency and clinical value of using phenotype information in clinical variant interpretation, we developed a quantitative method for curating genes for which certain phenotypes indicate a high prior probability that rare pathogenic variants will be detected in those genes. Here, we describe this novel curation approach and its validation within Sherloc, which assigns predetermined weight to different categories of phenotype evidence during variant interpretation. We also explore the clinical utility of this approach and its impact on reducing the number of VUS.
2. PATIENTS AND METHODS
2.1. Editorial policies and ethical considerations
Study data were de‐identified and approved for analysis by an independent institutional review board (WCG IRB protocol CR‐001‐02, Tracking ID 20161796). All patients provided informed consent (either written or verbal) before genetic screening tests were ordered. Relevant clinically reported variants and de‐identified clinical information were collected for analyses with institutional review board approval. The need for additional consent was waived.
2.2. Introduction to novel approach to phenotype data
In Sherloc, each type of evidence considered in variant interpretation (e.g., population frequency, functional data) is assigned predetermined points, and the points from all types of evidence are tallied to determine a variant's clinical classification (Nykamp et al., 2017). Classification as pathogenic or likely pathogenic (P/LP) requires a minimum of four points accumulated from evidence indicating a pathogenic effect. Classification as B/LB requires a minimum of three points toward a benign effect. Variants that do not reach the thresholds for P/LP or B/LB are categorized as VUS.
To improve Sherloc's handling of phenotype evidence, we first considered that some clinical, morphological, or biochemical traits (i.e., phenotypes) are frequently associated with a specific genetic condition and thus could be considered predictive for that condition. Broadly, we established two categories of predictive phenotype evidence to account for differences in the strength of association between a certain phenotype (or collection of phenotypes) and pathogenic variants in a given gene or set of genes. The first category is highly predictive phenotype evidence, which is nearly always associated with a genetic condition; in this category, a variant in the associated gene(s) is highly likely to explain the clinical phenotype. The second category is moderately predictive phenotype evidence, which is frequently but not always associated with a genetic condition; in this category, a variant in the associated gene(s) has only a moderate chance of explaining the clinical phenotype (Figure 1a).
FIGURE 1.
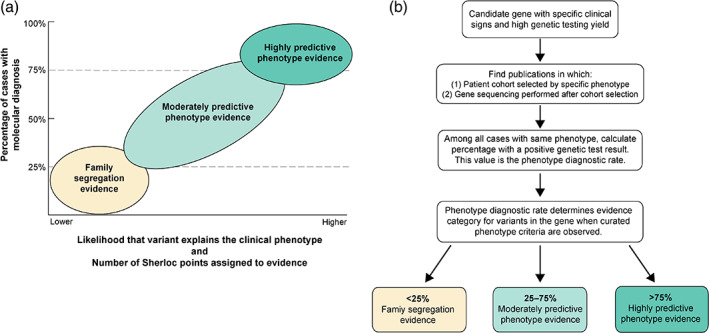
Model and methods for weighting phenotype evidence for variant interpretation. (a) In our model, phenotype evidence was categorized and weighted based on the likelihood that the cause of a condition had a known genetic etiology. When a high percentage of individuals with the same collection of clinical features were shown to have pathogenic variants indicating a molecular diagnosis, the phenotype was considered highly predictive. As shown in the lower left quadrant, the phenotype of an individual alone was less predictive of a known genetic basis, so classification could only rely on observation of the variant segregating in a family of individuals with similar clinical features, a separate type of evidence in the ACMG/AMP guidelines and Sherloc (Richards et al., 2015; Nykamp et al., 2017) (b) To determine the appropriate category of phenotype evidence for gene(s) associated with a condition, we followed a systematic curation workflow. Phenotype includes clinical signs and symptoms, either individually or in combination
2.3. Curation of predictive phenotype data
The systematic approach to using phenotypic evidence in variant interpretation begins with curating genes based on the likelihood that genetic testing of an individual with a specific set of clinical signs and symptoms (i.e., phenotype) will result in a molecular diagnosis in that gene (Figure 1a). To determine the likelihood that a given phenotype or collection of signs and symptoms would be associated with a molecular diagnosis, we reviewed the literature for clinical studies involving genetic testing of individuals who had the phenotype of interest (Figure 1b). The phenotype data and genetic testing results were extracted from the published studies and used to calculate the proportion of individuals with the phenotype of interest who had a molecular diagnosis. We referred to this proportion as the phenotype diagnostic rate (PDR). When the PDR was high (>75% of individuals with a given phenotype were found to have a positive molecular diagnosis or likely positive molecular result), we referred to the phenotype as “highly predictive.” When PDR was moderate (25%–75%), we referred to the phenotype as “moderately predictive.” Weaker gene‐phenotype associations were limited to family segregation evidence only. Additional details are provided in the Supplement, including an example evaluation of PDR in Table S1.
2.4. Predictive phenotype‐guided variant interpretation
We next integrated predictive phenotype evidence into Sherloc by establishing rules for applying predetermined numbers of points during variant interpretation. Predictive phenotype evidence was considered for VUS or LP variants when (1) the variant was in a gene with curated phenotype criteria and (2) sufficient clinical phenotype data were provided by the ordering clinician or had been described in the literature to be associated with that variant.
If the individual's phenotype or the published phenotype met highly predictive phenotype criteria, and all of the genes included in the PDR calculation for the suspected condition were sequenced, 1, 1.5, or 2 points toward pathogenicity were awarded depending on the inheritance pattern of the condition (autosomal dominant, X‐linked dominant, X‐linked recessive, or autosomal recessive), the zygosity of the variant (hemizygous, heterozygous, or homozygous), and the detection of additional variants (VUS or P/LP) in the same gene (Table 1). For genes associated with autosomal recessive inheritance, two heterozygous variants or one homozygous variant had to be present to apply highly predictive phenotype evidence, while one variant was sufficient for genes associated with dominant inheritance. Because two variants are less likely than one variant to arise by chance, a newly observed variant in an autosomal recessive gene was considered more likely to be the cause of an individual's condition than a variant in a gene associated with autosomal dominant inheritance. Therefore, variants in autosomal recessive genes were awarded more points toward pathogenicity for highly predictive phenotype evidence than variants in autosomal dominant genes if one of the variants was already classified as P/LP or if a variant was homozygous (Table 1).
TABLE 1.
Sherloc evidence codes and points awarded for variants observed in individuals who meet highly or moderately predictive phenotype criteria
| Predictive category | Sherloc evidence code | Evidence description | Pathogenic points awarded in Sherloc | Corresponding ACMG criteria category |
|---|---|---|---|---|
| Moderate | EV0228 | Variant previously identified in one individual with moderately predictive phenotype evidence | 0 | None |
| Moderate | EV0081 | Variant previously identified in two individuals with moderately predictive phenotype evidence | 1 | PS4 |
| Moderate | EV0080 | Variant previously identified in three individuals with moderately predictive phenotype evidence | 2 | PS4 |
| Moderate | EV0079 | Variant previously identified in four individuals with moderately predictive phenotype evidence | 3 | PS4 |
| High | EV0169 | In an AD gene, a rare heterozygous or hemizygous variant in one individual with highly predictive phenotype evidence | 2 | PP4 |
| High | EV0155 | In an AR gene, a rare heterozygous variant co‐occurring with heterozygous VUS in one individual with highly predictive phenotype evidence | 1 | PP4 |
| High | EV0154 | In an AR gene, a rare heterozygous variant co‐occurring with P/LP variant in same gene in one individual with highly predictive phenotype evidence | 1.5 | PP4 |
| High | EV0153 | In an AR gene, a rare homozygous variant in one individual with highly predictive phenotype evidence | 2 | PP4 |
Note: AD gene, a gene associated with a condition with autosomal dominant inheritance. AR gene, a gene associated with a condition with autosomal recessive inheritance.
For individuals who had a phenotype that met only moderately predictive criteria, we awarded 0, 1, 2, or 3 pathogenic evidence points depending on the number of previously observed individuals (either observed internally or published in the literature) whose variants met the curated phenotype criteria. Thus, each observed variant in an individual with highly predictive phenotype evidence was awarded twice the number of points as a variant in an individual with moderately predictive phenotype evidence. If neither highly nor moderately predictive phenotype criteria were curated for the gene, or the individual's phenotype as reported by the referring physician was insufficient to meet highly or moderately predictive phenotype criteria, pathogenic points were awarded only in the presence of family segregation data.
As an example of how this approach works, in studies of primary ciliary dyskinesia (PCD) that included clinical cohorts with at least two of four clinical features of PCD (i.e., unexplained neonatal respiratory distress in a term infant, year‐round daily cough beginning before 6 months of age, year‐round daily nasal congestion before 6 months of age, or organ laterality defect), the PDR was >75% when at least 26 known PCD genes were tested (Kim et al., 2014; Leigh et al., 2016; Marshall et al., 2015; Shapiro et al., 2018). Therefore, in our genetic testing context, if individuals had only one clinical feature of PCD or if fewer than these 26 genes were tested, highly predictive phenotype evidence would not be applied during variant interpretation. Instead, moderately predictive phenotype evidence would be applied.
2.5. Study cohort and genetic testing
We retrospectively reviewed DNA sequencing results from unrelated individuals who received diagnostic full‐gene sequencing and exonic deletion/duplication analyses between March 8, 2016 and October 7, 2019, for at least 1 of 187 genes curated for highly predictive phenotype criteria. Although phenotype data is not collected for individuals undergoing reproductive carrier screening, phenotype‐guided variant interpretation may inform carrier screening results when the variant identified has been previously interpreted through diagnostic testing. We therefore also reviewed results from unrelated individuals who received carrier screening in the same time period and had a positive result involving a variant previously interpreted with highly predictive phenotype evidence. Sequencing was performed as previously described with an average of 350× and a minimum of 50× depth of sequencing coverage (Lincoln et al., 2015; Truty et al., 2019).
2.6. Concordance between predictive phenotype criteria and current variant classifications
Given that predictive phenotype evidence was only counted toward pathogenicity (never toward a benign classification), we were able to measure the concordance of the predictive phenotype criteria by reviewing variant classification changes over time. We first identified variants originally classified as VUS as the result of variant interpretation that included predictive phenotype evidence (“historical VUS”). We then reviewed internal records for any subsequent reclassification of these VUS to P/LP or B/LB when new evidence (e.g., experimental findings or an independent clinical observation) became available. Reclassification of the historical VUS to P/LP indicated concordance of the criteria and reclassification to B/LB indicated discordance (Figure 2). The concordance of the predictive phenotype criteria was finally calculated as the proportion of concordant variants over the total number of historical VUS.
FIGURE 2.
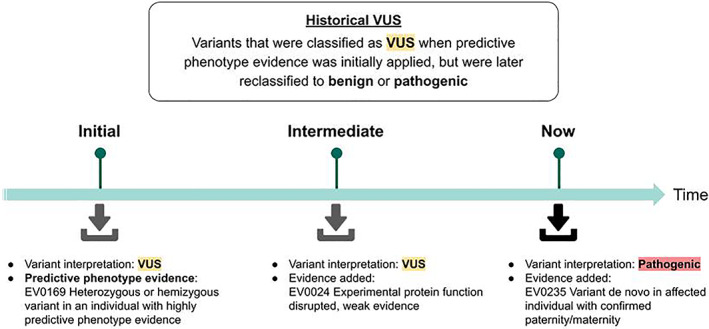
Evaluating predictive phenotype criteria concordance via a prospective performance approach. To start (“Initial”), the variant is identified in a patient with clinical features meeting curated highly predictive phenotype criteria. Highly predictive phenotype evidence is then used during clinical interpretation along with all other available lines of evidence, and the variant is originally classified as VUS. Over time ("Intermediate"), additional evidence is applied (e.g., new studies published in the literature reveal additional functional evidence), but the variant remains classified as a VUS. Finally ("Now"), additional evidence can be applied (e.g., the variant is reported in a new case study as de novo in an affected individual). The aggregate of the evidence now supports classification as pathogenic. This same methodology was used to examine the concordance of moderately predictive phenotype evidence
2.7. Utility of predictive phenotype evidence
To measure the utility of highly predictive phenotype evidence for variant classification, we first identified all variants for which highly predictive phenotype evidence had contributed to their clinical classifications via Sherloc. Using the collective recorded evidence for these variants, we then simulated their classifications using two other variant interpretation frameworks: (1) a version of Sherloc in which all highly predictive phenotype evidence was weighted as moderately predictive phenotype evidence (“Sherloc‐restricted” framework) and (2) the ACMG/AMP 2015 guidelines (“baseline ACMG/AMP” framework; Richards et al., 2015). Among the full set of variants, the proportions of P, LP, and VUS classifications from the three frameworks (i.e., Sherloc, Sherloc‐restricted, and baseline ACMG/AMP) were compared. To ensure that Sherloc and the Sherloc‐restricted framework could be equivalently compared with the ACMG/AMP framework, Sherloc, and Sherloc‐restricted interpretations were produced solely with evidence points, excluding the adjustments by expert review that are normally used in clinical practice. Additional details including an example of classifications resulting from the three interpretation frameworks (Table S2) are provided in the Supplement.
To assess the clinical impact of highly predictive phenotype evidence in both diagnostic testing and carrier screening, we first identified unrelated individuals who had at least one variant classified as P, LP, or VUS with highly predictive phenotype evidence (Sherloc) and then simulated results as if the variants had been interpreted within the Sherloc‐restricted and baseline ACMG/AMP frameworks. Within all three frameworks, diagnostic testing results were defined as (1) Positive, when P/LP variants were identified in a gene in which the zygosity of the variants was consistent with the established disease inheritance pattern; (2) Likely Positive, when one heterozygous P/LP variant and one heterozygous VUS was identified in the same gene for an autosomal recessive condition; (3) Carrier, when a heterozygous P/LP variant was identified in a gene associated with an autosomal recessive condition (or X‐linked condition in female individuals); and (4) Uncertain, when one or more VUS were identified in the absence of any P/LP variant. For example, in a male patient, a variant in ABCD1, which is associated with X‐linked adrenoleukodystrophy (MIM: 300100), would have a Positive result if classified as P within Sherloc but an Uncertain result if classified as a VUS within the Sherloc‐restricted framework. Carrier screening results were defined as Positive when a P/LP variant was identified and Negative when no P/LP variant was identified.
3. RESULTS
3.1. Accuracy of preemptive curation of genes for predictive phenotype evidence
We identified 890 unique variants that were historically classified as VUS when highly predictive phenotype evidence was applied during clinical interpretation of variants identified during diagnostic testing (Table 2). Of these, 275 were later reclassified; 270 (98.2%) were reclassified to P/LP. Only five of the historical VUS with highly predictive phenotype evidence were reclassified to B/LB after the addition of other clinical evidence; these VUS were found in SCN1A (n = 3), DNAH5 (n = 1), and RYR1 (n = 1). We also identified 11,942 unique variants originally classified as VUS when moderately predictive phenotype evidence was applied during clinical interpretation. Of these, 1808 were eventually reclassified: 1697 (93.9%) were reclassified to P/LP and 111 (6.1%) were reclassified to B/LB (Table 2).
TABLE 2.
Concordance of highly and moderately predictive criteria
| Highly predictive phenotype evidence | Moderately predictive phenotype evidence | |
|---|---|---|
| No. of variants classified as VUS when predictive phenotype evidence originally applied (historical VUS) | 890 | 11,942 |
| No. of historical VUS reclassified to P/LP | 270 | 1697 |
| No. of historical VUS reclassified to B/LB | 5 | 111 |
| Concordance | 98.2% | 93.9% |
3.2. Impact of predictive phenotype evidence on variant interpretation
The 187 genes curated for highly predictive phenotype criteria and included in this study were largely associated with inherited metabolic disorders (Table S3). Within the 187 genes, clinical sequencing identified 18,281 VUS and P/LP variants, of which 1505 (8%) had highly predictive phenotype evidence applied during the interpretation process. The remaining variants (i.e., the majority) were not eligible for highly predictive phenotype evidence criteria in Sherloc due to one of the following reasons: (1) available clinical information was lacking for the individual tested and any individuals reported in the literature, (2) available clinical information did not meet the highly predictive phenotype criteria, (3) the genotype and/or variant zygosity did not match the inheritance pattern requirements for the gene, (4) the variant had not been re‐evaluated since highly predictive phenotype criteria had been approved for the gene, or (5) the variant was already classified as P based on other lines of evidence and therefore did not require further evaluation.
We compared variant interpretation outcomes for the 1505 study variants under all three interpretation frameworks. When classified within Sherloc, 79.6% of variants (1198/1505) were classified as P/LP (Figure 3a). Under the simulated Sherloc‐restricted and baseline ACMG/AMP frameworks, the proportion classified as P/LP dropped to 53.5% (805/1505) and 45.5% (685/1505), respectively (Figure 3a). Concomitantly, the proportion of VUS increased from 20.4% (307/1505) within Sherloc to 46.5% (700/1505) and 54.5% (820/1505) within the Sherloc‐restricted and baseline ACMG/AMP frameworks, respectively (Figure 3a).
FIGURE 3.
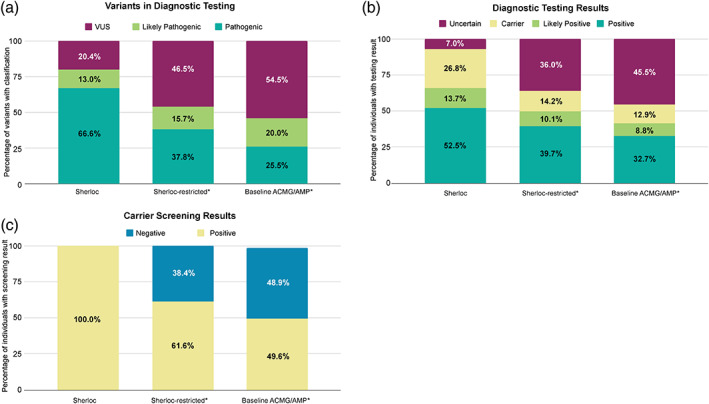
Impact of differentially weighted phenotype evidence on variant interpretation and patient results. (a) Percentage of variants observed in diagnostic testing classified as VUS and P/LP among 1505 variants interpreted under three frameworks: (1) Sherloc, which includes both highly predictive and moderately predictive phenotype evidence, (2) Sherloc with all predictive phenotype evidence weighted as moderately predictive (“Sherloc‐restricted”), and (3) baseline ACMG/AMP criteria. (b) Percentage of 3979 individuals with specific diagnostic testing results based on variants interpreted within the three frameworks. (c) Percentage of 2917 individuals with a positive carrier screening result involving a variant previously interpreted with highly predictive phenotype evidence in Sherloc, compared to carrier screenings results under Sherloc‐restricted and baseline ACMG/AMP. Asterisks indicate that the variant classifications and patient results shown are based on simulated variant interpretations
3.3. Impact of predictive phenotype evidence on patient results
We identified 3979 unrelated individuals who each had highly predictive phenotype evidence applied for at least one of the 1505 study variants identified during diagnostic testing. We then compared their genetic testing results under the three frameworks. When variants were classified within Sherloc, 66.2% of tested individuals (2634/3979) received a Positive or Likely Positive result, compared with only 49.8% (1982/3979) when the Sherloc‐restricted framework was used and 41.5% (1654/3979) when the baseline ACMG/AMP framework was used (Figure 3b).
Carrier screening also benefited from predictive phenotype‐guided interpretation. Out of the 302 genes on the lab's largest carrier screening panel, 48 had highly predictive phenotype evidence applied during variant interpretation in separate diagnostic tests. Among all Positive carrier screening results involving a variant interpreted with highly predictive phenotype evidence, only 61% (1783/2917) would be Positive if only moderately predictive evidence had been applied, and only 50% (1469/2917) would be Positive if only ACMG/AMP criteria had been applied (Figure 3c). The 2917 Positive carrier screening cases in this analysis accounted for 5.4% of the 53,695 Positive carrier screening cases reported during the study period. Thus, if strict ACMG/AMP criteria had been used instead of Sherloc with highly predictive phenotype evidence, 2.7% of Positive carrier results would have been missed.
4. DISCUSSION
Phenotype evidence can reduce uncertainty in variant classification when the phenotype is highly predictive for a specific condition. This is explained in the ACMG/AMP guidelines, but additional specificity is needed to guide appropriate and consistent use of phenotype evidence in clinical variant interpretation. To systematically incorporate this evidence type, we developed a novel, quantitative method for curating genes associated with distinctive phenotypes. We demonstrated that this methodology is valid and accurate, with 94%–98% of VUS reclassifications agreeing with the pathogenicity suggested by the curated phenotype criteria. Finally, based on studying a large cohort referred for clinical genetic testing, we showed that not incorporating phenotype information during variant interpretation can reduce diagnostic findings in certain genes associated with highly predictive phenotypes by 25%–37%.
Previous studies have suggested that a systematic way to assess phenotype information during the variant interpretation process would improve variant classification, including using phenotype‐informed prioritization of potential disease‐causative variants identified by exome sequencing (Cipriani et al., 2020; Masino et al., 2014; Thompson et al., 2019). This study demonstrates that clinician‐reported phenotype data can improve variant interpretation when those data are used in a refined interpretation framework that includes preemptively curated phenotype criteria. This improvement to variant interpretation benefits both individuals undergoing diagnostic testing and individuals undergoing carrier screening.
Although phenotype data is not collected from individuals undergoing carrier screening, phenotype‐guided variant interpretation can still have value because the clinical significance of a detected variant may have been previously interpreted in the context of diagnostic testing of affected individuals. Notably, nearly all conditions assessed in carrier screening are recessive conditions, and many conditions associated with highly predictive phenotypes are also recessive. This may partly explain why this method, when compared with other interpretation methods, doubled the number of positive carrier observations for variants that had predictive phenotype evidence applied in the context of diagnostic testing. Of note, the clinical impact of utilizing this method during carrier screening is likely an underestimate, as only 48 of 302 carrier genes in the study included variants in which highly predictive phenotype evidence had been applied. Expanding the number of carrier genes eligible for highly predictive phenotype evidence through the predictive phenotype curation process would likely further increase the impact of this evidence on the positive carrier rate.
Currently, phenotype information is used extensively to interpret genome and exome sequencing through the use of Human Phenotype Ontology algorithms that improve diagnostic yield. However, because phenotype information is not universally provided by clinicians when ordering targeted testing using gene panels (Son et al., 2018), the clinical utility of the test is limited. Clinical labs can reach out to clinicians after the initial test order to gather additional phenotype information, but this is an inefficient practice that is increasingly difficult to sustain as the number of tests grows. To fully realize the benefits of predictive phenotype evidence, clinicians should provide clinical labs with detailed clinical information at the time of the initial test order irrespective of whether the testing is performed with curated gene panels, exomes, or genomes. Using objective rules and preemptively weighted phenotype evidence during variant interpretation can reduce complexity for testing labs by eliminating subjectivity from the application of phenotype evidence, even as testing volume grows.
The approach described here for curating phenotype evidence and applying it during variant interpretation fits within the clinical genetic testing lab's broader approach to systematically and objectively use evidence in the clinical interpretation of novel variants. For phenotype evidence in particular, we incorporate the likelihood that the disease is caused by germline variants in a single gene (i.e., Mendelian disorders). Highly penetrant and early‐onset disorders, including many pediatric and inherited metabolic disorders, have a higher prior probability of receiving a molecular diagnosis from a germline genetic test than disorders such as cancer, which may be caused by inherited variants, somatic variants, and/or environmental factors. Therefore, the likelihood that clinical signs and symptoms are genetic in origin, and concomitantly the likelihood that a genetic test will have a high PDR, is much greater for certain pediatric and metabolic disorders than for cancer. We therefore award stronger evidence (i.e., more Sherloc points) for phenotype evidence associated with conditions with highly penetrant, early onset phenotypes and high PDRs than conditions with more diverse etiologies and low PDRs (Figure 1a). Moreover, the approach is highly accurate, with >98% concordance between the curated highly predictive phenotype criteria and current variant classifications. Notably, three of the five instances of discordance involved SCN1A, suggesting that the predictive phenotype criteria for this gene should be reviewed.
The approach described here is similar to efforts from various ClinGen working groups to curate genes and associated phenotypes to better inform variant interpretation (Gelb et al., 2018; Kelly et al., 2018; Lee et al., 2018; Mester et al., 2018; Zastrow et al., 2018). However, the present approach is more generalizable as it is not gene‐ or disease‐specific but rather uses an agnostic framework for curating any disease gene and quantitatively awarding evidence points toward pathogenicity during variant interpretation with Sherloc.
Preemptively curated phenotype evidence can be incorporated into any variant interpretation framework, enabling its use in many genetic testing modalities from single‐gene sequencing to panel testing, exome sequencing, and whole genome sequencing. Individuals presenting with inherited metabolic or pediatric conditions may benefit most from a variant interpretation framework that utilizes phenotype information, due to the strength of associations between phenotypes and genotypes and because clinicians may be more likely to supply phenotype information when the phenotype is more severe and a genetic etiology is strongly suspected.
This study was limited by the fact that many genetic test requisitions from clinicians come with incomplete phenotype data, precluding the application of predictive phenotype evidence when classifying variants observed in individuals who may actually have the relevant phenotypes and genotypes. In addition, by limiting the scope to disorders for which highly predictive phenotype evidence is available, the overall effect of curated phenotype evidence on VUS rates across all possible inherited conditions could not be assessed. Further, the lab does not reassess previously reported variants solely because the associated gene has been newly curated for predictive phenotype evidence. Therefore, the dataset may have contained more variants for which this type of evidence was relevant. Finally, very rare disorders with few or no instances described in the literature were not addressed, as multiple reported instances of a disease are required by the curation approach to establish that a gene is associated with a genetic condition.
Currently, the majority of genes with curated highly predictive phenotype evidence are associated with inherited metabolic diseases. In the future, we expect that more genes from a wide range of clinical areas will be curated by Invitae for predictive phenotype evidence, thus expanding the utility of this method across medical specialties. Additional future explorations include using Human Phenotype Ontology terms to analyze phenotype findings to assist in the application of predictive phenotype evidence.
5. CONCLUSION
Preemptively curating gene‐phenotype associations and systematically applying phenotype evidence for variant interpretation increases the clinical utility of genetic testing and reduces the frequency of uncertain results. These findings, coupled with the high concordance of highly predictive phenotype evidence, support weighting phenotype evidence according to its predictive value to improve the quality and accuracy of variant interpretation. In the future, the curation approach described in this study can be used for additional disease genes and phenotypes. In addition, the utility of phenotype evidence is expected to grow concomitantly with public gene‐phenotype curation databases such as the Gene Curation Coalition Database and Matchmaker Exchange (DiStefano et al., 2022; Philippakis et al. 2015) improvements in natural language processing algorithms for scouring the literature for gene‐phenotype associations, and machine‐learning approaches for modeling the predictive nature of specific phenotypes.
AUTHOR CONTRIBUTIONS
Conceptualization: Britt Johnson, Karen Ouyang, Lauren Frank, Rebecca Truty, Susan Rojahn, and Keith Nykamp. Data curation: Karen Ouyang, Rebecca Truty, Keith Nykamp, and Lauren Frank. Formal analysis: Karen Ouyang, Rebecca Truty, and Lauren Frank. Methodology: Britt Johnson, Karen Ouyang, Rebecca Truty, Susan Rojahn, and Keith Nykamp. Project administration: Britt Johnson, Ana Morales, and Keith Nykamp. Visualization: Karen Ouyang, Lauren Frank, Susan Rojahn, and Keith Nykamp. Writing – original draft: Britt Johnson, Karen Ouyang, Lauren Frank, Rebecca Truty, Susan Rojahn, Ana Morales, Swaroop Aradhya, and Keith Nykamp. Writing – review & editing: Britt Johnson, Karen Ouyang, Lauren Frank, Rebecca Truty, Susan Rojahn, Ana Morales, Swaroop Aradhya, and Keith Nykamp.
CONFLICT OF INTEREST
Britt Johnson, Karen Ouyang, Rebecca Truty, Susan Rojahn, Ana Morales, Swaroop Aradhya, and Keith Nykamp are employees and shareholders of Invitae.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENT
The authors thank Kerry Aradhya for scientific editing of this manuscript.
Johnson, B. , Ouyang, K. , Frank, L. , Truty, R. , Rojahn, S. , Morales, A. , Aradhya, S. , & Nykamp, K. (2022). Systematic use of phenotype evidence in clinical genetic testing reduces the frequency of variants of uncertain significance. American Journal of Medical Genetics Part A, 188A:2642–2651. 10.1002/ajmg.a.62779
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request. Additionally, all variants have been shared with ClinVar in a de‐identified manner: https://www.ncbi.nlm.nih.gov/clinvar/submitters/500031/
REFERENCES
- Amendola, L. M. , Jarvik, G. P. , Leo, M. C. , McLaughlin, H. M. , Akkari, Y. , Amaral, M. D. , Berg, J. S. , Biswas, S. , Bowling, K. M. , Conlin, L. K. , Cooper, G. M. , Dorschner, M. O. , Dulik, M. C. , Ghazani, A. A. , Ghosh, R. , Green, R. C. , Hart, R. , Horton, C. , Johnston, J. J. , … Rehm, H. L. (2016). Performance of ACMG‐AMP variant‐interpretation guidelines among nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. American Journal of Human Genetics, 99, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederbaum, S. (2015). Interpreting sequence variants in a clinical context. Genetics in Medicine, 17, 1012. [DOI] [PubMed] [Google Scholar]
- Cipriani, V. , Pontikos, N. , Arno, G. , Sergouniotis, P. I. , Lenassi, E. , Thawong, P. , Danis, D. , Michaelides, M. , Webster, A. R. , Moore, A. T. , Robinson, P. N. , Jacobsen, J. O. B. , & Smedley, D. (2020). An improved phenotype‐driven tool for rare Mendelian variant prioritization: Benchmarking exomiser on real patient whole‐exome data. Genes, 11(4), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStefano, M. T., Goehringer, S., Babb, L., Alkuraya, F. S., Amberger, J., Amin, M., Austin‐Tse, C., Balzotti, M., Berg, J. S., Birney, E., Bocchini, C., Bruford, E. A., Coffey, A. J., Collins, H., Cunningham, F., Daugherty, L. C., Einhorn, Y., Firth, H. V., Fitzpatrick, D. R., Foulger, R. E., … Rehm, H. L. (2022). The Gene Curation Coalition: A global effort to harmonize gene‐disease evidence resources. Genetics in Medicine, S1098‐3600(22)00746–8. 10.1016/j.gim.2022.04.017 [DOI] [PMC free article] [PubMed]
- Gelb, B. D. , Cavé, H. , Dillon, M. W. , Gripp, K. W. , Lee, J. A. , Mason‐Suares, H. , Rauen, K. A. , Williams, B. , Zenker, M. , Vincent, L. M. , & ClinGen RASopathy Working Group . (2018). ClinGen's RASopathy expert panel consensus methods for variant interpretation. Genetics in Medicine, 20, 1334–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, M. A. , Caleshu, C. , Morales, A. , Buchan, J. , Wolf, Z. , Harrison, S. M. , Cook, S. , Dillon, M. W. , Garcia, J. , Haverfield, E. , Jongbloed, J. D. H. , Macaya, D. , Manrai, A. , Orland, K. , Richard, G. , Spoonamore, K. , Thomas, M. , Thomson, K. , Vincent, L. M. , … Funke, B. (2018). Adaptation and validation of the ACMG/AMP variant classification framework for MYH7‐associated inherited cardiomyopathies: Recommendations by ClinGen's inherited cardiomyopathy expert panel. Genetics in Medicine, 20, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, R. H. , Hall, A. D. , Cutz, E. , Knowles, M. R. , Nelligan, K. A. , Nykamp, K. , Zariwala, M. A. , & Dell, S. D. (2014). The role of molecular genetic analysis in the diagnosis of primary ciliary dyskinesia. Annals of the American Thoracic Society, 11, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, K. , Krempely, K. , Roberts, M. E. , Anderson, M. J. , Carneiro, F. , Chao, E. , Dixon, K. , Figueiredo, J. , Ghosh, R. , Huntsman, D. , Kaurah, P. , Kesserwan, C. , Landrith, T. , Li, S. , Mensenkamp, A. R. , Oliveira, C. , Pardo, C. , Pesaran, T. , Richardson, M. , … Karam, R. (2018). Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Human Mutation, 39, 1553–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh, M. W. , Ferkol, T. W. , Davis, S. D. , Lee, H.‐S. , Rosenfeld, M. , Dell, S. D. , Sagel, S. D. , Milla, C. , Olivier, K. N. , Sullivan, K. M. , Zariwala, M. A. , Pittman, J. E. , Shapiro, A. J. , Carson, J. L. , Krischer, J. , Hazucha, M. J. , & Knowles, M. R. (2016). Clinical features and associated likelihood of primary ciliary dyskinesia in children and adolescents. Annals of the American Thoracic Society, 13, 1305–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln, S. E. , Kobayashi, Y. , Anderson, M. J. , Yang, S. , Desmond, A. J. , Mills, M. A. , Nilsen, G. B. , Jacobs, K. B. , Monzon, F. A. , Kurian, A. W. , Ford, J. M. , & Ellisen, L. W. (2015). A systematic comparison of traditional and multigene panel testing for hereditary breast and ovarian cancer genes in more than 1000 patients. The Journal of Molecular Diagnostics, 17, 533–544. [DOI] [PubMed] [Google Scholar]
- Marshall, C. R. , Scherer, S. W. , Zariwala, M. A. , Lau, L. , Paton, T. A. , Stockley, T. , Jobling, R. K. , Ray, P. N. , Knowles, M. R. , FORGE Canada Consortium , Hall, D. A. , Dell, S. D. , & Kim, R. H. (2015). Whole‐exome sequencing and targeted copy number analysis in primary ciliary dyskinesia. G3, 5(8), 1775–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino, A. J. , Dechene, E. T. , Dulik, M. C. , Wilkens, A. , Spinner, N. B. , Krantz, I. D. , Pennington, J. W. , Robinson, P. N. , & White, P. S. (2014). Clinical phenotype‐based gene prioritization: An initial study using semantic similarity and the human phenotype ontology. BMC Bioinformatics, 15, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester, J. L. , Ghosh, R. , Pesaran, T. , Huether, R. , Karam, R. , Hruska, K. S. , Costa, H. A. , Lachlan, K. , Ngeow, J. , Barnholtz‐Sloan, J. , Sesock, K. , Hernandez, F. , Zhang, L. , Milko, L. , Plon, S. E. , Hegde, M. , & Eng, C. (2018). Gene‐specific criteria for PTEN variant curation: Recommendations from the ClinGen PTEN expert panel. Human Mutation, 39, 1581–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health. (2022). ClinVar: Submitters and their submissions. Retrieved from: https://www.ncbi.nlm.nih.gov/clinvar/docs/submitter_list/
- Nykamp, K. , Anderson, M. , Powers, M. , Garcia, J. , Herrera, B. , Ho, Y.‐Y. , Kobayashi, Y. , Patil, N. , Thusberg, J. , Westbrook, M. , Invitae Clinical Genomics Group , & Topper, S. (2017). Sherloc: A comprehensive refinement of the ACMG‐AMP variant classification criteria. Genetics in Medicine, 19, 1105–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis, A. A., Azzariti, D. R., Beltran, S., Brookes, A. J., Brownstein, C. A., Brudno, M., Brunner, H. G., Buske, O. J., Carey, K., Doll, C., Dumitriu, S., Dyke, S. O., den Dunnen, J. T., Firth, H. V., Gibbs, R. A., Girdea, M., Gonzalez, M., Haendel, M. A., Hamosh, A., Holm, I. A., … Rehm, H. L. (2015). The Matchmaker Exchange: a platform for rare disease gene discovery. Human Mutation, 36(10), 915–921. doi: 10.1002/humu.22858. [DOI] [PMC free article] [PubMed]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera‐Muñoz, E. A. , Milko, L. V. , Harrison, S. M. , Azzariti, D. R. , Kurtz, C. L. , Lee, K. , Mester, J. L. , Weaver, M. A. , Currey, E. , Craigen, W. , Eng, C. , Funke, B. , Hegde, M. , Hershberger, R. E. , Mao, R. , Steiner, R. D. , Vincent, L. M. , Martin, C. L. , Plon, S. E. , … Berg, J. S. (2018). ClinGen variant curation expert panel experiences and standardized processes for disease and gene‐level specification of the ACMG/AMP guidelines for sequence variant interpretation. Human Mutation, 39, 1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, A. J. , Davis, S. D. , Polineni, D. , Manion, M. , Rosenfeld, M. , Dell, S. D. , Chilvers, M. A. , Ferkol, T. W. , Zariwala, M. A. , Sagel, S. D. , Josephson, M. , Morgan, L. , Yilmaz, O. , Olivier, K. N. , Milla, C. , Pittman, J. E. , Daniels, M. L. A. , Jones, M. H. , Janahi, I. A. , … American Thoracic Society Assembly on Pediatrics . (2018). Diagnosis of primary ciliary dyskinesia. An official American Thoracic Society clinical practice guideline. American Journal of Respiratory and Critical Care Medicine, 197, e24–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son, J. H. , Xie, G. , Yuan, C. , Ena, L. , Li, Z. , Goldstein, A. , Huang, L. , Wang, L. , Shen, F. , Liu, H. , Mehl, K. , Groopman, E. E. , Marasa, M. , Kiryluk, K. , Gharavi, A. G. , Chung, W. K. , Hripcsak, G. , Friedman, C. , Weng, C. , & Wang, K. (2018). Deep phenotyping on electronic health records facilitates genetic diagnosis by clinical exomes. American Journal of Human Genetics, 103, 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, R. , Papakonstantinou Ntalis, A. , Beltran, S. , Töpf, A. , de Paula, E. E. , Polavarapu, K. , C't Hoen, P. A. , Missier, P. , & Lochmüller, H. (2019). Increasing phenotypic annotation improves the diagnostic rate of exome sequencing in a rare neuromuscular disorder. Human Mutation, 40, 1797–1812. [DOI] [PubMed] [Google Scholar]
- Truty, R. , Paul, J. , Kennemer, M. , Lincoln, S. E. , Olivares, E. , Nussbaum, R. L. , & Aradhya, S. (2019). Prevalence and properties of intragenic copy‐number variation in Mendelian disease genes. Genetics in Medicine, 21, 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zastrow, D. B. , Baudet, H. , Shen, W. , Thomas, A. , Si, Y. , Weaver, M. A. , Lager, A. M. , Liu, J. , Mangels, R. , Dwight, S. S. , Wright, M. W. , Dobrowolski, S. F. , Eilbeck, K. , Enns, G. M. , Feigenbaum, A. , Lichter‐Konecki, U. , Lyon, E. , Pasquali, M. , Watson, M. , … Mao, R. (2018). Unique aspects of sequence variant interpretation for inborn errors of metabolism (IEM): The ClinGen IEM working group and the phenylalanine hydroxylase gene. Human Mutation, 39, 1569–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request. Additionally, all variants have been shared with ClinVar in a de‐identified manner: https://www.ncbi.nlm.nih.gov/clinvar/submitters/500031/