

Abstract
In artificial small‐molecule machines, molecular motors can be used to perform work on coupled systems by applying a mechanical load—such as strain—that allows for energy transduction. Here, we report how ring strain influences the rotation of a rotary molecular motor. Bridging the two halves of the motor with alkyl tethers of varying sizes yields macrocycles that constrain the motor's movement. Increasing the ring size by two methylene increments increases the mobility of the motor stepwise and allows for fine‐tuning of strain in the system. Small macrocycles (8–14 methylene units) only undergo a photochemical E/Z isomerization. Larger macrocycles (16–22 methylene units) can perform a full rotational cycle, but thermal helix inversion is strongly dependent on the ring size. This study provides systematic and quantitative insight into the behavior of molecular motors under a mechanical load, paving the way for the development of complex coupled nanomachinery.
Keywords: Macrocycles, Molecular Motors, Photochemistry, Strained Molecules
The influence of ring strain on the 360° operation of a light‐driven rotary molecular motor was investigated by constraining the motor unit in macrocyclic structures of various sizes. It was found that two of the four steps of a rotational cycle are significantly affected by chemical strain.
Introduction
Biological molecular machines (BMMs)[ 1 , 2 ] perform a variety of vital tasks in living organisms, such as driving the formation of energy carriers (ATP synthase), transporting cellular cargo (kinesin) or synthesizing proteins (ribosome). Taking BMMs as an example, a key challenge for artificial molecular machines (AMMs)[ 3 , 4 , 5 , 6 , 7 , 8 , 9 ] is to perform work on a coupled system, i.e. transducing the energy that is put into the system into other forms of energy, by avoiding heat dissipation as efficiently as possible.[ 5 , 8 , 10 , 11 ] In recent years, molecular machinists developed AMMs that can execute more and more sophisticated tasks, such as multifunctional catalysis,[ 12 , 13 , 14 ] contracting or moving macroscopic objects,[ 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 ] penetrating and affecting cell walls,[ 23 , 24 , 25 ] transporting and assembling molecular entities,[ 26 , 27 , 28 , 29 ] shifting chemical equilibria,[ 30 , 31 ] performing coupled motions[ 32 , 33 , 34 , 35 , 36 , 37 ] or pumping molecular entities.[ 38 , 39 , 40 , 41 , 42 , 43 ]
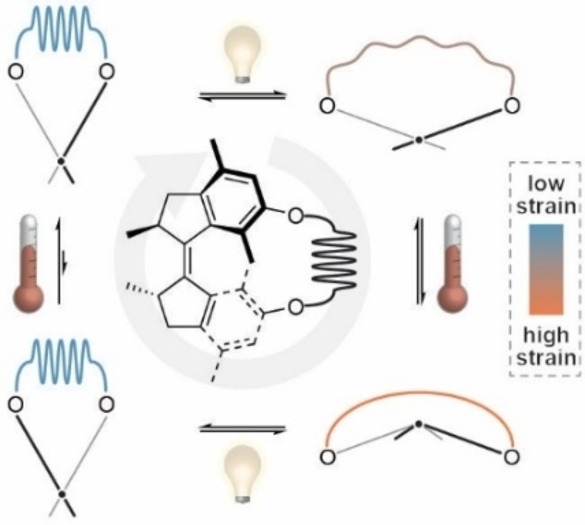
As demonstrated in the examples above, photochemical molecular motors[ 44 , 45 , 46 , 47 , 48 , 49 , 50 ] can be crucial components in AMMs. They are utilized as energy converters able to transduce light into other forms of energy. [11] The working mechanism of Feringa‐type molecular motors is based on light‐induced configurational isomerization of a chiral overcrowded alkene, which can adopt two different helicities in both of its stereoisomers (E and Z, Figure 1A). Upon light illumination, the Z stable (s) molecule undergoes an E/Z isomerization to form an E metastable (m) isomer with inverted helicity, which then relaxes in a thermal step by helix inversion (THI) to the Es isomer. This process corresponds to a directed 180° rotation. To close a full 360° rotational cycle the molecule needs to undergo a second E/Z isomerization (this time from the Es to Zm isomer) followed by a subsequent THI from the Zm to Zs diastereomer. Constant exposure to light leads to continuous unidirectional rotary motion, the direction of which is governed by a chiral sp3‐hybridized carbon center in the allylic position of the molecule.
Figure 1.

Operational routine, synthesis and structural characterization of motor macrocycles. A) Full rotational cycle of unconstrained motor OMe. Starting from the Zs isomer, the motor undergoes an E/Z isomerization upon UV light illumination, followed by a subsequent THI that converts OMe ‐Em into OMe‐Es. Subsequent UV light illumination forms the Zm isomer, which undergoes another THI to form the starting isomer Zs. B) Full rotational cycle of a constrained motor C2n in which an alkyl tether functions as a molecular spring. In the Zs isomer, the tether is not strained by the motor. When illuminated with UV light, the motor is converted into the Em isomer in which the tether is subjected to chemical strain which is further increased by a subsequent THI that forms C2n ‐Es. If the tether is long enough to skip over the motor core, the system can undergo another light‐induced E/Z isomerization to form the Zm isomer, leading to a relaxation of chemical strain in the tether. Further relaxation by a THI yields the initial motor C2n‐Zs. C) Macrocyclization of dihydroxy motor OH under high‐dilution conditions by Williamson ether synthesis. D) X‐ray structures of OMe‐Zs, OMe‐Es, C8‐Zs, C12‐Zs and C12‐Em. Solvent molecules, hydrogen and disordered atoms are omitted for clarity. Ellipsoids are drawn at 50 % probability.
In the case of our molecular motors, the challenge is to couple them to an external load, so that their light‐driven unidirectional motion can be harnessed to perform work on a coupled system. [8] One possible way to achieve this target is by constraining the motion of the motor, i.e. applying a mechanical load, so that parts of the motor movement are converted into, for instance, coupled motion[ 33 , 34 , 35 , 36 , 37 , 51 ] or chemical strain[ 17 , 31 , 52 , 53 , 54 , 55 , 56 , 57 , 58 , 59 ] of a secondary molecular entity. Detailed mechanistic studies of photochemical molecular motors that perform work against a mechanical load, applied by connecting their contra‐rotating halves with molecular tethers, were reported by the Dube,[ 34 , 36 ] Giuseppone [59] and our group. [31] While the two latter AMMs[ 31 , 59 ] are bridged bismacrocyclic molecules that can induce twists in the tethers, the system by Dube and co‐workers consists of a hemithioindigo motor and a biaryl unit both embedded within one macrocycle.[ 34 , 36 ] The light‐driven, unidirectional motion of the hemithioindigo actuator induces a directed, coupled rotation of the biaryl moiety which can act as a hurdle that has to be overcome by the motor if the activation barrier for atropisomerization is sufficiently high. To get an even more systematic insight into the behavior of a molecular motor that is subjected to varying amounts of mechanical stress, we wanted to investigate a paradigmatic system which allows for facile and stepwise adjustment of ring strain.
We accomplished our goal by linking the two contra‐rotating parts of a first‐generation Feringa motor with an alkyl tether, thereby forming macrocyclic structure C2n (with n=4–11). By systematically increasing the length of the alkyl chain—and thus the size of the macrocycles C8−C22—the degree of mechanical constraint on the movement of the motor can be precisely tuned and studied. Related strategies were successfully applied for hydrazone [56] and stilbene[ 53 , 54 , 55 , 57 ] photoswitches to investigate the influence of strain on their switchability or the reactivity of remote chemical bonds. It is important to note that switches usually undergo only one isomerization process, have no thermal ratcheting step and are therefore not unidirectional.[ 60 , 61 ] In stark contrast, a full motor rotation comprises four distinct isomerization steps, each of which can be potentially influenced by mechanical strain.
Results and Discussion
In our system, the alkyl tether functions as a molecular spring that is fully relaxed when motor C2n is in its Zs form (Figure 1B). Upon UV light illumination, the macrocyclized rotary motor undergoes an E/Z isomerization, leading to the formation of the Em isomer and to the build‐up of chemical strain in the system, which is even further increased in the subsequent THI step that yields the Es isomer. Assuming that the alkyl tether is long enough to skip over one half of the motor core (Figure 1B), illumination with UV light induces an E/Z double bond isomerization which yields the Zm isomer that can undergo another THI to restore the initial Zs motor. We hypothesized that only tethers with sufficient length would enable a full motor rotation, [58] while tethers that are too short only allow for the first photochemical E/Z isomerization, thus preventing full rotation of the motor. We further anticipated that mechanical constraint should have a measurable effect on the THI steps.[ 34 , 36 ]
Constrained motors C8−C22‐Zs were synthesized by macrocyclizing the Zs isomer of dihydroxy molecular motor OH with hydrocarbon chains in a Williamson ether synthesis under high‐dilution conditions (Figure 1C).[ 62 , 63 ] The length of the alkyl chains ranges from octyl to docosyl (C8H16 to C22H44) and was gradually increased by two methylene unit increments. We chose an even number of methylene units to avoid any influence of odd‐even effects. [64] Molecular motors C8−C22 were characterized by NMR spectroscopy, high‐resolution mass spectrometry and in part by X‐ray crystallography (Figure 1D). [65]
Indeed, we experimentally observed that mechanical strain has a significant effect on the switchability of the motor unit, as macrocycles C8−C14 only undergo a photochemical E/Z isomerization and show no subsequent THI (Table 1 and Supporting Figures S1–S4, S10–S13 and S24–S27 and Tables S1–S3). 1H NMR spectroscopy revealed that the photostationary state (PSS) is dependent on the macrocycle size (Figures 2A, B and Supporting Figures S1–S4). Motor C8‐Zs forms the Em isomer in 50±2 %, while the larger macrocycles C10 and C12−C14 reach up to 74±2 % and 81±2 %, respectively (Figure 2A, Table 1 and Supporting Table S1). Similarly, the activation barrier for the thermal E/Z back isomerization increases with increasing ring size (Table 1 and Supporting Table S3 and Figures S10–S13). Macrocycle C8‐Em rapidly isomerizes back to C8‐Zs with an activation barrier of ΔG C8 ≠ (Em→Zs)=18.8±0.2 kcal mol−1. For motors C10‐Em and C12‐Em the activation barrier increases dramatically with ΔG C10 ≠ (Em→Zs)=25.3±0.1 kcal mol−1 and ΔG C12 ≠ (Em→Zs)=29.0±0.1 kcal mol−1, respectively. The thermal E/Z isomerization of isomer C14‐Em is so slow that the activation energy for the process could only be estimated to be ΔG C14 ≠ (Em→Zs)≥32 kcal mol−1. Interestingly, formation of the Es isomer was not observed by UV/Vis and NMR spectroscopy for motors C8−C14‐Em, indicating that ring strain induced by the alkyl tethers inhibits the THI (Figure 2B).
Table 1.
PSSs and energy differences (ΔG) between stable and metastable states and activation parameters (Gibbs free energy of activation (ΔG ≠), enthalpy of activation (ΔH ≠) and entropy of activation (ΔS ≠)) for THI and double bond isomerizations of macrocycles C8–22 and reference compound OMe. PSSs were determined by 1H NMR spectroscopy in toluene‐d 8 at −20 °C and are given as the amount of product formed in the indicated (photo)reactions. Energies were determined by 1H NMR spectroscopy in toluene‐d 8 (1 mM) and are reported for 20 °C in kcal mol−1. Activation parameters were determined by UV/Vis spectroscopy in octane (20 μM) and are reported for 20 °C. ΔH ≠ and ΔG ≠ are given in kcal mol−1; ΔS ≠ is given in cal K−1 mol−1.
|
|
PSS |
ΔG |
ΔG ≠ (ΔH ≠, ΔS ≠) |
|||||
|---|---|---|---|---|---|---|---|---|
|
|
(Zs→Em) |
(Es→Zm) |
(Em→Zs) |
(Em→Es) |
(Zm→Zs) |
(Em→Zs) |
(Em→Es) |
(Zm→Zs) |
|
|
|
|
|
|
|
|
|
|
|
C8 |
50±2 %[a] |
– |
≤ −2.0 |
– |
– |
18.8±0.2 (18.2, −2.16) |
– |
– |
|
C10 |
74±2 % |
– |
≤ −2.0 |
– |
– |
25.3±0.1 (26.3, 3.56) |
– |
– |
|
C12 |
81±2 % |
– |
≤ −2.0 |
– |
– |
29.0±0.1[c] (29.8, 2.22) |
– |
– |
|
C14 |
81±2 % |
– |
≤ −2.0 |
– |
– |
≥32[c,d] |
– |
– |
|
C16 |
83±2 % |
67±4 % |
– |
+0.9±0.1 |
≤ −2.0 |
– |
22.1±0.1 (14.1, −27.5) |
23.6±0.2[e] (19.9, −12.5) |
|
C18 |
80±2 % |
65±4 % |
– |
−0.5±0.1 |
≤ −2.0 |
– |
20.4±0.1 (18.1, −7.70) |
23.7±0.1 (19.9, −12.8) |
|
C20 |
81±2 % |
63±4 % |
– |
−1.0±0.1 |
≤ −2.0 |
– |
20.3±0.1 (17.3, −9.95) |
23.7±0.1 (19.3, −15.0) |
|
C22 |
80±2 % |
65±4 % |
– |
−1.4±0.1 |
≤ −2.0 |
– |
20.1±0.1 (18.6, −4.99) |
23.7±0.1 (19.5, −14.4) |
|
OMe |
78±2 %[b] |
70±4 % |
– |
≤ −2.0 |
≤ −2.0 |
– |
18.6±0.3 (15.7, −9.99) |
23.4±0.2 (19.5, −13.3) |
[a] PSS determined at −40 °C. [b] PSS determined at −46 °C. [c] Energy given at 110 °C. [d] Energy estimated from a single relaxation at 110 °C. [e] Activation parameters determined by NMR spectroscopy in toluene‐d 8.
Figure 2.

Photochemical E/Z isomerization and THI. A) The amount of Em isomer at PSS after UV light illumination increases with increasing ring size. The partial 1H NMR spectra (toluene‐d 8, 1 mM, −20 °C, 500 MHz) show the composition at PSS after illuminating pure C8–22‐Zs and OMe‐Zs samples with UV light. Only signals of the aromatic protons in position a are shown. B) The ratio of Es to Em isomer at PSS after UV light illumination increases with increasing ring size. The partial 1H NMR spectra (toluene‐d 8, 1 mM, −20 °C, 500 MHz) show the composition of pure C8–22‐Zs and OMe‐Zs samples that were illuminated with UV light to PSS and subsequently allowed to reach thermal equilibrium. Motor C14 was partially equilibrated at 100 °C for 3 d. Only signals of the aromatic protons in position a are shown.
To further investigate this behavior we performed single‐crystal X‐ray analyses of motors OMe‐Es, C12‐Zs and C12‐Em (Figures 1D and 3A, B). The crystal structures suggest that skipping of the alkyl tether over the motor core is sterically unlikely (see space‐filling models of C12‐Zs and C12‐Em in Figures 3A, B) and would form a highly strained species (for computational results see Figure 4). We also assume for C8 and C10 that the alkyl tether is unlikely to skip over the motor core as both macrocycles are smaller than C12. More importantly, a comparison with the reference compound OMe‐Es indicates that the THI would induce additional ring strain in the system, since an inversion of the xylenyl moieties stretches the oligomeric tether even further (Figure 3B). In the case of the (S,S) C12‐Em macrocycle, the alkyl tether as well as the oxygen atoms that connect it with the motor core face the Si side of the motor (with respect to the overcrowded double bond). Upon THI to form the Es isomer, the xylenyl units invert, pushing the oxygen atoms to the Re side of the motor. This movement would lead to an increase in ring strain in the system and is a likely reason why the THI from Em to Es in macrocycles C8−C12 (and also C14, see Figure 4) does not occur. We also would like to emphasize that only thanks to the relative mechanochemical stabilization of the C12‐Em isomer, we could obtain the first unambiguous structural characterization of the usually thermally labile Em isomer by X‐ray analysis. Additionally, the alkyl tether allows for tracking of the motor movement (Figure 3A). The alkyl chain follows the rotation of the motor and is located on the Si side of the (S,S) C12‐Em motor (Re side for (R,R) C12‐Em) after the photoreaction. Therefore, the only plausible direction of rotation of C12‐Zs to form C12‐Em is clockwise for the (S,S) and counterclockwise for the (R,R) diastereomer. This gives unprecedented structural insight into the mechanism of molecular rotary motors based on the Feringa design.
Figure 3.

Unidirectional motor rotation and inhibition of helix inversion. A) The crystal structures of (S,S) C12‐Zs and (S,S) C12‐Em show that only a clockwise rotation of the bottom half of the motor (indicated by the red arrow) can position the dodecyl tether on the Si side of the molecule. The box shows the schematic structures of motor macrocycles C2n‐Zs and C2n‐Em with a view along the central double bond and the influence of an E/Z isomerization on the ring strain in the system. B) The crystal structure of (S,S) C12‐Em shows that the alkyl tether is facing the Si side of the motor with respect to the central double bond. The oxygen atoms point in the same direction, thus minimizing strain in the system. The crystal structure of (S,S) OMe‐Es indicates that a THI requires the oxygen atoms to move to the Re side of the motor. Therefore, a THI from (S,S) C12‐Em (Si) to (S,S) C12‐Es (Re) would increase the ring strain in the molecule. Red arrows indicate the direction of movement of the motor core to interconvert Em to Es and vice versa. The box shows the schematic structures of motor macrocycles C2n‐Em and C2n‐Es with a view along the central double bond and the influence of a THI on the ring strain in the system. All X‐ray structures are depicted with a space‐filling overlay. Hydrogen and disordered atoms are omitted for clarity. Ellipsoids are drawn at 50 % probability.
Figure 4.

Box plots for the computed relative Gibbs free energies of C2n‐Em (Si), C2n‐Em (Re), C2n‐Es (Si) and C2n‐Es (Re). The computations lead to two main conclusions: i) in case of C12−C16‐Em the energetically least favorable species are the ones with the alkyl tether on the Re side of the motor and ii) the relative energy difference between having the alkyl tether on the Re or Si side of the motor decreases with increasing ring size. Gibbs free energies were calculated at the r2SCAN‐3c level of theory. The accuracy of the computational method is ≥1 kcal mol−1, see reference [70].
By further increasing the ring size of the macrocycles, full rotation in motors C16−C22 was observed (Table 1 and Supporting Figures S5–S9, S14–S23 and S28–S32 and Tables S1–S3). According to 1H NMR spectroscopy, the length of the tether from C16 onward has no measurable effect on the photochemical steps in the rotational cycle (Supporting Figures S5–S9). The PSS ratio for the photoreaction of C16−C22‐Es and OMe‐Es to the respective Zm isomer is similar and leads to the formation of Zm in ≈66±4 % (Figure 2A). Also, the PSS ratio for the photochemical double bond isomerization from the Zs to the respective Em diastereomer is not affected by larger macrocycles and is the same for motors C12−C22 and OMe forming the Em isomer in ≈80±2 % (Figure 2A). Looking at the thermal steps, we did not observe a significant dependence of the THI from C16−C22‐Zm and OMe‐Zm to the respective Zs isomer on the length (or presence) of the alkyl tethers. All reactions are exergonic and irreversible with ΔG (Zm→Zs)≤−2.0 kcal mol−1 and ΔG ≠ (Zm→Zs)≈23.7±0.1 kcal mol−1. However, the THI from C16−C22‐Em to the respective Es isomer does depend on the size of the macrocycle (Figure 2B). The THI repositions the allylic methyl groups from an energetically less favored pseudo‐equatorial into a more favorable pseudo‐axial position (Figures 1A, B and 3B). While this process releases tension in the motor unit, ring strain hampers the THI and destabilizes the Es relative to the Em isomers (Table 1). By increasing the length of the alkyl tether from hexadecyl to docosyl the formation of Es becomes increasingly favored energetically, ranging from ΔG C16 (Em→Es)=0.9±0.1 kcal mol−1 to ΔG C22 (Em→Es)=−1.4±0.1 kcal mol−1 with activation energies of ΔG C16 ≠ (Em→Es)=22.1±0.1 kcal mol−1 and ΔG C22 ≠ (Em→Es)=20.1±0.1 kcal mol−1 (a similar dependence of THI activation barriers in hemithioindigo motors on ring strain was also observed by Dube and co‐workers).[ 34 , 36 ] Consequently, the THI from C16‐Em to C16‐Es is endergonic (please note: C16‐Em is thermodynamically more stable than C16‐Es), with a (comparably) low activation entropy of ΔS C16 ≠ (Em→Es)=−27.5 cal K−1 mol−1 suggesting that the transition state is particularly structured and conformationally restricted. Nonetheless, the rotation for C16 is still unidirectional, since the THI from C16‐Zm to C16‐Zs is irreversible (ΔG C16 (Zm→Zs)≤−2.0 kcal mol−1) and thus energetically more favorable than the THI from C16‐Em to C16‐Es. By comparing these energy differences with the uncyclized model compound OMe (ΔG OMe (Em→Es)≤−2.0 kcal mol−1 and ΔG OMe ≠ (Em→Es)=18.6±0.3 kcal mol−1) we can show that ring strain makes the formation of the Es isomer kinetically and thermodynamically less favorable.
The fact that molecular motors C16−C22 perform a full rotational cycle implies that a skipping of the tether over the motor core is kinetically feasible. [58] The mechanism of such a skipping event could be mechanistically elucidated for hemithioindigo motors by using a biaryl unit as an “energy hurdle”. [36] However, we were not able to directly measure such a “skipping rope” process experimentally (only one distinct species per motor diastereomer was observed). Therefore, we performed a computational study to elucidate the relative thermodynamic stability of motors C2n‐Em and C2n‐Es with the alkyl tether on the Re or Si side of the motor core, which provides further information about the distribution of such species in equilibrium.
Motor structures C2n‐Em (Si), C2n‐Em (Re), C2n‐Es (Si) and C2n‐Es (Re) were pre‐screened via a preliminary metadynamics run of 2 ns (2 fs step, 298.15 K using a Berendsen thermostat) using the GFN force field implemented in xTB 6.4.1. [66] Subsequently, 50 geometries were randomly extracted and re‐optimized with the composite functional r2SCAN‐3c, [67] as implemented in the Orca 5.0.1 package, [68] and the thermochemical data were calculated at the same level of theory. Finally, the relative Gibbs free energies (with respect to the most stable C2n‐Em (Si) geometry) were computed taking into account the electronic energies calculated at the r2SCAN‐3c, pW6B95/def2‐TZVP,gCP(TZ) or ωB97X‐V/def2‐TZVP,gCP(TZ) levels of theory (Figure 4 and Supporting Data File).[ 69 , 70 ] The calculations indicate that positioning the alkyl chain on the Re side of motors C12−C16‐Em is thermodynamically unlikely, as the Em (Re) species has the highest median energy in these cases (note that the structures of C8‐ and C10‐Es (Re) and ‐Em (Si) could not be modeled, as they were not found to be minima on the potential energy surface of these motors). Furthermore, our computational analysis shows the general trend that increasing the ring size of motors C2n‐Em and C2n‐Es decreases the relative energy differences between species in which the alkyl chain faces the Re or Si side of the molecule. In case of C18−C22‐Em and C18−C22‐Es, the energies of the computed structures are comparable indicating that the alkyl tether can be localized on the Re and Si side of the motor.
Conclusion
Our systematic study shows that chemical strain has a significant impact on the thermal and photochemical steps in a 360° rotational cycle of a first‐generation molecular motor. We observed that the PSS from Zs to Em, the thermal double bond isomerization from Em to Zs and THI from Em to Es in motor macrocycles C2n are dependent on ring strain. The alkyl tethers in macrocycles C8−C14 are so short that the motor unit can only undergo a photochemical E/Z isomerization from Zs to Em, while subsequent THI was not observed. Instead, motors C16−C22 can perform a 360° rotation, but ring strain hampers the formation of the Es isomer by THI. Furthermore, we could use the relative stabilization of the Em isomer of motor C12 to gain unprecedented structural insight into the operational routine of a rotary molecular motor by single‐crystal X‐ray analysis. We anticipate that our findings will serve as a blueprint and open up design space for the development of more complex molecular machinery.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
We gratefully acknowledge the generous support from the Alexander von Humboldt Foundation (Feodor Lynen Research Fellowship to M.K.), the Marie Skłodowska‐Curie Actions (Individual Fellowship 838280 to S.C.), the Horizon 2020 Framework Program (ERC Advanced Investigator Grant No. 694345 to B.L.F.) and the Ministry of Education, Culture and Science of the Netherlands (Gravitation Program No. 024.001.035 to B.L.F.). We would like to thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high performance computing cluster.
M. Kathan, S. Crespi, A. Troncossi, C. N. Stindt, R. Toyoda, B. L. Feringa, Angew. Chem. Int. Ed. 2022, 61, e202205801; Angew. Chem. 2022, 134, e202205801.
Contributor Information
Dr. Michael Kathan, Email: michael.peter.kathan@chemie.hu-berlin.de.
Prof. Dr. Ben L. Feringa, Email: b.l.feringa@rug.nl.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Schliwa M., Molecular Motors, Wiley-VCH, Weinheim, 2006. [Google Scholar]
- 2. Goodsell D. S., The Machinery of Life, Springer, New York, 2009. [Google Scholar]
- 3. Balzani V., Credi A., Raymo F. M., Stoddart J. F., Angew. Chem. Int. Ed. 2000, 39, 3348–3391; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 3484–3530. [Google Scholar]
- 4. Sauvage J.-P., Molecular Machines and Motors, Springer, Berlin, 2001. [Google Scholar]
- 5. Browne W. R., Feringa B. L., Nat. Nanotechnol. 2006, 1, 25–35. [DOI] [PubMed] [Google Scholar]
- 6. Kay E. R., Leigh D. A., Zerbetto F., Angew. Chem. Int. Ed. 2007, 46, 72–191; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 72–196. [Google Scholar]
- 7. Balzani V., Credi A., Venturi M., Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld, Wiley-VCH, Weinheim, 2008. [Google Scholar]
- 8. Aprahamian I., ACS Cent. Sci. 2020, 6, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Feng Y., Ovalle M., Seale J. S. W., Lee C. K., Kim D. J., Astumian R. D., Stoddart J. F., J. Am. Chem. Soc. 2021, 143, 5569–5591. [DOI] [PubMed] [Google Scholar]
- 10. Coskun A., Banaszak M., Astumian R. D., Stoddart J. F., Grzybowski B. A., Chem. Soc. Rev. 2012, 41, 19–30. [DOI] [PubMed] [Google Scholar]
- 11. Astumian R. D., Chem. Sci. 2017, 8, 840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang J., Feringa B. L., Science 2011, 331, 1429–1432. [DOI] [PubMed] [Google Scholar]
- 13. Vlatković M., Bernardi L., Otten E., Feringa B. L., Chem. Commun. 2014, 50, 7773–7775. [DOI] [PubMed] [Google Scholar]
- 14. Pizzolato S. F., Štacko P., Kistemaker J. C. M., van Leeuwen T., Feringa B. L., Nat. Catal. 2020, 3, 488–496. [Google Scholar]
- 15. Eelkema R., Pollard M. M., Vicario J., Katsonis N., Ramon B. S., Bastiaansen C. W. M., Broer D. J., Feringa B. L., Nature 2006, 440, 163. [DOI] [PubMed] [Google Scholar]
- 16. Iamsaard S., Aßhoff S. J., Matt B., Kudernac T., Cornelissen J. J. L. M., Fletcher S. P., Katsonis N., Nat. Chem. 2014, 6, 229–235. [DOI] [PubMed] [Google Scholar]
- 17. Li Q., Fuks G., Moulin E., Maaloum M., Rawiso M., Kulic I., Foy J. T., Giuseppone N., Nat. Nanotechnol. 2015, 10, 161–165. [DOI] [PubMed] [Google Scholar]
- 18. Foy J. T., Li Q., Goujon A., Colard-Itté J.-R., Fuks G., Moulin E., Schiffmann O., Dattler D., Funeriu D. P., Giuseppone N., Nat. Nanotechnol. 2017, 12, 540–545. [DOI] [PubMed] [Google Scholar]
- 19. Chen J., Leung F. K. C., Stuart M. C. A., Kajitani T., Fukushima T., Van Der Giessen E., Feringa B. L., Nat. Chem. 2018, 10, 132–138. [DOI] [PubMed] [Google Scholar]
- 20. Colard-Itté J.-R., Li Q., Collin D., Mariani G., Fuks G., Moulin E., Buhler E., Giuseppone N., Nanoscale 2019, 11, 5197–5202. [DOI] [PubMed] [Google Scholar]
- 21. Ryabchun A., Li Q., Lancia F., Aprahamian I., Katsonis N., J. Am. Chem. Soc. 2019, 141, 1196–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hou J., Mondal A., Long G., de Haan L., Zhao W., Zhou G., Liu D., Broer D. J., Chen J., Feringa B. L., Angew. Chem. Int. Ed. 2021, 60, 8251–8257; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 8332–8338. [Google Scholar]
- 23. Koçer A., Walko M., Meijberg W., Feringa B. L., Science 2005, 309, 755–758. [DOI] [PubMed] [Google Scholar]
- 24. García-López V., Chen F., Nilewski L. G., Duret G., Aliyan A., Kolomeisky A. B., Robinson J. T., Wang G., Pal R., Tour J. M., Nature 2017, 548, 567–572. [DOI] [PubMed] [Google Scholar]
- 25. Mutter N. L., Volarić J., Szymanski W., Feringa B. L., Maglia G., J. Am. Chem. Soc. 2019, 141, 14356–14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lewandowski B., De Bo G., Ward J. W., Papmeyer M., Kuschel S., Aldegunde M. J., Gramlich P. M. E., Heckmann D., Goldup S. M., D'Souza D. M., Fernandes A. E., Leigh D. A., Science 2013, 339, 189–193. [DOI] [PubMed] [Google Scholar]
- 27. Chen J., Wezenberg S. J., Feringa B. L., Chem. Commun. 2016, 52, 6765–6768. [DOI] [PubMed] [Google Scholar]
- 28. Kassem S., Lee A. T. L., Leigh D. A., Markevicius A., Solà J., Nat. Chem. 2016, 8, 138–143. [DOI] [PubMed] [Google Scholar]
- 29. Kassem S., Lee A. T. L., Leigh D. A., Marcos V., Palmer L. I., Pisano S., Nature 2017, 549, 374–378. [DOI] [PubMed] [Google Scholar]
- 30. Sell H., Gehl A., Plaul D., Sönnichsen F. D., Schütt C., Köhler F., Steinborn K., Herges R., Commun. Chem. 2019, 2, 62. [Google Scholar]
- 31. Kathan M., Crespi S., Thiel N. O., Stares D. L., Morsa D., de Boer J., Pacella G., van den Enk T., Kobauri P., Portale G., Schalley C. A., Feringa B. L., Nat. Nanotechnol. 2022, 17, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Muraoka T., Kinbara K., Aida T., Nature 2006, 440, 512–515. [DOI] [PubMed] [Google Scholar]
- 33. Štacko P., Kistemaker J. C. M., van Leeuwen T., Chang M.-C., Otten E., Feringa B. L., Science 2017, 356, 964–968. [DOI] [PubMed] [Google Scholar]
- 34. Uhl E., Thumser S., Mayer P., Dube H., Angew. Chem. Int. Ed. 2018, 57, 11064–11068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 11231–11235. [Google Scholar]
- 35. Yu J. J., Zhao L. Y., Shi Z. T., Zhang Q., London G., Liang W. J., Gao C., Li M. M., Cao X. M., Tian H., Feringa B. L., Qu D.-H., J. Org. Chem. 2019, 84, 5790–5802. [DOI] [PubMed] [Google Scholar]
- 36. Uhl E., Mayer P., Dube H., Angew. Chem. Int. Ed. 2020, 59, 5730–5737; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 5779–5786. [Google Scholar]
- 37. Bach N. N., Josef V., Maid H., Dube H., Angew. Chem. Int. Ed. 2022, 61, e202201882; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2022, 134, e202201882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ragazzon G., Baroncini M., Silvi S., Venturi M., Credi A., Nat. Nanotechnol. 2015, 10, 70–75. [DOI] [PubMed] [Google Scholar]
- 39. Cheng C., McGonigal P. R., Schneebeli S. T., Li H., Vermeulen N. A., Ke C., Stoddart J. F., Nat. Nanotechnol. 2015, 10, 547–553. [DOI] [PubMed] [Google Scholar]
- 40. Qiu Y., Zhang L., Pezzato C., Feng Y., Li W., Nguyen M. T., Cheng C., Shen D., Guo Q. H., Shi Y., Cai K., Alsubaie F. M., Astumian R. D., Stoddart J. F., J. Am. Chem. Soc. 2019, 141, 17472–17476. [DOI] [PubMed] [Google Scholar]
- 41. Qiu Y., Song B., Pezzato C., Shen D., Liu W., Zhang L., Feng Y., Guo Q., Cai K., Li W., Chen H., Nguyen M. T., Shi Y., Cheng C., Astumian R. D., Li X., Stoddart J. F., Science 2020, 368, 1247–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Feng L., Qiu Y., Guo Q.-H., Chen Z., Seale J. S. W., He K., Wu H., Feng Y., Farha O. K., Astumian R. D., Stoddart J. F., Science 2021, 374, 1215–1221. [DOI] [PubMed] [Google Scholar]
- 43. Amano S., Fielden S. D. P., Leigh D. A., Nature 2021, 594, 529–534. [DOI] [PubMed] [Google Scholar]
- 44. Koumura N., Zijistra R. W. J., Van Delden R. A., Harada N., Feringa B. L., Nature 1999, 401, 152–155. [DOI] [PubMed] [Google Scholar]
- 45. Koumura N., Geertsema E. M., Van Gelder M. B., Meetsma A., Feringa B. L., J. Am. Chem. Soc. 2002, 124, 5037–5051. [DOI] [PubMed] [Google Scholar]
- 46. Greb L., Lehn J. M., J. Am. Chem. Soc. 2014, 136, 13114–13117. [DOI] [PubMed] [Google Scholar]
- 47. Kistemaker J. C. M., Štacko P., Visser J., Feringa B. L., Nat. Chem. 2015, 7, 890–896. [DOI] [PubMed] [Google Scholar]
- 48. Roke D., Wezenberg S. J., Feringa B. L., Proc. Natl. Acad. Sci. USA 2018, 115, 9423–9431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. García-López V., Liu D., Tour J. M., Chem. Rev. 2020, 120, 79–124. [DOI] [PubMed] [Google Scholar]
- 50. Pooler D. R. S., Lubbe A. S., Crespi S., Feringa B. L., Chem. Sci. 2021, 12, 14964–14986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Costil R., Holzheimer M., Crespi S., Simeth N. A., Feringa B. L., Chem. Rev. 2021, 121, 13213–13237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hugel T., Holland N. B., Cattani A., Moroder L., Seitz M., Gaub H. E., Science 2002, 296, 1103–1106. [DOI] [PubMed] [Google Scholar]
- 53. Huang Z., Yang Q.-Z., Khvostichenko D., Kucharski T. J., Chen J., Boulatov R., J. Am. Chem. Soc. 2009, 131, 1407–1409. [DOI] [PubMed] [Google Scholar]
- 54. Yang Q.-Z., Huang Z., Kucharski T. J., Khvostichenko D., Chen J., Boulatov R., Nat. Nanotechnol. 2009, 4, 302–306. [DOI] [PubMed] [Google Scholar]
- 55. Akbulatov S., Tian Y., Huang Z., Kucharski T. J., Yang Q.-Z., Boulatov R., Science 2017, 357, 299–303. [DOI] [PubMed] [Google Scholar]
- 56. Li Q., Qian H., Shao B., Hughes R. P., Aprahamian I., J. Am. Chem. Soc. 2018, 140, 11829–11835. [DOI] [PubMed] [Google Scholar]
- 57. Olsson S., Pérez Ó. B., Blom M., Gogoll A., Beilstein J. Org. Chem. 2019, 15, 2408–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liu Y., Zhang Q., Crespi S., Chen S., Zhang X.-K., Xu T.-Y., Ma C.-S., Zhou S.-W., Shi Z.-T., Tian H., Feringa B. L., Qu D.-H., Angew. Chem. Int. Ed. 2021, 60, 16129–16138; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 16265–16274. [Google Scholar]
- 59. Gao C., Jentzsch A. V., Moulin E., Giuseppone N., J. Am. Chem. Soc. 2022, 144, 9845–9852. [DOI] [PubMed] [Google Scholar]
- 60. Feringa B. L., Browne W. R., Molecular Switches, Wiley-VCH, Weinheim, 2011. [Google Scholar]
- 61. Kathan M., Hecht S., Chem. Soc. Rev. 2017, 46, 5536–5550. [DOI] [PubMed] [Google Scholar]
- 62. Vriesema B. K., Buter J., Kellogg R. M., J. Org. Chem. 1984, 49, 110–113. [Google Scholar]
- 63. Martí-Centelles V., Pandey M. D., Burguete M. I., Luis S. V., Chem. Rev. 2015, 115, 8736–8834. [DOI] [PubMed] [Google Scholar]
- 64. Pradeilles J. A., Zhong S., Baglyas M., Tarczay G., Butts C. P., Myers E. L., Aggarwal V. K., Nat. Chem. 2020, 12, 475–480. [DOI] [PubMed] [Google Scholar]
- 65.Deposition Numbers 2165821, 2165822, 2165823, 2165824, 2165825 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 66. Bannwarth C., Ehlert S., Grimme S., J. Chem. Theory Comput. 2019, 15, 1652–1671. [DOI] [PubMed] [Google Scholar]
- 67. Grimme S., Hansen A., Ehlert S., Mewes J. M., J. Chem. Phys. 2021, 154, 064103. [DOI] [PubMed] [Google Scholar]
- 68. Neese F., Wennmohs F., Becker U., Riplinger C., J. Chem. Phys. 2020, 152, 224108. [DOI] [PubMed] [Google Scholar]
- 69. Goerigk L., Hansen A., Bauer C., Ehrlich S., Najibi A., Grimme S., Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. [DOI] [PubMed] [Google Scholar]
- 70.M. Bursch, J.-M. Mewes, A. Hansen, S. Grimme, ChemRxiv 2022, 10.26434/chemrxiv-2022-n304h. This content is a preprint and has not been peer-reviewed. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.