

Abstract
Prion diseases are transmissible, neurodegenerative disorders associated with misfolding of the prion protein. Previous studies show that reduction of microglia accelerates central nervous system (CNS) prion disease and increases the accumulation of prions in the brain, suggesting that microglia provide neuroprotection by phagocytosing and destroying prions. In Csf1r ΔFIRE mice, the deletion of an enhancer within Csf1r specifically blocks microglia development, however, their brains develop normally and show none of the deficits reported in other microglia‐deficient models. Csf1r ΔFIRE mice were used as a refined model in which to study the impact of microglia‐deficiency on CNS prion disease. Although Csf1r ΔFIRE mice succumbed to CNS prion disease much earlier than wild‐type mice, the accumulation of prions in their brains was reduced. Instead, astrocytes displayed earlier, non‐polarized reactive activation with enhanced phagocytosis of neuronal contents and unfolded protein responses. Our data suggest that rather than simply phagocytosing and destroying prions, the microglia instead provide host‐protection during CNS prion disease and restrict the harmful activities of reactive astrocytes.
Keywords: central nervous system, microglia, neurodegeneration, prion disease, reactive astrocyte
Main Points
CNS prion disease is accelerated in mice completely lacking microglia.
The rate of prion accumulation in the brain was unaltered in absence of microglia.
Microglia provide host‐protection during CNS prion disease independent of prion clearance.
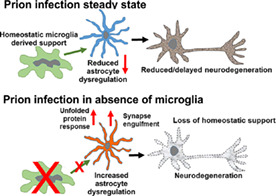
1. INTRODUCTION
The parenchymal macrophages of the central nervous system (CNS) are known as microglia (Hortega, 1919) and their proliferation and survival is dependent upon signaling via the colony stimulating factor 1 receptor (CSF1R) (Hume et al., 2020). Microglia have been attributed essential functions in the development and homeostasis of the CNS including synaptogenesis, neurogenesis, and maturation of neuronal circuits (Prinz et al., 2019). However, mice with a Csf1r hypomorphic mutation (Csf1r ΔFIRE) (Rojo et al., 2019), with conditional Csf1r deletion (using Iba1‐cre) (Nakayama et al., 2018) and rats with a Csf1r null mutation (Pridans et al., 2018) each lack microglia entirely but have normal CNS development. These findings indicate that developmental roles of microglia are redundant as studies reveal their functions can be carried out by other cells when microglia are absent (Damisah et al., 2020; Guo et al., 2019; Patkar et al., 2021). There is much greater evidence that microglia contribute to neuropathology (Prinz et al., 2019). Neurodegenerative diseases associated with mutations in microglia‐expressed genes such as CSF1R in humans have been referred to as microgliopathies (Hume et al., 2020).
Prion diseases, or transmissible spongiform encephalopathies, are fatal progressive neurodegenerative diseases to which there are no cures. Infectious prions are considered to result from the misfolding of the host's cellular prion protein (PrPC) into an abnormal disease‐associated isoform (PrPSc) (Prusiner, 1982). The accumulation of PrPSc within the brain is accompanied by the impairment of neuronal dendritic spines and synapse structures, glial cell activation, vacuolar (spongiform) degeneration and ultimately neurodegeneration. Inhibiting the proliferation and pro‐inflammatory responses of microglia via CSF1R inhibition decelerated CNS prion disease (Gómez‐Nicola et al., 2013). Conversely, the partial depletion or deficiency in microglia was reported to enhance the accumulation of prions in the brain and accelerate the onset of clinical disease (Carroll et al., 2018; Zhu et al., 2016). However, none of these studies resulted in 100% microglial ablation nor addressed the potential confounding effects of ablative cell death or bystander effects, such as impact upon other non‐microglial CSF1R‐sensitive mononuclear phagocyte populations. For example although the CSF1R‐targeting kinase inhibitor PLX5622 has been widely used to ablate the microglia in the brain, such kinase inhibitors also impact peripheral CSF1R‐dependent macrophages (Hume & Macdonald, 2012). Since the ablation of peripheral macrophages enhances prion accumulation in the secondary lymphoid tissues (Beringue et al., 2000; Maignien et al., 2005), effects on peripheral macrophage populations in the above studies also cannot be excluded.
To address the above concerns we investigated CNS prion disease in Csf1r ΔFIRE mice which have a complete and specific lack of microglia in the brain but retain brain‐associated macrophages (Rojo et al., 2019). We show that microglial‐deficiency in Csf1r ΔFIRE mice was associated with accelerated prion disease in the absence of increased PrPSc accumulation or prion‐seeding activity. Instead, earlier astrocyte activation was associated with increased engulfment of neuronal contents and unfolded protein responses without induction of genes associated with neurotoxic (A1) or neuroprotective (A2) reactive astrocyte polarization (Liddelow et al., 2017). These data indicate that microglia provide neuroprotection during CNS prion disease independently of PrPSc clearance, and restrict the harmful effects of reactive astrocyte activation. Identification of the mechanisms by which the microglia provide neuroprotection during CNS prion disease may reveal novel targets for therapeutic intervention in these and other neurodegenerative disorders.
2. MATERIALS AND METHODS
2.1. Ethics statement
Ethical approvals for the in vivo mouse experiments were obtained from The Roslin Institute's and University of Edinburgh's ethics committees. These experiments were also performed under the authority of a UK Home Office Project License and in accordance with the guidelines and regulations of the UK Home Office “Animals (scientific procedures) Act 1986.” Appropriate care was provided to minimize harm and suffering, and anesthesia was administered where necessary. Mice were humanely culled at the end of the experiments by cervical dislocation.
2.2. Mice
Csf1r ΔFIRE/WT mice produced in‐house (Rojo et al., 2019) were crossed to produce homozygous Csf1r ΔFIRE (Csf1r ΔFIRE/ΔFIRE) or Csf1r WT (Csf1r WT/WT) littermates. Offspring were genotyped as described (Rojo et al., 2019). Pups were weaned and co‐housed under specific pathogen‐free conditions. Food and water were provided ad libitum.
2.3. Prion infection
Mice were infected at 10 weeks old via intracerebral injection with 20 μl of a 1.0% (wt/vol) brain homogenate prepared from mice terminally infected with ME7 scrapie prions. Mice were culled at the intervals indicated after exposure, or observed for signs of clinical prion disease as described elsewhere (Brown & Mabbott, 2014) and culled at a standard clinical end‐point. Survival times were calculated as the interval between injection and positive clinical assessment of terminal prion disease. Groups of age‐matched Csf1r ΔFIRE mice and Csf1r WT mice were used throughout the study.
2.4. Gait analysis
Gait analysis was performed weekly using the CatWalkXT (Noldus) from 8 weeks of age until positive clinical assessment of prion disease. Uninfected mice of both genotype were monitored weekly from 8 to 30 weeks of age as controls.
2.5. Neuropathological analysis
Clinical prion disease diagnosis was confirmed by histopathological assessment of vacuolation (spongiform pathology) in the brain. Coronal sections of paraffin‐embedded brain tissue were cut at 6 μm thickness, de‐paraffinized, and stained with hematoxylin & eosin and scored for spongiform vacuolar degeneration as described previously (Fraser & Dickinson, 1967). For the construction of lesion profiles, sections were scored for the presence and severity (scale 0–5) of prion‐disease‐specific vacuolation in nine gray matter and three white matter areas: G1, dorsal medulla; G2, cerebellar cortex; G3, superior colliculus; G4, hypothalamus; G5, thalamus; G6, hippocampus; G7, septum; G8, retrosplenial and adjacent motor cortex; G9, cingulate and adjacent motor cortex; W1, inferior and middle cerebellar peduncles; W2, decussation of superior cerebellar peduncles; and W3, cerebellar peduncles.
2.6. Immunohistochemistry
Paraffin‐embedded sections (thickness 6 μm) were deparaffinized, pre‐treated by autoclaving in distilled water at 121°C for 15 min, and for PrP‐immunostaining immersed in 98% formic acid for 10 min, endogenous peroxidases were quenched by immersion in 4% H2O2 in methanol for 5 min. Sections were incubated overnight with primary antibodies (see Table 1). Primary antibody binding was detected using biotinylated goat anti‐species specific antibodies (Jackson Immunoresearch, West Grove, PA) where necessary and visualized using the Elite ABC/HRP kit (Vector Laboratories, Peterborough, UK) and diaminobenzidine (DAB) between stringent washing steps. Sections were lightly counterstained with hematoxylin and imaged on a Nikon Ni.1 Brightfield Compound upright microscope, 4×/10×/20×/air lenses, Zeiss 105c color camera & Zen 2 software for image capture. For fluorescence immunohistochemistry primary antibodies were detected with species‐specific Alexa‐Fluor 488 or 594 conjugated secondary antibodies. Phosphorylated PERK (PERK‐P) staining was detected using biotinylated goat anti‐rabbit specific antibodies (Jackson Immunoresearch, West Grove, PA) and visualized using the Elite ABC/HRP kit (Vector Laboratories, Peterborough, UK) and Tyramide Alexa‐Fluor488 (Biotium) and imaged on a Zeiss LSM 710 Confocal Microscope with 6 Laser Lines (405/458/488/514/543/633 nm)/2 PMT's + 32 channel Quasar detector. 10×/20×/40 × 1.3 na oil/60 × 1.4na oil lenses using Zen Software.
TABLE 1.
Primary antibodies
| Target | Antibody | Supplier/reference |
|---|---|---|
| β‐Actin | Mouse monoclonal C4 | Santa Cruz Biotechnology |
| C3 | Rat monoclonal RMC11H9 | Connex |
| CD44 | Biotinylated rat anti‐mouse/human monoclonal IM7 | Biolegend |
| eIF2a | Mouse monoclonal L57A5 | Cell Signaling Technology |
| Gephryin | Mouse monoclonal 45/Gephryin | BD Biosciences |
| GFAP | Rabbit anti bovine polyclonal | Dako |
| Iba1 | Rabbit polyclonal | Wako |
| Lcn2 | Goat polyclonal | R&D Systems |
| NeuN | Mouse monoclonal A60 | Millipore |
| PERK | Rabbit monoclonal C33E10 | Cell Signaling Technology |
| Phopsho‐Eif2a (Ser51) | Rabbit monoclonal 119A11 | Cell Signaling Technology |
| Phospho‐PERK (Thr980) | Rabbit monoclonal 16F8 | Cell Signaling Technology |
| PrP | Mouse monoclonal BH1 | McCutcheon et al. (2014) |
| PrP | Mouse monoclonal 6H4 | Prionics |
| PSD95 | Goat polyclonal | Abcam |
2.7. Western blot analysis
Brain homogenates (10% wt/vol) were prepared in NP40 lysis buffer (1% NP40, 0.5% sodium deoxycholate, 150 mM NaCl, 50 mM TrisHCl [pH 7.5]). For the detection of PrPSc a sample of homogenate was incubated at 37°C for 1 h with 20 μg/ml proteinase K (PK) and digestion halted by addition of 1 mM phenylmethylsulfonyl fluoride. Samples were denatured at 98°C for 15 min in 1× SDS sample buffer (Life Technologies) and separated via electrophoresis through 12% Tris‐glycine polyacrylamide gels (Nupage, Life Technologies) and transferred to polyvinylidene difluoride PVDF membranes by semi‐dry electroblotting. Primary antibodies (Table 1) were detected by horseradish peroxidase‐conjugated goat anti‐species specific antibody (Jackson Immunoresearch) and visualized via chemiluminescence (BM Chemiluminescent substrate kit, Roche, Burgess Hill, UK) as described previously (Bradford et al., 2017).
2.8. Image analyses
Image analysis was performed using ImageJ software (http://imagej/nih.gov/ij) (Schneider et al., 2012). The magnitude of PrPd, GFAP, and CD44 immunostaining on DAB stained sections was compared as previously described (Bradford et al., 2019). Briefly, the optical density (OD) values for immunostaining were calculated using ImageJ software following H‐DAB deconvolution. Mean gray OD values were measured from DAB grayscale images (scaled 0–255) and expressed as a % relative intensity by dividing by the maximum value (255). Immunofluorescent images were analyzed using ImageJ as previously described (McCulloch et al., 2011). Briefly intensity thresholds were applied and then the number of pixels of each color counted and presented as a proportion of the total pixel area under analysis (% area coverage). The preferential co‐localization of fluorochromes was determined as previously described (McCulloch et al., 2011) by comparing the observed distribution of colors with those predicted by the null hypothesis that each element of positive staining was randomly and independently distributed. Values significantly greater (P < .05) than the null hypothesis confirm significant co‐localization of fluorochromes. The assessment of relative synaptic protein phagocytosis was calculated as the % of PSD95 or gephyrin staining co‐localized with GFAP relative to total of each synpaptic protein. Western blot images were subjected to densitometric analyzed by ImageJ. Briefly lanes and bands were identified, threshold levels set and area under the curve measurements taken (pixels). For PrPC and PrPSc relative expression levels were calculated as a percentage relative to a control normal brain PrPC measurement.
2.9. Real‐time quaking induced conversion (RT‐QuiC)
Brain homogenates were diluted at 10–3 vol/vol in PBS. RT‐QuIC reaction mix prepared as follows: 10 mM phosphate buffer (pH 7.4), 170 mM NaCl (total 300 mM including phosphate buffer), 0.1 mg/ml recombinant PrPc (Bank Vole 23‐230, [Orrú et al., 2015] construct kindly provided by Byron Caughey, Rockey Mountain Laboratories, Montana, USA), 10 μM Thioflavin‐T (ThT), and 10 μM ethylenediaminetetraacetic acid tetrasodium salt (EDTA). Reactions were performed in quadruplicate. Aliquots of the reaction mix (98 μl) were loaded into each well of a black 96‐well plate with a clear bottom (Thermo Scientific) and seeded with 2 μl of diluted brain homogenate. Samples were incubated in a FLUOstar® OMEGA plate reader (BMG LABTECH Ltd.) at 42°C for 80 h with intermittent shaking cycles: 1 min shake (double orbital, 700 rpm), 1 min rest. Fluorescence measurements (450 nm excitation and 480 nm emission; bottom read), referred to as relative fluorescent units (rfu) were taken every 15 min. A baseline rfu of ~38,000 for unseeded and initial BH seeded reactions were recorded, with saturation occurring at 260,000 rfu. All 4 quadruplicates of the 8 test samples, displayed a significant rise in rfu over time; a sample was considered “positive for PrP seeding” if replicates crossed a threshold of fluorescence set at 50,000 rfu based on the mean ± 10 SD (36,941 ± 8348) of the unseeded negative control samples analyzed. The mean time for each quadruplicate reading to reach the 50,000 rfu threshold was calculated and plotted.
2.10. Gene expression analysis via RT‐qPCR
Total RNA was isolated from brain using RNABee (AMSBio, Abingdon, UK) and RNeasy Mini kit (Qiagen). RNA was Dnase treated (Promega) to remove genomic DNA. Reverse transcription of polyA mRNA from 5 μg total DNA‐free RNA was performed using Superscript First Strand Synthesis (Invitrogen) with Oligo‐dT primers. Quantitative PCR (qPCR) were performed using SYBR master mix (Rox) (Roche) on an MX3005pro (Stratagene) using the primer sequences detailed (Table 2). Gene expression relative to naïve Csf1r WT mice was calculated using the ΔΔCT method (Livak & Schmittgen, 2001) using Rpl19 as a reference gene.
TABLE 2.
Oligonucleotide primers
| Gene | Forward primer | Reverse primer |
|---|---|---|
| Aif1 | GGATCAACAAGCAATTCCTCGA | CTGAGAAAGTCAGAGTAGCTGA |
| B3gnt5 | CGTGGGGCAATGAGAACTAT | CCCAGCTGAACTGAAGAAGG |
| Ccl2 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| Ccr2 | AGCACATGTGGTGAATCCAA | TGCCATCATAAAGGAGCCA |
| Cd44 | ACCTTGGCCACCACTCCTAA | GCAGTAGGCTGAAGGGTTGT |
| Cd44v6 | CTAATAGTACAGCAGAAGCAGCAGCTA | CCTGCCATCCGTTCTGAAA |
| Csf1r | AGGCAGGCTGGAATAATCTGACCT | CGTCACAGAACAGGACATCAGAGC |
| Cx3cr1 | CAGCATCGACCGGTACCTT | GCTGCACTGTCCGGTTGTT |
| Gbp2 | GGGGTCACTGTCTGACCACT | GGGAAACCTGGGATGAGATT |
| Gfap | AGAAAGGTTGAATCGCTGGA | CGGCGATAGTCGTTAGCTTC |
| Itgam | TGGCCTATACAAGCTTGGCTTT | AAAGGCCGTTACTGAGGTGG |
| Psmb8 | CAGTCCTGAAGAGGCCTACG | CACTTTCACCCAACCGTCTT |
| Ptx3 | AACAAGCTCTGTTGCCCATT | TCCCAAATGGAACATTGGAT |
| Srgn | GCAAGGTTATCCTGCTCGGA | TGGGAGGGCCGATGTTATTG |
| Tmem119 | GTGTCTAACAGGCCCCAGAA | AGCCACGTGGTATCAAGGAG |
| Tnf | TGTGCTCAGAGCTTTCAACAA | CTTGATGGTGGTGCATGAGA |
| Rpl19 | GAAGGTCAAAGGGAATGTGTTCA | CCTTGTCTGCCTTCAGCTTGT |
2.11. Statistical analyses
Statistical analyses were performed in GraphPad Prism 6.01 (GraphPad Software Inc.). Survival curve analysis was performed by Log‐rank [Mantel Cox] Test. Image and gene expression analyses were performed by Student's t‐test (two groups) or ANOVA (four groups). Results are expressed as dot plots of individual animal observations with median values indicated (bar). CatWalkXT analysis was performed using two‐way ANOVA and expressed as group mean with 95% confidence interval. Values of P < .05 were accepted as significant.
3. RESULTS
3.1. Csf1r ΔFIRE mice rapidly succumb to prion disease in the absence of microglia
To determine the role of microglia in prion disease, groups of homozygous microglia‐deficient Csf1r ΔFIRE transgenic mice and wild‐type (Csf1r WT) littermate controls were injected intracerebrally (IC) with the ME7 strain of mouse adapted scrapie prions. As expected, all the Csf1r WT mice displayed clinical signs of prion disease from approximately 140 days after injection and succumbed to terminal disease with a mean survival time of 167 ± 5 days. In Csf1r ΔFIRE mice, clinical manifestations of prion disease were evident by 98 days after infection and progressed rapidly resulting in a mean survival time of 124 ± 2 days (Figure 1a).
FIGURE 1.

Csf1r ΔFIRE mice rapidly succumb to prion disease. (a) Survival curve following intracerebral injection of ME7 prions into Csf1r WT or Csf1r ΔFIRE mice (N = 5–6 mice/group). Log‐rank Mantel Cox test, P = .0018. (b) Catwalk XT automated gait analysis weekly assessment of hind base of stance in age‐matched uninfected Csf1r WT or Csf1r ΔFIRE mice. Points represent group mean and error bars 95% confidence interval. (c) Weekly assessment of hind base of stance in prion‐infected Csf1r WT or Csf1r ΔFIRE mice. (d) Weekly assessment of right hind (RH) paw print area in age‐matched uninfected mice. Two‐way ANOVA. (e) Weekly assessment of right hind (RH) paw print area in prion‐infected mice. (f) Weekly assessment of right front (RF) paw intensity in age‐matched uninfected mice. (g) Weekly assessment of right front (RF) paw intensity in prion‐infected mice. *P < .05; **P < .005; ****P < .0001; Two‐way ANOVA, Sidak's multiple comparisons test. Panels B‐G, N = 6–10 mice/group
3.2. Longitudinal gait analysis during prion infection
CNS prion disease in mice is associated with profound motor‐coordination disturbances (Heitzman & Corp, 1968). We therefore used longitudinal gait analysis to determine whether microglia‐deficiency affected the onset of motor disturbances during CNS prion disease (Figure 1b–g). Our analyses revealed no significant impact of the complete absence of microglia in the cerebellum of Csf1r ΔFIRE mice on motor function analyzed at any time point in the absence of the prion challenge (Figure 1b,d,f). As expected, various motor functions were rapidly impacted in prion disease. The base of stance (BOS, or distance between the hind paws) increased gradually with age in uninfected mice regardless of genotype (Figure 1b) but diverged by 10 days post infection (dpi) with prions and was maintained until 63 dpi (9 weeks) in Csf1r ΔFIRE mice and 108 dpi (15 weeks) in Csf1r WT mice (Figure 1c). At the onset of clinical signs of prion disease at 101 dpi Csf1r ΔFIRE mice were hyperactive and continued to perform Catwalk Gait analysis with ease until the terminal stage. In contrast, Csf1r WT mice at the onset of clinical symptoms at 143 dpi were severely ataxic and unable to cross the Catwalk within the time‐period required for data acquisition (Figure 1c).
Due to the potential effects of IC injection of prions into the right hemisphere on the contralateral paws, we analyzed the effects on footfall using only the unilateral right front and hind paws. In uninfected mice, hind paw area remained unchanged with no statistically significant differences between Csf1r WT and Csf1r ΔFIRE mice (Figure 1d). A significant increase in hind paw area was observed at 10 dpi in prion‐infected Csf1r ΔFIRE mice with a further large increase at 38 dpi. In contrast Csf1r WT mice did not experience a large increase in right hind paw area until 129 dpi, 3 weeks before commencement of clinical signs, these data are indicative of a more rapid response to prion infection in Csf1r ΔFIRE mice (Figure 1e).
In age‐matched uninfected mice, front paw intensity remained unchanged with no statistically significant differences between Csf1r WT and Csf1r ΔFIRE mice (Figure 1f). Concurrent with changes in footprint area, footfall intensity was increased in the prion‐infected mice (Figure 1g). Front footfall intensity increased significantly from 3 dpi in Csf1r WT mice and this increase was maintained almost throughout the duration of the prion infection until the terminal stage. In contrast, footfall intensity in the prion‐infected Csf1r ΔFIRE mice commenced at 10 dpi and was maintained until onset of clinical symptoms at 101 dpi (Figure 1g).
3.3. Detection of microgliosis in prion‐infected WT mice
The brains of terminal prion‐infected Csf1r WT mice displayed abundant, activated microglia (allograft inhibitory factor‐1‐positive [AIF1+] cells), whereas these cells and other potential AIF1+ CNS‐infiltrating mononuclear phagocyte populations remained absent in uninfected and terminally‐affected Csf1r ΔFIRE mice, despite evidence of prion‐induced vacuolation and synaptic loss as revealed by co‐staining with the post‐synaptic protein PSD95 (Figure 2a,b) (Rojo et al., 2019). RT‐qPCR analysis confirmed that Aif1 (Figure 2c) and Csf1r (Figure 2d) mRNA expression was significantly increased in the brains of terminally‐affected Csf1r WT mice when compared to age‐matched uninfected controls, but remained almost undetectable in the brains of Csf1r ΔFIRE mice even at the terminal stage of prion disease. Expression of other important microglia genes including Itgam (encoding CD11b; Figure 2e), Cx3cr1 (Figure 2f) and Tmem119 (Figure 2g) were also significantly increased in Csf1r WT mice, but absent in Csf1r ΔFIRE mice at the terminal stage of prion infection. Together, these data show that onset of CNS prion disease was accelerated in Csf1r ΔFIRE mice in the complete absence of microglia. The monocyte chemokine receptor Ccr2 (Figure 2h) was significantly increased following prion infection in Csf1r WT mice, but not in Csf1r ΔFIRE mice, despite significantly increased expression of the monocyte chemoattractant Ccl2 in brains of infected Csf1r WT mice and Csf1r ΔFIRE mice (Figure 2i). Notably, the Csf1r ΔFIRE mice are not monocyte‐deficient but their monocytes lack CSF1R expression (Rojo et al., 2019). The IHC and expression profiling indicates that the Csf1r ΔFIRE mutation also prevents monocyte recruitment into the injured brain. Why monocytes aren't recruited into the brains of Csf1r ΔFIRE mice is uncertain. Studies by Gómez‐Nicola and colleagues have similarly shown that CNS prion disease was not associated with significant monocytic recruitment in wild‐type mice, and the absence circulating monocytes in Ccr2 −/− mice had little, if any, impact on the microgliosis or the progression of CNS disease (Gómez‐Nicola et al., 2014).
FIGURE 2.
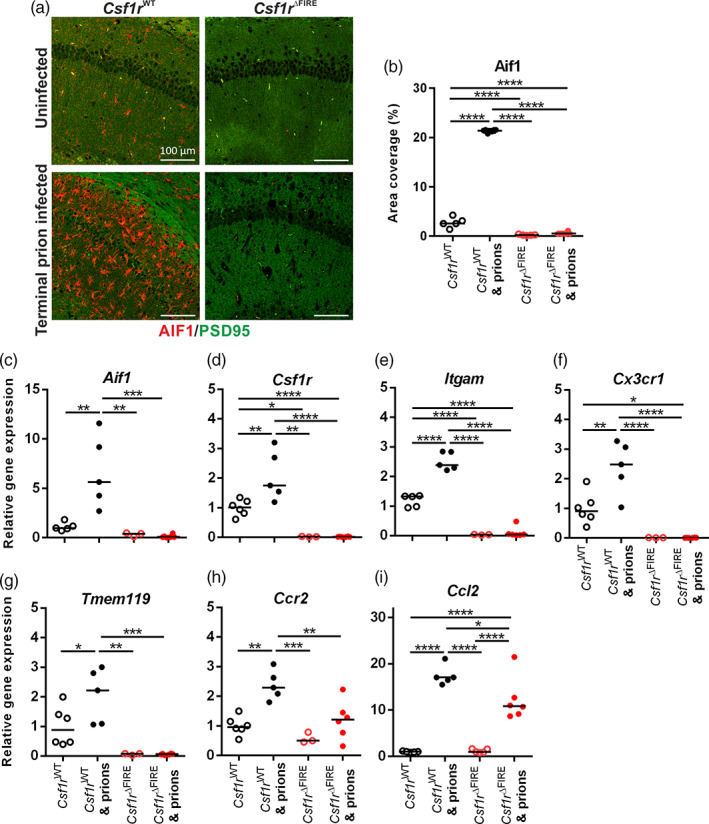
Csf1r ΔFIRE mice succumb to prion disease in the absence of microglia. (a) Immunohistochemical assessment of AIF1 (red) in hippocampus CA1 of terminal prion infected or age‐matched uninfected Csf1r WT or Csf1r ΔFIRE mice. Sections were counterstained to detect the post‐synaptic protein PSD95 (green). Scale bars = 100 μm. (b) AIF1 immunostaining quantitation expressed as % area coverage in hippocampus CA1. (c–i) RT‐qPCR of (c) Aif1, (d) Csf1r, (e) Itgam, (f) Cx3cr1, (g) Tmem119, (h) Ccr2, and (i) Ccl2 mRNA in uninfected or terminal prion‐infected brains from Csf1r WT or Csf1r ΔFIRE mice. Points show individual mice. Horizontal bar = median. *P < .05; **P < .01; ****P < .0001; ANOVA. N = 5–6 mice/group
3.4. Unaltered neuronal loss but reduced prion accumulation in the brains of microglia‐deficient mice
Assessment of hippocampal CA1 pyramidal cells in hematoxylin and eosin stained brain sections (Figure 3a) revealed no difference in neuronal density or the frequency of pyknotic (apoptotic) neuronal nuclei between terminal prion‐infected Csf1r WT and Csf1r ΔFIRE mice despite the difference in time of onset of pathology (Figure 3b,c, respectively). The prion‐specific vacuolation was also comparable in most brain areas of terminal prion‐infected Csf1r WT and Csf1r ΔFIRE mice, except for a significant reduction of vacuolation in the cerebellar cortex (G2), inferior and middle cerebellar peduncles (W1) and decussation of superior cerebellar peduncles (W2) of brains from prion‐infected Csf1r ΔFIRE mice (Figure 3d). This suggested the pathological impact of prion infection upon the cerebellum was reduced in the Csf1r ΔFIRE mice at the terminal stage of prion disease.
FIGURE 3.

Microglia‐deficiency effects on prion‐specific vacuolation and prion accumulation. (a) Hematoxylin and eosin stained hippocampus CA1 of terminal prion infected or age‐matched uninfected Csf1r WT or Csf1r ΔFIRE mice. Scale bars = 200 μm. (b) Hippocampal CA1 pyramidal cell density in terminal prion infected Csf1r WT or Csf1r ΔFIRE mice. Student's t‐test. (c) Assessment of neuronal condition expressed as percentage of total neurons pyknotic in terminal prion infected Csf1r WT or Csf1r ΔFIRE mice. Student's t‐test. (d) Lesion profile analysis of prion‐infected brains. Points represent the mean vacuolation score, error bars = ± SEM. Two‐way ANOVA, Sidak's multiple comparisons test. **P < .005; ****P < .0001. (e) Microarray analysis of relative gene expression of Prnp in the brain. Student's t‐test. (f) Microarray analysis of relative gene expression of Csf1 in the brain. Student's t‐test, ****P < .0001. (g) Western blot analysis of uninfected Csf1r WT and Cs1fr ΔFIRE mouse brain, probed with anti‐PrP antibody clone BH1. Relative protein sizes indicated in kilodaltons (kDa). (h) Quantitation of relative brain PrPC expression in the brains of uninfected Csf1r WT and Cs1fr ΔFIRE mice. Student's t‐test. (i) Western blot analysis of terminal prion‐infected Csf1r WT and Cs1fr ΔFIRE mouse brain, probed with anti‐PrP antibody clone BH1. Relative protein sizes indicated in kilodaltons (kDa). (j) Quantitation of relative PrPSc accumulation in the brains of terminal prion‐infected Csf1r WT and Cs1fr ΔFIRE mice. Students t‐test. ***P < .001. Panels a–d, N = 5–6 mice/group. Panels e and f, 3 mice/group. Panels g–j, N = 3–6 mice/group. Panels b, c, e, f, h, and j. points show individual mice, horizontal bar = median.
The relative expression level of PrPC can directly influence prion disease duration (Manson et al., 1994). Previous expression profiling of the cortex of Csf1r ΔFIRE compared to Csf1r WT mice revealed no impacts on expression of Prnp mRNA (which encodes PrPC) or any other neuron‐associated transcripts (Rojo et al., 2019). Expression of Prnp mRNA in the hippocampus in published mRNA microarray data GEO dataset GSE108207 (Rojo et al., 2019) (Figure 3e) was similar in each mouse strain, despite loss of Csf1r expression (Figure 3f). Whole brain PrPC protein expression (Figure 3g,h) was also similar between naïve Csf1r ΔFIRE mice and Csf1r WT mice. Partial‐deficiency or temporary ablation of microglia during CNS prion infection was reported to accelerate the accumulation of prion‐disease‐specific PrPSc in the brain (Carroll et al., 2018; Zhu et al., 2016). By contrast, PrPSc accumulation was reduced in the brains of terminally prion‐infected Csf1r ΔFIRE compared to Csf1r WT mice (Figure 3i,j).
3.5. Altered neuropathology in the absence of microglia during CNS prion disease
Consistent with data presented in Figure 3i,j, immunostaining for prion disease‐associated PrP (PrPd) in the brains of Csf1r ΔFIRE mice at the terminal stage was approximately 50% of the intensity detected in Csf1r WT mice (Figure 4a,b). Since the accumulation of PrPSc within the brain increases as the infection proceeds (Tatzelt et al., 1999), this finding is most likely a consequence of their significantly shortened survival times, and implies that microglia deficiency produces hyper‐sensitivity to the accumulation of PrPSc.
FIGURE 4.
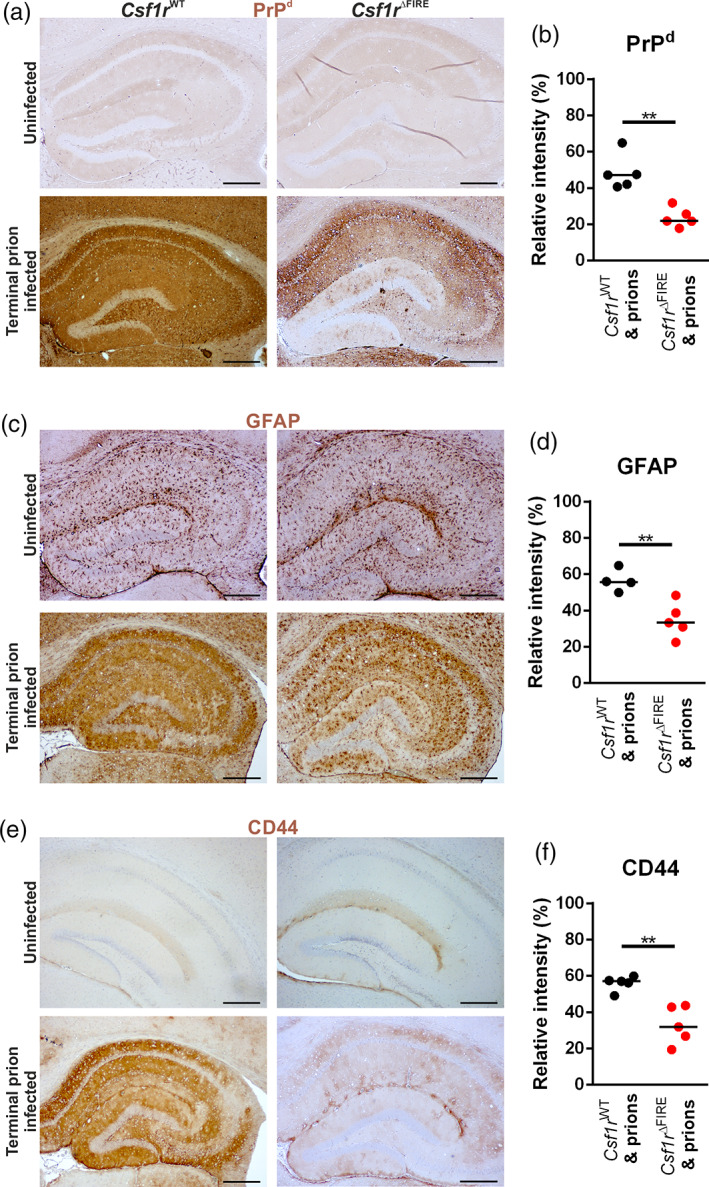
Microglial deficiency reduces terminal neuropathology. (a, c, e) Immunohistochemical assessment of (a) PrPd accumulation, (c) GFAP expression and (e) CD44 expression (brown) in the hippocampus of terminal prion infected or age‐matched uninfected Csf1r WT and Cs1fr ΔFIRE mice. DAB (brown) immunostaining lightly counterstained with hematoxylin (blue). Scale bars = 500 μm. (b) PrPd immunostaining quantified by relative intensity. (d) GFAP immunostaining quantified by relative intensity. (f) CD44 immunostaining quantified by relative intensity. Points show individual mice. Horizontal bar = median. Student's t‐test, **P < .005. N = 4–5 mice/group
CNS prion disease is accompanied by extensive reactive astrocytosis characterized by high levels of expression of glial fibrillary acidic protein (GFAP), CD44 and the CD44v6 alternative splice variant (Bradford et al., 2019). Microglia and microglial‐derived factors have been shown to induce reactive astrocytosis in a range of neurodegenerative conditions (Kunyu et al., 2019; Liddelow et al., 2017; Vainchtein & Molofsky, 2020). Despite the absence of microglia, reactive astrocytes expressing high levels of GFAP (Figure 4c,d) and CD44 (Figure 4e,f) were increased in the brains of prion‐infected Csf1r ΔFIRE mice but the level of GFAP+ and CD44+ immunostaining was lower than in infected Csf1r WT mice. As astrocyte activation also increases temporally during CNS prion infection (Bradford et al., 2019; Hwang et al., 2009), this again is most likely a consequence of the Csf1r ΔFIRE mice succumbing to terminal prion disease significantly earlier than infected Csf1r WT mice. In summary, these data reveal that although CNS prion disease duration is shorter in microglia‐deficient Csf1r ΔFIRE mice, this is not accompanied by increased neuronal vacuolation, prion accumulation, or upregulation of GFAP or CD44 at the terminal stage, when compared to infected Csf1r WT mice.
3.6. Absence of induction of neurotoxic “A1” or neuroprotective “A2” reactive astrocyte‐associated genes in the brains of prion‐infected microglia‐deficient mice
Reactive astrocytes may be classified into distinct functional subclasses; an A1 subclass with neurotoxic activity and A2 astrocytes considered neurotrophic (Liddelow et al., 2017). Microglia‐derived factors have been implicated in the induction of pan‐ and A1‐reactive astrocyte‐associated genes (Liddelow et al., 2017). Consistent with the immunohistochemistry data presented in Figure 4, high levels of mRNA encoding the pan‐reactive astrocyte‐associated genes Gfap, Cd44, and Cd44v6 were detected in the brains of prion‐infected Csf1r WT mice (Figure 5a–c, respectively). The LPS‐mediated induction of expression of pan‐reactive astrocyte‐associated genes including Gfap and Cd44 was reported to be blocked in microglia‐deficient Csf1r −/− mice (Liddelow et al., 2017). However, because of the limited viability of Csf1r −/− mice, these studies were performed at postnatal day 8, and these mice are also deficient in peripheral macrophage populations. In the Csf1r ΔFIRE mice, the expression of Gfap, Cd44, and Cd44v6 was upregulated in response to prion infection despite the complete absence of microglia. These data demonstrate CNS prion‐induced reactive astrocyte activation is not dependent on the presence of microglia.
FIGURE 5.

Microglia‐deficiency alters astrocyte response to prions. RT‐qPCR analysis of (a) Gfap, (b) Cd44, (c) Cd44v6, (d) Gbp2, (e) Psmb8, (f) Srgn, (g) Tnf, (h) B3gnt5, and (i) Ptx3 mRNA in the brains of terminal prion infected or age‐matched uninfected Csf1r WT or Csf1r ΔFIRE mice. Points show individual mice. Horizontal bar = median. *P < .05; **P < .005; ***P < .001; ****P < .0001; ANOVA. N = 3–6 mice/group
At the terminal stage of prion disease, the reactive astrocytes display a dysregulated transcriptional signature including expression of both A1 and A2 astrocyte‐associated genes (Donaldson et al., 2020; Hartmann et al., 2019). The expression of the neurotoxic A1 astrocyte‐associated genes Gbp2, Psmb8, and Srgn was upregulated in the brains of terminal prion‐infected Csf1r WT mice, but absent in Csf1r ΔFIRE mice (Figure 5d–f). Microglia‐derived cytokines including tumor necrosis factor (TNFα) are important inducers of neurotoxic A1 reactive astrocyte activation. Indeed, Tnf was elevated in the brains of prion‐infected Csf1r WT mice but absent in Csf1r ΔFIRE mice, coincident with the lack of induction of A1 reactive astrocyte‐associated gene expression. Consistent with previous data from the brains of mice infected with ME7 scrapie prions (Donaldson et al., 2020), neuroprotective A2 astrocyte‐associated genes (B3gnt5 and Ptx3) were not induced in the brains of infected Csf1r WT or Csf1r ΔFIRE mice (Figure 5h,i). Together these data show that CNS prion disease in microglia‐deficient Csf1r ΔFIRE mice is accompanied by dysregulated reactive astrocytosis that lacks evidence of a typical neurotoxic A1 or neuroprotective A2 transcriptional profile.
3.7. Csf1r ΔFIRE mice display accelerated onset of vacuolation but unaltered kinetics of prion accumulation
To determine how disease progression was affected by the absence of microglia, brains were collected from groups of Csf1r WT and Csf1r ΔFIRE mice at 98 dpi prior to the histopathological detection of neuronal loss. Prion‐specific vacuolation was already more severe in prion‐infected Csf1r ΔFIRE mice in multiple brain regions, including dorsal medulla, superior colliculus, hypothalamus and cerebellar peduncles (Figure 6a; vacuolation scoring areas G1, G3, G4, and W3). However, within the hippocampus little evidence of prion‐specific vacuolation (Figure 6a, vacuolation scoring area G6; Figure 6c upper panels) or neuronal loss (Figure 6b) was observed in brains from each group at this time. The early “synaptic” patterned PrPd deposition and mild reactive astrocytosis also presented to a similar extent in the hippocampus of infected Csf1r ΔFIRE and Csf1r WT mice at this time point (Figure 6c, middle and lower panels, respectively).
FIGURE 6.
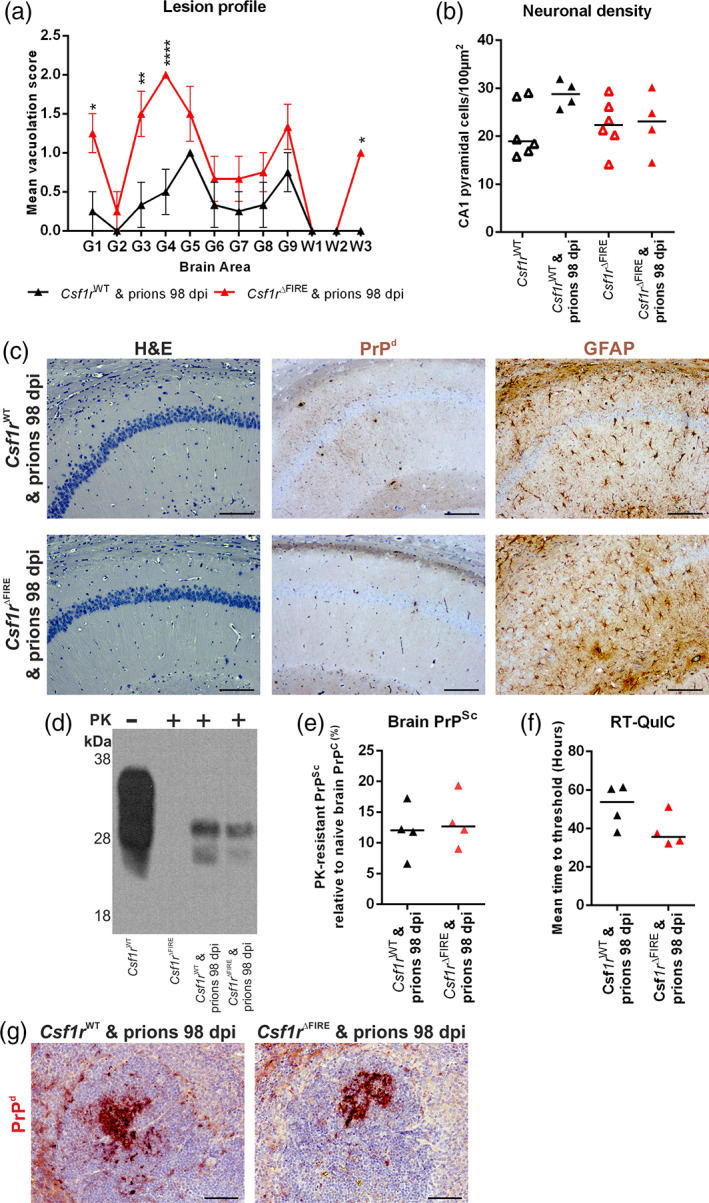
Microglial deficiency accelerates prion vacuolation but not brain or peripheral prion accumulation. (a) Lesion profile analysis of prion‐infected brains at 98 dpi (N = 4 mice/group). Points represent the mean vacuolation score, error bars = SEM. *P < .05; **P < .01; ****P < .0001; two‐way ANOVA, Sidak's multiple comparisons test. (b) Hippocampal CA1 pyramidal neuron density was assessed in 98 dpi prion‐infected mice and age‐matched uninfected (N = 4–6 mice/group). Points show individual mice, bar = median. Not significantly different, ANOVA. (c) Hematoxylin and eosin (H&E) stained sections used for vacuolation and neuronal density analyses. Immunohistochemical analysis of PrPd accumulation and GFAP expression in 98 dpi prion‐infected Csf1r WT and Csf1r ΔFIRE hippocampus CA1. Scale bars = 200 μm. (d) Western blot analysis as indicated to determine the relative amount of PrPSc accumulation in the brains of mice from each group at 98 dpi with prions. (e) Quantitation of PrPSc levels in brains of 98 dpi prion‐infected Csf1r WT and Csf1r ΔFIRE mice. Points show individual mice, bar = median. Not significantly different, Student's t‐test. (F) Relative prion seeding activities in brains at 98 dpi with prions were quantified in vitro by RT‐QuIC expressed as mean time to threshold. Points show individual mice, bar = median. Not significantly different, Student's t‐test. (g) Immunohistochemical analysis of PrPd accumulation in spleens of prion‐infected Csf1r WT and Csf1r ΔFIRE mice at 98 dpi. PrPd immunostaining (red) counterstained with hematoxylin (blue). Scale bar = 100 μm. Panels c–g, N = 4 mice/group
The levels of PrPSc in the brains of Csf1r ΔFIRE or Csf1r WT mice at 98 dpi were indistinguishable (Figure 6d,e). In parallel, the highly sensitive real‐time quaking‐induced conversion (RT‐QuIC) assay was used to quantify the relative prion seeding activity present within the brains of each group (Atarashi et al., 2011). Consistent with data presented in Figure 6c–e, the relative levels of prion seeding activity were also similar in the brains of infected Csf1r ΔFIRE mice and Csf1r WT mice (Figure 6f).
After IC injection, some of the infectious prions from the inoculum spread to the spleen via the bloodstream where they accumulate on stromal follicular dendritic cells (FDC) (Brown et al., 1999). Following accumulation within the spleen and other secondary lymphoid organs, the prions can subsequently spread back to the brain (Brown et al., 2012; Brown & Mabbott, 2014). In the absence of peripheral macrophages, the accumulation of prions in secondary lymphoid tissues is enhanced (Beringue et al., 2000; Maignien et al., 2005). Since certain peripheral macrophages will also have been ablated in the previous studies (Carroll et al., 2018; Lei et al., 2020; Zhu et al., 2016) it is plausible that this may have increased the burden of prions in the spleen and other secondary lymphoid organs, and by doing so, enhanced their rate of spread to the brain. However, such an effect was unlikely to responsible for the accelerated prion disease in Csf1r ΔFIRE mice, as a similar abundance of prion‐specific PrPd was detected on FDC in the spleens of Csf1r ΔFIRE mice and Csf1r WT mice (Figure 6g). This is consistent with the demonstration that spleen macrophage populations are not affected in Csf1r ΔFIRE mice (Rojo et al., 2019).
3.8. Accelerated onset of reactive astrocyte activation in the absence of microglia
The increased prion‐specific vacuolation in multiple brain regions by 98 dpi (Figure 6a), for example within the superior colliculus and hypothalamus (vacuolation scoring areas G3 and G4, respectively) in the Csf1r ΔFIRE mice, was not accompanied by evidence of loss of neuronal nuclear antigen NeuN+ neurons within these regions in either Csf1r ΔFIRE or Csf1r WT mice at this time (Figure 7a,b). Instead, the increased vacuolation (Figure 7c) in the intermediate gray layer (motor associated area) of the superior colliculus of prion‐infected Csf1r ΔFIRE mice compared to Csf1r WT mice at 98 dpi was accompanied by increased expression of the pan‐astrocytic activation marker CD44 (Figure 7d,e) (Bradford et al., 2019) and increased frequency of GFAP+ morphologically reactive astrocytes (Figure 8a,b).
FIGURE 7.

Accelerated astrocyte activation in the absence of microglia. (a) Superior colliculus (G3) and (b) hypothalamus (G4) neuronal density was assessed via quantitation of the density of NeuN+ cells in 98 dpi prion‐infected or age‐matched uninfected Csf1r WT and Csf1r ΔFIRE mice. Not significantly different, ANOVA. N = 2–5 mice/group. (c) Hematoxylin and eosin (H&E) stained superior colliculus in 98 dpi prion‐infected Csf1r WT and Csf1r ΔFIRE mice. Scale bars = 100 μm. (d) Immunohistochemical assessment of CD44 expression in 98 dpi prion‐infected Csf1r WT and Cs1fr ΔFIRE superior colliculus. Scale bars = 200 μm. (e) Quantitation of CD44% area coverage in superior colliculus. Points show individual mice. Horizontal bar = median. *P < .05, Student's t test. N = 4 mice/group
FIGURE 8.
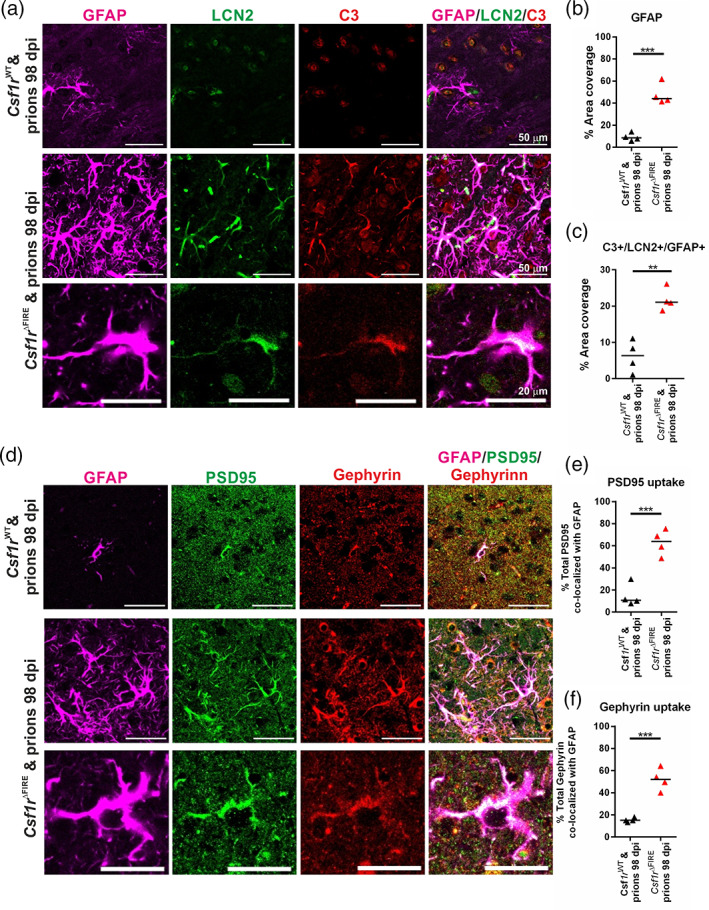
Increased astrocyte synaptic phagocytosis in the absence of microglia (a) Immunofluorescent assessment of GFAP (violet), lipocalin2 (LCN2, green) and complement component C3 (red) in 98 dpi prion‐infected Csf1r WT and Cs1fr ΔFIRE superior colliculus. Scale bars = 50 or 20 μm as indicated. (b) Quantitation of GFAP % area coverage in superior colliculus. (c) Quantitation of C3+/LCN2+/GFAP+ astrocytes. (d) Immunofluorescent assessment of GFAP (violet), and the post‐synaptic proteins PSD95 (green) and gephyrin (red) in 98 dpi prion‐infected Csf1r WT and Cs1fr ΔFIRE superior colliculus. Scale bars = 50 or 20 μm as indicated. (E) quantitation of PSD95 uptake by astrocytes expressed as % of total PSD95 colocalized with GFAP. (F) Quantitation of gephyrin uptake by astrocytes expressed as % of total gephryin colocalized with GFAP. Points show individual mice. Horizontal bar = median. **P < .01; ***P < .001, Student's T test. N = 4 mice/group
The innate immune proteins complement component C3 and neutrophil gelatinase‐associated lipocalin/lipocalin‐2 (NGAL/LCN2) have previously been shown to be upregulated by neurotoxic astrocytes in response to prion infection (Hartmann et al., 2019; Kushwaha et al., 2021; Smith et al., 2020). We observed a greater abundance of C3+/LCN2+/GFAP+ morphologically reactive astrocytes within the superior colliculus in Csf1r ΔFIRE compared to Csf1r WT mice at 98 dpi (Figure 8a,c).
Astrocytes in the steady state prune synapses to help maintain neural circuitry (Chung et al., 2013). However, abnormal astrocyte synaptic engulfment has been implicated in the pathogenesis of some neurodegenerative disorders (reviewed in Lee & Chung, 2019), and synaptic alterations are considered to contribute to the early behavioral changes observed during CNS prion disease (Cunningham et al., 2003). We therefore assessed the localization of the post‐synaptic proteins gephyrin and post‐synaptic density protein 95 (PSD95) in relation to GFAP+ astrocytes (Figure 8d). The co‐localization of both post‐synaptic marker proteins in punctate inclusions within GFAP+ morphologically reactive astrocytes was increased in the superior colliculus of Csf1r ΔFIRE compared to Csf1r WT mice at 98 dpi (Figure 8d). Morphometric analyses suggested over half of the total amount of these synaptic proteins detected in Csf1r ΔFIRE mice were within astrocytes (Figure 8e,f). Furthermore, additional analyses suggested that the preferential co‐localization of PSD95 and gephyrin within the GFAP+ reactive astrocytes of prion‐infected Csf1r ΔFIRE mice was highly significant compared to the null hypothesis that the immunostaining was randomly distributed (PSD95/GFAP, P < .0002; Gephyrin/GFAP, P < 7 × 10−5). Together, these data reveal a statistically significant increase in synaptic engulfment by reactive astrocytes in the brains of prion‐infected Csf1r ΔFIRE mice compared to Csf1r WT mice at 98 dpi within this region.
3.9. Accelerated onset of unfolded protein response in the absence of microglia
Accumulation of misfolded PrPSc in the brain triggers the unfolded protein response in reactive astrocytes (Smith et al., 2020). Specifically, phosphorylation of protein kinase‐like endoplasmic reticulum kinase (PERK) causes the transient shutdown of protein synthesis via phosphorylation of eukaryotic translation initiation factor 2A (eIF2α). Inhibition of PERK‐eIF2α signaling in astrocytes alleviated prion‐induced neurodegeneration (Smith et al., 2020).
Levels of PERK and eIF2α expression were assessed in brains of age‐matched uninfected mice and revealed no difference in uninfected mice Csf1r WT and Csf1r ΔFIRE mice (Figure 9a–c). Similarly, in uninfected mice we were unable to detect phosphorylated PERK and eIF2α (Figure 9a). However, the levels of phosphorylated PERK and eIF2α were statistically significantly increased in the brains of infected Csf1r ΔFIRE mice when compared to infected Csf1r WT mice at 98 dpi (Figure 9d–f). Immunohistochemical analysis also revealed earlier expression of phosphorylated PERK expression in GFAP+ reactive astrocytes and neurons in infected Csf1r ΔFIRE mice, particularly within the superior colliculus (Figure 9g), coincident with the increased vacuolation and reactive astrocytosis in this region at 98 dpi (Figure 7c). Conversely, little if any, phosphorylated PERK expression in GFAP+ reactive astrocytes was detected in infected Csf1r WT mice at 98 dpi (Figure 6g).
FIGURE 9.
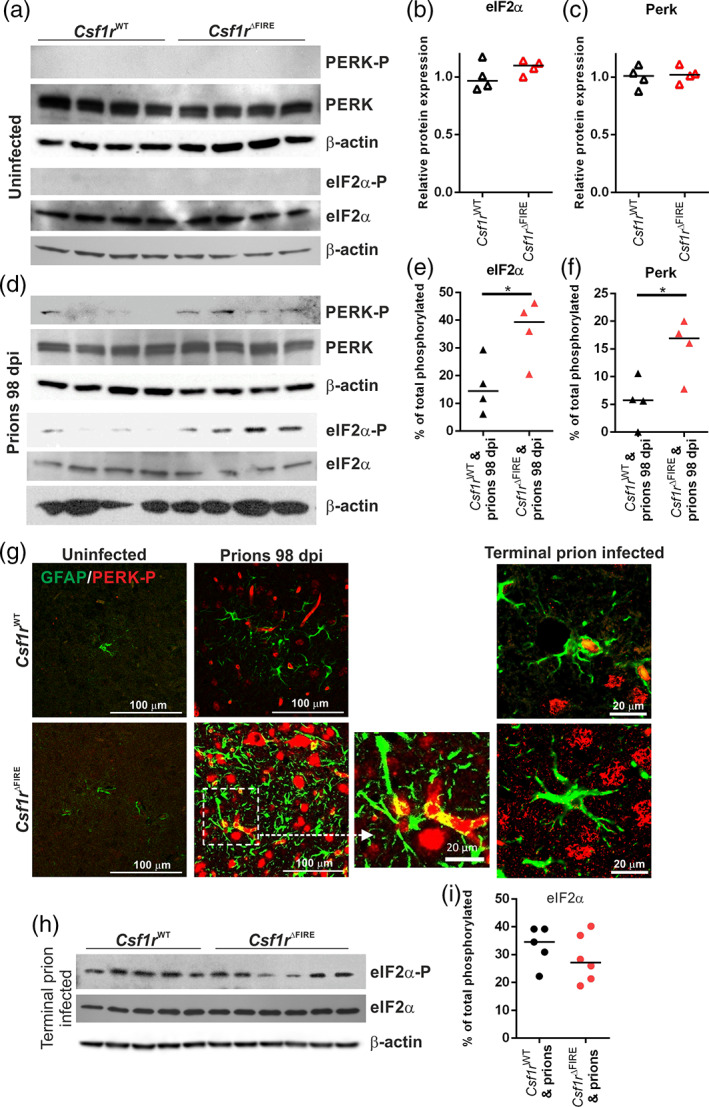
Increased unfolded protein response pathway is associated with earlier astrocyte activation. (a) Western blot analyses of age‐matched uninfected Csf1r WT and Cs1fr ΔFIRE mouse brains for unfolded protein response components as indicated, β‐Actin displayed as a loading control. (b) Quantitation of relative expression levels of eIF2α in uninfected Csf1r WT and Cs1fr ΔFIRE mouse brains. Not significantly different, Student's t‐test. (c) Quantitation of relative expression levels of PERK uninfected Csf1r WT and Cs1fr ΔFIRE mouse brain. Not significantly different, Student's t‐test. (d) Western blot analysis of 98 dpi prion‐infected Csf1r WT and Cs1fr ΔFIRE mouse brain for unfolded protein response components as indicated. (e) Quantitation of the percentage of total phosphorylated eIF2α in 98 dpi prion‐infected Csf1r WT and Cs1fr ΔFIRE mouse brain. *P < .05, Student's t‐test. (f) Quantitation of the percentage of total phosphorylated PERK in 98 dpi prion‐infected Csf1r WT and Cs1fr ΔFIRE mouse brain. *P < .05, Student's t‐test. (g) Immunohistochemical analysis of phosphorylated PERK (PERK‐P; red) and GFAP (green) in 98 dpi prion infected, terminal prion infected and age‐matched uninfected Csf1r WT and Cs1fr ΔFIRE superior colliculus (G3). Scale bars = 100 μm or 20 μm as indicated. (h) Western blot analysis of terminal prion‐infected brain homogenates probed for unfolded protein response components as indicated, β‐Actin displayed as a loading control. (i) Quantitation of the percentage of total phosphorylated eIF2α in terminal prion‐infected Csf1r WT and Cs1fr ΔFIRE mouse brains. Not significantly different, Student's t‐test. Points show individual mice. Panels A‐G, N = 4 mice/group. Horizontal bar = median. Panels H&I, N = 5–6 mice/group
However, by the terminal stage of prion infection similar levels of phosphorylated eIF2α were detected in the brains of each mouse group despite the Csf1r ΔFIRE mice succumbing to clinical prion disease earlier (Figure 9h,i). Thus, these data suggest that the earlier astrocyte activation and neuronal vacuolation in the prion‐infected Csf1r ΔFIRE mice was accompanied by an increased unfolded protein response.
4. DISCUSSION
In this study, we investigated prion neuropathogenesis in microglia‐deficient Csf1r ΔFIRE mice. Spongiform vacuolation and neuronal loss at the terminal stage were indistinguishable in Csf1r WT and Csf1r ΔFIRE mice and the onset of pathology was not correlated with the accumulation of misfolded prions, which are in any case not directly neurotoxic (Benilova et al., 2020). Microglia deficiency did not lead to the increased accumulation of prions in the brain, suggesting that microglial degradation of prions (if it occurs) can be compensated by other cells such as reactive astrocytes. We conclude that the non‐redundant function of microglia is to moderate the harmful effects of dysregulated reactive astrocytes and/or to provide supportive factors to neurons (Sariol et al., 2020). Consistent with that interpretation, microglia can suppress astrocyte phagocytic activity and astrocytes are capable of complete, though slower, clearance of neurons in the absence of microglia (Damisah et al., 2020). Previous studies have used a CSF1R kinase inhibitor to infer the role of microglia in CNS prion disease and reported that overall expression of A1‐ and A2‐ reactive astrocyte‐associated transcripts in the brain was enhanced upon microglial depletion (Carroll et al., 2018; Carroll et al., 2020). However, use of CSF1R inhibitors can lead to partial depletion of microglia, impact other kinases (e.g. KIT, FLT3), cause localized microglial cell death and impact monocytes and macrophages outside the brain. So, the impacts on pathology should be interpreted with caution (Hume et al., 2020).
During the early stage of prion infection, the reactive astrocytes were more abundant in the brains of Csf1r ΔFIRE mice. Although there was no induction of A1 neurotoxic astrocyte‐associated genes, the reactive astrocytes displayed signs of enhanced engulfment of neuronal synapses. The observation of activated astrocytes engulfing synapses in the superior colliculus (G3) region of the brains of Csf1r ΔFIRE mice at 98 dpi with prions was coincident with the commencement of overt clinical signs in these mice at this time. These observations strengthen the hypothesis that loss of neuronal connectivity underlies neurological symptoms and precedes complete loss of neurons (Brown et al., 2001; Cunningham et al., 2003; Jeffrey et al., 2000). The engulfment of damaged synapses and neurons by reactive astrocytes could provide a clearance mechanism to protect surrounding undamaged neurons and synapses, as neuronal damage is required for astrocyte‐mediated toxicity (Guttenplan et al., 2020).
Independent studies have shown that the reactive astrocytes in the prion infected brain express complement component C3 and LCN2 highly (Hartmann et al., 2019; Kushwaha et al., 2021; Smith et al., 2020). In the current study the onset of the expression of C3 and LCN2 in reactive astrocytes was accelerated in the brains of infected Csf1r ΔFIRE mice compared to infected Csf1r WT mice. Further studies are required to determine whether complement component C3 and LCN2 contribute to the development of the neuropathology in the prion disease‐affected brain, or whether they are simply indicative markers of dysregulated reactive astrocytic activation. Hartmann and colleagues showed that the abolishment of C3+ astrocytes in mice deficient in TNFα, interleukin‐1α and complement component C1qa coincided with accelerated CNS prion disease (Hartmann et al., 2019). However, independent studies have shown that deficiency in complement component C3 does not affect the development of CNS prion disease (Klein et al., 2001), and TNFα was undetectable in the brains of prion‐infected Csf1r ΔFIRE mice. Studies from other CNS disorders suggest that the secretion of LCN2 from reactive astrocytes may contribute to the neuropathology by enhancing neuroinflammation or neurotoxicity (reviewed in Lim et al., 2021). The expression of LCN2 may also play a role in the phagocytosis of neuronal material by the reactive astrocytes (Wan et al., 2022).
The phosphorylated activation of PERK and eIF2α in the unfolded protein response pathway is also upregulated in reactive astrocytes during CNS prion disease (Smith et al., 2020), and the onset of this activation was similarly accelerated in the brains of microglia‐deficient prion‐infected Csf1r ΔFIRE mice. Targeted blockade of this pathway specifically in astrocytes has proved beneficial during prion disease (Smith et al., 2020). Our data from microglia‐deficient Csf1r ΔFIRE mice indicate that the microglia employ mechanisms to protect the neurons in the brain against prion infection by restricting both phagocytosis and unfolded protein response in astrocytes. A similar role for microglia has recently been described in the suppression of ATP‐mediated excitoxicity in neurons (Badimon et al., 2020).
In conclusion, our data indicate that the microglia provide neuroprotection independently of PrPSc clearance during prion disease and restrict the harmful activities of reactive astrocytes. Since astrocytes can contribute to both prion replication (Krejciova et al., 2017; Raeber et al., 1997) and synaptic loss in infected brains, preventing these activities would have therapeutic potential (Smith et al., 2020). Of course, since microglia have been attributed essential functions in CNS development and homeostasis (reviewed in Prinz et al., 2019) we cannot entirely exclude the possibility that the absence of microglia in Csf1r ΔFIRE mice may have rendered their neurons more vulnerable to prion‐mediated damage. However, CNS development appears normal in Csf1r ΔFIRE mice despite the complete absence of microglia (Rojo et al., 2019). We also cannot exclude the possibility that the microglia play a role in modulating prion particle toxicity. Abnormal prion accumulations within the brain may comprise a mixture of fibrillar and smaller oligomers of PrPSc. However, since the smaller, non‐fibrillar, PrPSc particles are more pathological than larger fibrillary aggregates (Silviera et al., 2005), the engulfment and partial digestion of fibrillary PrPSc aggregates by the microglia may instead enhance their toxicity in the brain.
Further studies are now required to identify the molecular mechanisms by which the microglia provide neuroprotection during CNS prion disease. The previous characterization of the Csf1r ΔFIRE mice included mRNA expression profiling of the hippocampus which identified 85 transcripts that were significantly depleted when compare to wild‐type mice, and were presumably not compensated by astrocytes or other cells (Rojo et al., 2019). That list does not include most endosomal and lysosome‐associated genes that are more highly expressed by microglia and by inference must be upregulated by other cells in Csf1r ΔFIRE mice. An overlapping gene list was generated by expression profiling multiple brain regions in the Csf1rko rat (Pridans et al., 2018). Amongst the most down‐regulated transcripts are the three subunits of C1q, which have been implicated in regulating astrocyte function (Clarke et al., 2018; Liddelow et al., 2017) and neurodegeneration (Cho, 2019) and have complex roles in neuronal development and homeostasis (Vukojicic et al., 2019). These Csf1r‐dependent genes provide a short list of non‐redundant pathways that may be used by microglia to provide this neuroprotection and restrict the reactive astrocyte activation in prion disease. Paradoxically, given the focus of the literature on harmful functions of microglia, enhancing their functions may provide novel intervention treatments against these devastating neurodegenerative disorders.
AUTHOR CONTRIBUTIONS
Barry M. Bradford, David A. Hume, Clare Pridans, and Neil A. Mabbott conceived the study; Neil A. Mabbott obtained funding; Barry M. Bradford and Neil A. Mabbott designed the experiments; Barry M. Bradford and Lynne I. McGuire performed the experiments and acquired data; all authors interpreted these data and contributed to the final version of this report.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGMENTS
We thank University of Edinburgh Biological and Veterinary Services, including Darren Smith & Fraser Laing, Pathology Services Group including Aileen Boyle, The Roslin Institute Histology Suite and Bioimaging facility, including Bob Fleming and Graeme Robertson for helpful advice and technical support. This work was supported by funding from the RS Macdonald Charitable Trust, Edinburgh Neuroscience and project (BB/S005471/1) and Institute Strategic Programme Grant funding from the Biotechnology and Biological Sciences Research Council (grant numbers BBS/E/D/20002173 and BBS/E/D/10002071). For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) license to any author accepted manuscript version arising from this submission.
Bradford, B. M. , McGuire, L. I. , Hume, D. A. , Pridans, C. , & Mabbott, N. A. (2022). Microglia deficiency accelerates prion disease but does not enhance prion accumulation in the brain. Glia, 70(11), 2169–2187. 10.1002/glia.24244
Funding information Biotechnology and Biological Sciences Research Council, Grant/Award Numbers: BBS/E/D/10002071, BBS/E/D/20002173, BB/S005471/1; Edinburgh Neuroscience, RS Macdonald Charitable Trust
Contributor Information
Barry M. Bradford, Email: barry.bradford@roslin.ed.ac.uk.
Neil A. Mabbott, Email: neil.mabbott@roslin.ed.ac.uk.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Atarashi, R. , Sano, K. , Satoh, K. , & Nishida, N. (2011). Real‐time quaking‐induced conversion. Prion, 5(3), 150–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badimon, A. , Strasburger, H. J. , Ayata, P. , Chen, X. , Nair, A. , Ikegami, A. , Hwang, P. , Chan, A. T. , Graves, S. M. , Uweru, J. O. , Ledderose, C. , Kutlu, M. G. , Wheeler, M. A. , Kahan, A. , Ishikawa, M. , Wang, Y.‐C. , Loh, Y.‐H. E. , Jiang, J. X. , Surmeier, D. J. , … Schaefer, A. (2020). Negative feedback control of neuronal activity by microglia. Nature, 586, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova, I. , Reilly, M. , Terry, C. , Wenborn, A. , Schmidt, C. , Marinho, A. T. , Risse, E. , Al‐Doujaily, H. , Wiggins De Oliveira, M. , Sandberg, M. K. , Wadsworth, J. D. F. , Jat, P. S. , & Collinge, J. (2020). Highly infectious prions are not directly neurotoxic. Proceedings of the National Academy of Sciences USA, 117(38), 23815–23822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beringue, V. , Demoy, M. , Lasmezas, C. I. , Gouritin, B. , Weingarten, C. , Deslys, J. P. , Andreux, J. P. , Couvreur, P. , & Dormont, D. (2000). Role of spleen macrophages in the clearance of scrapie agent early in pathogenesis. Journal of Pathology, 190(4), 495–502. [DOI] [PubMed] [Google Scholar]
- Bradford, B. M. , Reizis, B. , & Mabbott, N. A. (2017). Oral prion disease pathogenesis is impeded in the specific absence of CXCR5‐expressing dendritic cells. Journal of Virology, 91(10), e00124–e00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford, B. M. , Wijaya, C. A. W. , & Mabbott, N. A. (2019). Discrimination of prion strain targeting in the central nervous system via reactive astrocyte heterogeneity in CD44 expression. Frontiers in Cellular Neuroscience, 13, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, D. , Belichenko, P. , Sales, J. , Jeffrey, M. , & Fraser, J. R. (2001). Early loss of dendritic spines in murine scrapie revealed by confocal analysis. Neuroreport, 12(1), 179–183. [DOI] [PubMed] [Google Scholar]
- Brown, K. L. , Gossner, A. , Mok, S. , & Mabbott, N. A. (2012). The effects of host age on the transport of complement‐bound complexes to the spleen and the pathogenesis of intravenous scrapie infection. Journal of Virology, 86(1), 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, K. L. , & Mabbott, N. A. (2014). Evidence of subclinical prion disease in aged mice following exposure to bovine spongiform encephalopathy. Journal of General Virology, 95(1), 231–243. [DOI] [PubMed] [Google Scholar]
- Brown, K. L. , Stewart, K. , Ritchie, D. , Mabbott, N. A. , Williams, A. , Fraser, H. , Morrison, W. I. , & Bruce, M. E. (1999). Scrapie replication in lymphoid tissues depends on prion protein‐expressing follicular dendritic cells. Nature Medicine, 5(11), 1308–1312. [DOI] [PubMed] [Google Scholar]
- Carroll, J. A. , Race, B. , Williams, K. , Striebel, J. , & Chesebro, B. (2018). Microglia are critical in host defense against prion disease. Journal of Virology, 92(15), e00549–e00518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll, J. A. , Race, B. , Williams, K. , Striebel, J. , & Chesebro, B. (2020). RNA‐seq and network analysis reveal unique glial gene expression signatures during prion infection. Molecular Brain, 13(1), 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, K. (2019). Emerging roles of complement protein C1q in neurodegeneration. Aging and Disease, 10(3), 652–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, W. S. , Clarke, L. E. , Wang, G. X. , Stafford, B. K. , Sher, A. , Chakraborty, C. , Joung, J. , Foo, L. C. , Thompson, A. , Chen, C. F. , Smith, S. J. , & Barres, B. A. (2013). Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature, 504(7480), 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L. E. , Liddelow, S. A. , Chakraborty, C. , Münch, A. E. , Heiman, M. , & Barres, B. A. (2018). Normal aging induces A1‐like astrocyte reactivity. Proceedings of the National Academy of Sciences USA, 115(8), E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, C. , Deacon, R. , Wells, H. , Boche, D. , Waters, S. , Diniz, C. P. , Scott, H. , Rawlins, J. N. , & Perry, V. H. (2003). Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. European Journal of Neuroscience, 17(10), 2147–2155. [DOI] [PubMed] [Google Scholar]
- Damisah, E. C. , Hill, R. A. , Rai, A. , Chen, F. , Rothlin, C. V. , Ghosh, S. , & Grutzendler, J. (2020). Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Science Advances, 6(26), eaba3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson, D. S. , Bradford, B. M. , Else, K. J. , & Mabbott, N. A. (2020). Accelerated onset of CNS prion disease in mice co‐infected with a gastrointestinal helminth pathogen during the preclinical phase. Scientific Reports, 10(1), 4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, H. , & Dickinson, A. G. (1967). Distribution of experimentally induced scrapie lesions in the brain. Nature, 216(5122), 1310–1311. [DOI] [PubMed] [Google Scholar]
- Gómez‐Nicola, D. , Fransen, N. L. , Suzzi, S. , & Perry, V. H. (2013). Regulation of microglial proliferation during chronic neurodegeneration. Journal of Neuroscience, 33(6), 2481–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Nicola, D. , Schetters, S. T. T. , & Perry, V. H. (2014). Differential role of CCR2 in the dynamics of microglia and perivascular macrophages during prion disease. Glia, 62(7), 1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, L. , Bertola, D. R. , Takanohashi, A. , Saito, A. , Segawa, Y. , Yokota, T. , Ishibashi, S. , Nishida, Y. , Yamamoto, G. L. , Franco, J. , Honjo, R. S. , Kim, C. A. , Musso, C. M. , Timmons, M. , Pizzino, A. , Taft, R. J. , Lajoie, B. , Knight, M. A. , Fischbeck, K. H. , … Ikegawa, S. (2019). Bi‐allelic CSF1R mutations cause skeletal dysplasia of dysosteosclerosis‐pyle disease spectrum and degenerative encephalopathy with brain malformation. American Journal of Human Genetics, 104(5), 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttenplan, K. A. , Stafford, B. K. , El‐Danaf, R. N. , Adler, D. I. , Münch, A. E. , Weigel, M. K. , Huberman, A. D. , & Liddelow, S. A. (2020). Neurotoxic reactive astrocytes drive neuronal death after retinal injury. Cell Reports, 31(12), 107776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann, K. , Sepulveda‐Falla, D. , Rose, I. V. L. , Madore, C. , Muth, C. , Matschke, J. , Butovsky, O. , Liddelow, S. , Glatzel, M. , & Krasemann, S. (2019). Complement 3+−astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathologica Communications, 7(1), 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzman, R. J. , & Corp, C. R. (1968). Behaviour in emergence and open‐field tests of normal and scrapie mice. Research in Veterinary Science, 9(6), 600–601. [PubMed] [Google Scholar]
- Hortega, R. (1919). El tercer elemento de los centros nerviosos. Boletin de la Sociedad Española de Biologia, 9, 69–120. [Google Scholar]
- Hume, D. A. , Caruso, M. , Ferrari‐Cestari, M. , Summers, K. M. , Pridans, C. , & Irvine, K. M. (2020). Phenotypic impacts of CSF1R deficiencies in humans and model organisms. Journal of Leukocyte Biology, 107(2), 205–219. [DOI] [PubMed] [Google Scholar]
- Hume, D. A. , & Macdonald, K. P. A. (2012). Therapeutic applications of macrophage colony‐stimulating factor‐1 (CSF‐1) and antagonists of CSF‐1 receptor (CSF‐1R) signaling. Blood, 119(8), 1810–1820. [DOI] [PubMed] [Google Scholar]
- Hwang, D. , Lee, I. Y. , Yoo, H. , Gehlenborg, N. , Cho, J. H. , Petritis, B. , Baxter, D. , Pitstick, R. , Young, R. , Spicer, D. , Price, N. D. , Hohmann, J. G. , Dearmond, S. J. , Carlson, G. A. , & Hood, L. E. (2009). A systems approach to prion disease. Molecular Systems Biology, 5, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey, M. , Halliday, W. G. , Bell, J. , Johnston, A. R. , Macleod, N. K. , Ingham, C. , Sayers, A. R. , Brown, D. A. , & Fraser, J. R. (2000). Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie‐infected murine hippocampus. Neuropathology and Applied Neurobiology, 26(1), 41–54. [DOI] [PubMed] [Google Scholar]
- Klein, M. A. , Kaeser, P. S. , Schwarz, P. , Weyd, H. , Xenarios, I. , Zinkernagel, R. M. , Carroll, M. C. , Verbeer, J. S. , Botto, M. , Walport, M. J. , Molina, H. , Kalinke, U. , Acha‐Orbea, H. , & Aguzzi, A. (2001). Complement facilitates early prion pathogenesis. Nature Medicine, 7(4), 488–492. [DOI] [PubMed] [Google Scholar]
- Krejciova, Z. , Alibhai, J. , Zhao, C. , Krencik, R. , Rzechorzek, N. M. , Ullian, E. M. , Manson, J. , Ironside, J. W. , Head, M. W. , & Chandran, S. (2017). Human stem cell‐derived astrocytes replicate human prions in a PRNP genotype‐dependent manner. Journal of Experimental Medicine, 214(12), 3481–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunyu, L. I. , Jialin Zheng, J. L. , & Song, Q. (2019). Reactive astrocytes in neurodegenerative diseases. Aging and Disease, 10(3), 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha, R. , Sinha, A. , Makarava, N. , Molesworth, K. , & Baskakov, I. V. (2021). Non‐cell autonomous astrocyte‐mediated neuronal toxicity in prion diseases. Acta Neuropathologica Communications, 9(1), 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, E. , & Chung, W.‐S. (2019). Glial control of synapse number in healthy and diseased brain. Frontiers in Cellular Neuroscience, 13, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, F. , Cui, N. , Zhou, C. , Chodosh, J. , Vavvas, D. G. , & Paschalis, E. I. (2020). CSF1R inhibition by a small‐molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proceedings of the National Academy of Sciences USA, 117(38), 23336–23338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow, S. A. , Guttenplan, K. A. , Clarke, L. E. , Bennett, F. C. , Bohlen, C. J. , Schirmer, L. , Bennett, M. L. , Munch, A. E. , Chung, W. S. , Peterson, T. C. , Wilton, D. K. , Frouin, A. , Napier, B. A. , Panicker, N. , Kumar, M. , Buckwalter, M. S. , Rowitch, D. H. , Dawson, V. L. , Dawson, T. M. , … Barres, B. A. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541(7638), 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, D. , Jeong, J.‐H. , & Song, J. (2021). Lipocalin 2 regulates iron homeostasis, neuroinflammation and insulin resistance in the brains of patients with dementia: Evidence from the current literature. CNS Neuroscience & Therapeutics, 27, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak, K. J. , & Schmittgen, T. D. (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods, 25(4), 402–408. [DOI] [PubMed] [Google Scholar]
- Maignien, T. , Shakweh, M. , Calvo, P. , Marce, D. , Sales, N. , Fattal, E. , Deslys, J. P. , Couvreur, P. , & Lasmezas, C. I. (2005). Role of gut macrophages in mice orally contaminated with scrapie or BSE. International Journal of Pharmacology, 298(2), 293–304. [DOI] [PubMed] [Google Scholar]
- Manson, J. C. , Clarke, A. R. , Mcbride, P. A. , Mcconnell, I. , & Hope, J. (1994). PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration, 3(4), 331–340. [PubMed] [Google Scholar]
- Mcculloch, L. , Brown, K. L. , Bradford, B. M. , Hopkins, J. , Bailey, M. , Rajewsky, K. , Manson, J. C. , & Mabbott, N. A. (2011). Follicular dendritic cell‐specific prion protein (PrPc) expression alone is sufficient to sustain prion infection in the spleen. PLoS Pathogens, 7(12), e1002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon, S. , Langeveld, J. P. M. , Tan, B. C. , Gill, A. C. , De Wolf, C. , Martin, S. , Gonzalez, L. , Alibhai, J. , Blanco, A. R. A. , Campbell, L. , Hunter, N. , & Houston, E. F. (2014). Prion protein‐specific antibodies that detect multiple TSE agents with high sensitivity. PLoS One, 9(3), e91143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama, H. , Abe, M. , Morimoto, C. , Iida, T. , Okabe, S. , Sakimura, K. , & Hashimoto, K. (2018). Microglia permit climbing fiber elimination by promoting GABAergic inhibition in the developing cerebellum. Nature Communications, 9(1), 2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orrú, C. D. , Groveman, B. R. , Raymond, L. D. , Hughson, A. G. , Nonno, R. , Zou, W. , Ghetti, B. , Gambetti, P. , & Caughey, B. (2015). Bank vole prion protein as an apparently universal substrate for RT‐QuIC‐based detection and discrimination of prion strains. PLoS Pathogens, 11(6), e1004983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patkar, O. L. , Caruso, M. , Teakle, N. , Keshvari, S. , Bush, S. J. , Pridans, C. , Belmer, A. , Summers, K. M. , Irvine, K. M. , & Hume, D. A. (2021). Analysis of homozygous and heterozygous Csf1r knockout in the rat as a model for understanding microglial function in brain development and the impacts of human CSF1R mutations. Neurobiology of Disease, 151, 105268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pridans, C. , Raper, A. , Davis, G. M. , Alves, J. , Sauter, K. A. , Lefevre, L. , Regan, T. , Meek, S. , Sutherland, L. , Thomson, A. J. , Clohisey, S. , Bush, S. J. , Rojo, R. , Lisowski, Z. M. , Wallace, R. , Grabert, K. , Upton, K. R. , Tsai, Y. T. , Brown, D. , … Hume, D. A. (2018). Pleiotropic impacts of macrophage and microglial deficiency on development in rats with targeted mutation of the Csf1r locus. Journal of Immuology, 201(9), 2683–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz, M. , Jung, S. , & Priller, J. (2019). Microglia biology: One century of evolving concepts. Cell, 179(2), 292–311. [DOI] [PubMed] [Google Scholar]
- Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science, 216(4542), 136–144. [DOI] [PubMed] [Google Scholar]
- Raeber, A. J. , Race, R. E. , Brandner, S. , Priola, S. A. , Sailer, A. , Bessen, R. A. , Mucke, L. , Manson, J. , Aguzzi, A. , Oldstone, M. B. , Weissmann, C. , & Chesebro, B. (1997). Astrocyte‐specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO Journal, 16(20), 6057–6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo, R. , Raper, A. , Ozdemir, D. D. , Lefevre, L. , Grabert, K. , Wollscheid‐Lengeling, E. , Bradford, B. , Caruso, M. , Gazova, I. , Sánchez, A. , Lisowski, Z. M. , Alves, J. , Molina‐Gonzalez, I. , Davtyan, H. , Lodge, R. J. , Glover, J. D. , Wallace, R. , Munro, D. A. D. , David, E. , … Pridans, C. (2019). Deletion of a Csf1r enhancer selectively impacts CSF1R expression and development of tissue macrophage populations. Nature Communications, 10(1), 3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariol, A. , Mackin, S. , Allred, M.‐G. , Ma, C. , Zhou, Y. , Zhang, Q. , Zou, X. , Abrahante, J. E. , Meyerholz, D. K. , & Perlman, S. (2020). Microglia depletion exacerbates demyelination and impairs remyelination in a neurotropic coronavirus infection. Proceedings of the National Academy of Sciences USA, 117(39), 24464–24474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, C. A. , Rasband, W. S. , & Eliceiri, K. W. (2012). NIH image to ImageJ: 25 years of image analysis. Nature Methods, 9(7), 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silviera, J. R. , Raymond, G. J. , Hughson, A. G. , Race, R. E. , Sim, V. L. , Hayes, S. F. , & Caughey, B. (2005). The most infectious prion particles. Nature, 437, 257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, H. L. , Freeman, O. J. , Butcher, A. J. , Holmqvist, S. , Humoud, I. , Schatzl, T. , Hughes, D. T. , Verity, N. C. , Swinden, D. P. , Hayes, J. , De Weerd, L. , Rowitch, D. H. , Franklin, R. J. M. , & Mallucci, G. R. (2020). Astrocyte unfolded protein response induces a specific reactivity state that causes non‐cell‐autonomous neuronal degeneration. Neuron, 105(5), 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatzelt, J. , Groth, D. F. , Torchia, M. , Prusiner, S. B. , & Dearmond, S. J. (1999). Kinetics of prion protein accumulation in the CNS of mice with experimental scrapie. Journal of Neuropathology & Experimental Neurology, 58(12), 1244–1249. [DOI] [PubMed] [Google Scholar]
- Vainchtein, I. D. , & Molofsky, A. V. (2020). Astrocytes and microglia: In sickness and in health. Trends in Neurosciences, 43(3), 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukojicic, A. , Delestrée, N. , Fletcher, E. V. , Pagiazitis, J. G. , Sankaranarayanan, S. , Yednock, T. A. , Barres, B. A. , & Mentis, G. Z. (2019). The classical complement pathway mediates microglia‐dependent remodeling of spinal motor circuits during development and in SMA. Cell Reports, 29(10), 3087–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, T. , Zhu, W. , Zhao, Y. , Zhang, X. , Ye, R. , Zuo, M. , Xu, P. , Huang, Z. , Zhang, C. , Xie, Y. , & Liu, X. (2022). Astrocytic phagocytosis contributes to demyelination after focal cortical ischemia in mice. Nature Communications, 13, 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, C. , Hermann, U. S. , Falsig, J. , Abakumova, I. , Nuvolone, M. , Schwarz, P. , Frauenknecht, K. , Rushing, E. J. , & Aguzzi, A. (2016). A neuroprotective role for microglia during prion diseaes. Journal of Experimental Medicine, 213(6), 1047–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.