

Summary
Castleman disease (CD) describes a group of rare, potentially fatal lymphoproliferative disorders. To determine factors associated with mortality in CD, we analysed data from deceased patients in the ACCELERATE registry and compared them with matched controls. We analysed demographic, treatment and laboratory data from all deceased CD patients, matched controls and a subgroup of idiopathic multicentric Castleman disease (iMCD) patients. Of the 140 patients in ACCELERATE with a confirmed CD diagnosis, 10 had died. There were 72 patients with confirmed iMCD; six were deceased. The deceased CD cohort had more hospitalisations per year, higher overall hospitalisations and more days hospitalised per month, and received more treatment regimens per year than the matched‐control group. Analysis of laboratory values showed a significantly decreased absolute lymphocyte count at months 3 and 6 in the deceased cohort compared with controls. Among iMCD patients, there was a higher proportion of iMCD‐TAFRO (thrombocytopenia, anasarca, fever, reticulin myelofibrosis, renal dysfunction and organomegaly) cases in the deceased group. The deceased iMCD group had significantly lower immunoglobulin M, international normalised ratio and platelet count. These data demonstrate that there may be differences between patients who have fatal and non‐fatal outcomes, and provide preliminary suggestions for parameters to evaluate further.
Keywords: ACCELERATE, disease course, idiopathic multicentric Castleman disease, mortality, registry
INTRODUCTION
Castleman disease (CD) is a rare, poorly understood lymphoproliferative disorder, classified first as unicentric (UCD) or multicentric (MCD) based on whether one or multiple regions of enlarged lymph nodes are involved. 1 , 2 MCD, unlike UCD, cannot be cured by excision of the enlarged nodes, follows a more serious and protracted disease course, and requires tailored specialist care and therapy. 3 , 4 , 5
MCD is subdivided into three groups based on aetiology. 6 Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes (POEMS)‐associated MCD involves excessive cytokine production by a monoclonal plasma cell population. Human herpesvirus‐8 (HHV8)‐associated MCD is a lymphoproliferative disorder caused by uncontrolled proliferation of HHV8‐infected plasma cells/plasmablasts and HHV8‐driven cytokine dysregulation, 7 , 8 , 9 whereas idiopathic MCD (iMCD) involves a similar cytokine storm — in which interleukin (IL)‐6 is a key driver in many cases — but has an unknown aetiology and an emerging understanding of pathogenesis. 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17
iMCD is further subdivided into iMCD‐TAFRO (thrombocytopenia, anasarca, fever, reticulin myelofibrosis, renal dysfunction and organomegaly) and iMCD‐not otherwise specified (iMCD‐NOS). 18 , 19 iMCD‐TAFRO tends to have a more severe clinical course and worse outcomes than iMCD‐NOS. 18 Given that iMCD is the most severe and least understood subtype of CD, identifying unique laboratory or clinical factors that may indicate patients who are at higher risk of mortality is needed.
Siltuximab, an anti‐IL6 monoclonal antibody (mAb), is the only US Food and Drug Administration (FDA)‐ or European Medicines Agency (EMA)‐approved treatment for iMCD; it is recommended for first‐line use. 20 , 21 , 22 Corticosteroids are commonly used alongside siltuximab, which is effective in approximately 34%–45% of iMCD patients. Immunomodulatory/immunosuppressive agents are often used in refractory patients, and chemotherapy is often needed in the most severe cases. 20 , 23 , 24 , 25
There is a paucity of data describing the natural history of all forms of CD and limited data related to outcomes and prognosis. 4 , 26 ACCELERATE is an international natural history registry established in 2016 to collect real‐world data on patients with CD. 20 , 27 Patients who have received a pathological CD diagnosis can enrol online (www.CDCN.org/ACCELERATE) from anywhere in the world or through one of nine sites in Europe. Their medical history is then collected and extracted into the study database. One of the goals of ACCELERATE is to better understand the characteristics of deceased patients and outcomes associated with CD. 25 , 27 , 28 , 29 , 30 , 31 , 32
We conducted analyses of deceased patients in the ACCELERATE registry and compared them with matched controls (patients who had a similar disease type, age and severity at presentation but a non‐fatal outcome), with the aim of determining factors associated with mortality in CD. We also examined a subgroup consisting of deceased iMCD patients compared with all other iMCD patients in the registry to determine if there are any unique factors or prognostic markers associated with mortality in this group, for which there is no known aetiological trigger for disease.
METHODS
Study design and patient population
ACCELERATE (NCT02817997) inclusion criteria require a reference pathology report suggesting ‘Castleman disease’ and ability to provide informed consent. There are no age restrictions. The enrolment and study process have been previously described. 27 Diagnostic, medical and treatment data are subsequently reviewed by an expert panel of clinicians and pathologists, referred to as the Certification and Access Subcommittee (CAS), to assess the likelihood of an accurate CD diagnosis. Selected patients and controls needed to have pathological and clinical features determined by the CAS to be consistent with CD. At the time of this analysis, 372 patients met enrolment criteria, 140 of whom had been reviewed and graded as likely to have CD by the CAS. All patients in these analyses were required to have a certified CAS grade and confirmed CD diagnosis.
In these analyses, we describe the characteristics and disease course for enrolled patients in four groups: (1) the deceased CD cohort (n = 10): CD patients who died prior to analysis; (2) the matched‐control CD cohort (n = 19): patients best matched to the deceased cohort in terms of CD subtype (UCD, HHV8‐MCD, POEMS‐associated, iMCD), age and disease severity who were still alive at the time of analysis; (3) the deceased iMCD cohort (n = 6): patients with a confirmed iMCD diagnosis who died prior to analysis; and (4) the non‐deceased iMCD group (n = 66): all patients with a confirmed iMCD diagnosis who were still alive at the time of analysis. Patients in the deceased iMCD cohort are also included in the deceased CD cohort, so comparisons between these groups are not possible.
For the matched‐control CD cohort, three criteria were used to match patients to those with a fatal outcome. The primary matching criterion was diagnosis subtype, followed by age and disease severity. Disease severity was assessed and matched through a modified CHAP [C‐reactive protein (CRP), haemoglobin, albumin, performance status] score. As performance status was not universally reported, we modified the score to only include the first three features: CRP, haemoglobin and albumin (CHA). 34 CHA score measured closest to diagnosis was used, and both total scores and individual components of the CHA score were used to compare and match patients. We matched two control patients to each individual deceased patient.
Statistical analyses
Due to the limited patient numbers and varying datasets, descriptive analyses were performed on demographic and clinical data at first patient‐reported symptom onset, at diagnosis and throughout the disease course. The number of regimens, regimens per year and days from first patient‐reported symptom onset to first CD treatment were calculated.
Exploratory analyses were conducted using laboratory data. To delineate differences in the profile of laboratory measures, we performed two‐tailed t‐tests with Welch correction to determine statistical differences between the deceased and non‐deceased groups for the different cohorts (deceased CD versus matched‐control CD, and deceased iMCD versus non‐deceased iMCD) at the time of diagnosis. Repeated‐measures linear mixed modelling was used to look at longitudinal changes in the laboratory measures.
For the quantitative analysis, the mean ± standard deviation (SD) was reported. A p value of <0.05 was considered significant. All analyses were performed using GraphPad Prism 9.0 (GraphPad Inc.). Kaplan–Meier survival curves were also developed to analyse survival in the general CD population and iMCD population. However, one iMCD patient did not have a first patient‐reported symptom date and therefore was not included in the survival curve analysis.
RESULTS
Cohort baseline characteristics
Of the 372 patients who met inclusion criteria for enrolment in ACCELERATE at the time of this analysis, 140 had been reviewed by the CAS and given a grade consistent with a CD diagnosis (CAS‐confirmed). Ten of these patients had died at the time of this analysis. Using the three specified matching criteria, it was possible to match 19 patients to the deceased cohort according to our prespecified 2:1 matching approach. The 2:1 matching was not possible for POEMS‐associated MCD patients, as there were only three suitable matches for the two patients in the deceased cohort. Table 1 lists the baseline characteristics for the deceased CD and matched‐control CD cohorts. For the deceased CD cohort, the median (range) age at diagnosis was 47.9 (13.5–80.8) years (50.0% male), and the median age at death was 56.2 (13.7–80.9) years. The median (range) time from diagnosis to death was 226.0 (16–3516) days. One patient was diagnosed with UCD, eight with HHV8‐negative MCD (two were POEMS‐associated MCD, and six were iMCD) and one with HHV8‐positive MCD. Owing to the small sample size, differences in mortality based on CD subtype could not be studied. Three were children at the time of diagnosis, with an age range of 13.5–17.0 years.
TABLE 1.
Patient demographics and characteristics (deceased CD and matched‐control CD cohorts)
| Patient ID | Diagnosis | Histopathology | Sex | Age at diagnosis |
|---|---|---|---|---|
| Deceased CD group | ||||
| 1 | UCD | Hyaline vascular | Male | 13.5 |
| 2 | iMCD | Hypervascular | Female | 15.2 |
| 3 | iMCD | Hypervascular | Male | 17.0 |
| 4 | iMCD | NS | Female | 43.4 |
| 5 | iMCD | Hypervascular | Female | 65.5 |
| 6 | iMCD | Hypervascular | Female | 61.3 |
| 7 | iMCD | Mixed | Male | 66.7 |
| 8 | POEMS‐associated MCD | Plasmacytic | Male | 52.3 |
| 9 | POEMS‐associated MCD | NS | Male | 40.5 |
| 10 | HHV8‐associated MCD | Plasmablastic | Female | 80.8 |
| Matched‐control CD cohort | ||||
| 11 | UCD | Mixed | Male | 48.5 |
| 12 | UCD | NS | Female | 19.0 |
| 13 | iMCD | Hypervascular | Male | 13.9 |
| 14 | iMCD | Hypervascular | Female | 21.7 |
| 15 | iMCD | Hypervascular | Female | 18.5 |
| 16 | iMCD | Hypervascular | Male | 2.6 |
| 17 | iMCD | Mixed | Female | 45.7 |
| 18 | iMCD | Hypervascular | Female | 39.8 |
| 19 | iMCD | Hypervascular | Female | 65.9 |
| 20 | iMCD | Mixed | Male | 47.6 |
| 21 | iMCD | Hypervascular | Male | 65.2 |
| 22 | iMCD | Hypervascular | Female | 40.3 |
| 23 | iMCD | Plasmacytic | Female | 53.7 |
| 24 | iMCD | Mixed | Female | 50.7 |
| 25 | POEMS‐associated MCD | Mixed | Male | 43.0 |
| 26 | POEMS‐associated MCD | Mixed | Male | 39.7 |
| 27 | POEMS‐associated MCD | NS | Male | 56.7 |
| 28 | HHV8‐associated MCD | Plasmablastic | Male | 68.1 |
| 29 | HHV8‐associated MCD | Plasmablastic | Female | 64.2 |
Abbreviations: CD, Castleman disease; HHV8, human herpesvirus 8; iMCD, idiopathic multicentric Castleman disease; NS, not stated; POEMS, polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy and skin changes; UCD, unicentric Castleman disease.
There were 72 CAS‐confirmed iMCD patients, of which six (8.3%) died prior to analysis. The median (range) age at diagnosis for the deceased iMCD group was 52.4 (15.2–66.7) years (50.0% male) and 37.3 (2.6–71.3) years (53.0% male) for the remaining non‐deceased iMCD group (Table 2). The median (range) time from final diagnosis to death was 63.5 (16–2952) days. The most common histopathological subtype was hypervascular (66.7% in the deceased group and 59.1% in the non‐deceased iMCD group). In the deceased iMCD group, 83.3% were iMCD‐TAFRO cases, whereas in the non‐deceased iMCD group, 51.5% of cases were iMCD‐TAFRO. Overall, there were 33 iMCD‐NOS patients (32 lived; one died) and 39 iMCD‐TAFRO patients (34 lived, five died). The odds ratio and p value of dying among iMCD‐TAFRO cases were 4.7 and p = 0.17.
TABLE 2.
Patient characteristics of the non‐deceased iMCD and deceased iMCD cohorts
| Characteristic | Deceased iMCD group (n = 6) | Non‐deceased iMCD group (n = 66) |
|---|---|---|
| Age at diagnosis, years: median (range) | 52.4 (15.2–66.7) | 37.3 (2.6–71.3) |
| Male, % | 50.0 | 53.0 |
| TAFRO cases, % | 83.3 | 51.5 |
| Histopathology, % | ||
| Hypervascular | 66.7 | 59.1 |
| Plasmacytic | 0.0 | 7.6 |
| Mixed | 16.7 | 28.8 |
| Not stated | 16.7 | 4.5 |
| Nationality, % | ||
| American | 66.7 | 89.4 |
| Other | 33.3 | 10.6 |
Abbreviations: iMCD, idiopathic multicentric Castleman disease; TAFRO, thrombocytopenia, anasarca, fever, reticulin myelofibrosis, renal dysfunction and organomegaly.
Time to treatment
For the deceased CD cohort, the median time to first treatment from first patient‐reported symptom onset was 47.0 days. For the matched‐control CD cohort, the median time to first treatment was 214.0 days. There was substantial variability in time between first patient‐reported symptom onset and first treatment for both the deceased CD cohort (23–884 days) and the matched‐control CD group. Four matched‐control CD patients received a CD treatment before they reported their first CD symptoms. For an additional patient, no treatments were reported. There were no significant differences between the two cohorts.
Treatment regimens
In the deceased CD cohort, eight (80.0%) patients were first treated with a mAb‐containing regimen (e.g. tocilizumab, siltuximab or rituximab). Other first‐line treatments included cyclophosphamide, prednisolone and valganciclovir. Among patients with iMCD (n = 6), three (50.0%) were treated with siltuximab first line.
Regimens were similar in the matched‐control cohort. One patient had no treatment recorded. Of the matched‐control CD patients with an iMCD diagnosis, one (8.3%) was given siltuximab first line. One POEMS‐associated MCD patient was given siltuximab first line.
Overall, the deceased CD cohort received 50 regimens, with only 22 (44.0%) containing a targeted therapy (rituximab, siltuximab or tocilizumab) (Supplementary Table S2). The matched‐control CD cohort received 69 regimens, with 35 (50.7%) containing a targeted therapy. Although rituximab was not given to any deceased HHV8‐positive MCD patients as first‐line therapy, all HHV8‐positive MCD patients received a rituximab‐containing regimen in their disease course.
The deceased CD cohort was treated with significantly more regimens per year than the matched‐control CD cohort (2.0 vs 0.5 respectively; p < 0.05). The deceased CD cohort had a similar number of total regimens throughout the duration of follow‐up to that of the matched‐control CD group (3.5 vs 3.0), despite a longer follow‐up (p = 0.054) in the matched‐control CD group.
In the iMCD group (both deceased iMCD and non‐deceased iMCD cohorts), 44 (61.1%) patients received siltuximab therapy at some point in their treatment course, of which 39 (54.1%) patients received siltuximab as their only IL6‐blocking therapy and five (6.9%) patients received both siltuximab and tocilizumab at points during their treatment. Ten (13.9%) patients did not receive any anti‐IL6 blocking therapy and 18 (25.0%) patients received tocilizumab only.
Hospitalisations
Patients in the deceased CD cohort had significantly more hospitalisations per year than those in the matched‐control CD group (2.16 vs 0.59 respectively; p = 0.028) (Table S1). A similar trend was observed in the number of hospitalisations (3.30 vs 2.32; p = 0.290) and days hospitalised per month (2.16 vs 0.86; p = 0.076). The mean number of days hospitalised since diagnosis trended towards being greater in the deceased CD cohort than in the matched‐control CD cohort (64.70 vs 25.84), but this was not significant (p = 0.076). The duration of follow‐up (years) was also lower in the deceased than in the matched‐control cohort (3.64 vs 7.15; p = 0.054).
Mortality
Mortality data from the MCD literature suggest that the five‐year overall survival rate has historically been 51%–77%. 25 , 27 , 28 , 29 , 30 , 31 , 32 A substantially lower proportion of patients in the ACCELERATE registry died within five years of diagnosis than in the literature, likely owing to reporting bias. Deceased patients comprise 8.3% of the iMCD patients enrolled into the ACCELERATE cohort (n = 72), with a mean survival of 1744 days. Immediate cause of death was cardiopulmonary events (n = 2), pneumonia (n = 3), polyneuropathy and extensive paraneoplastic pemphigus (n = 1), and unknown (n = 3). Preliminary cause of death was reported in two patients (multi‐organ failure and cardiorespiratory failure). Kaplan–Meier survival estimates showed that nearly all deaths occurred within 1000 days of first patient‐reported symptoms in both the general CD (Figure 3A) and iMCD cohorts (Figure 3B). Median survival was not reached in either cohort.
FIGURE 3.
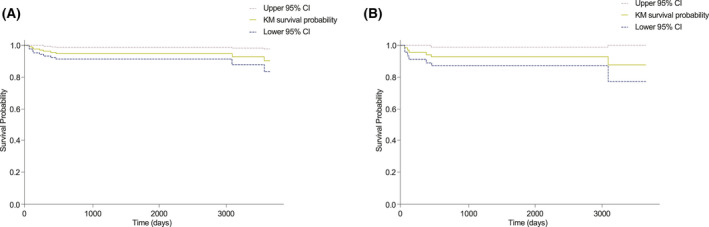
Kaplan–Meier survival estimates for the Castleman disease patients with a Certification and Access Subcommittee‐approved diagnosis in (A), the general Castleman disease cohort (n = 139) and (B), the idiopathic multicentric Castleman disease cohort (n = 71). CI, Confidence interval; KM, Kaplan–Meier
Laboratory data analysis
We assessed differences in the profile of 25 laboratory measures at the time of diagnosis between the matched‐control CD and deceased CD groups (see Table S2).
CD analysis (matched‐control CD cohort, n = 19; deceased CD cohort, n = 10)
There were no significant differences in laboratory values around the time of diagnosis between the deceased CD and matched‐control CD groups, with the exception of uric acid levels, which were significantly decreased in the deceased cohort compared with the matched‐control group (t = 2.194, p = 0.041) (Table S2) but still within the normal range. Next, we sought to understand if changes in any laboratory values over time were associated with mortality. A mixed longitudinal measures analysis evaluated the repeated measures for several laboratory tests. Absolute lymphocyte count (ALC) was the only test that demonstrated significant differences at any time. ALC was found to be significantly decreased at months 3 (t = 3.713, p = 0.0334) and 6 (t = 4.668, p = 0.0047) after diagnosis in the deceased CD cohort compared with the matched‐control CD cohort (Figure 2).
FIGURE 2.
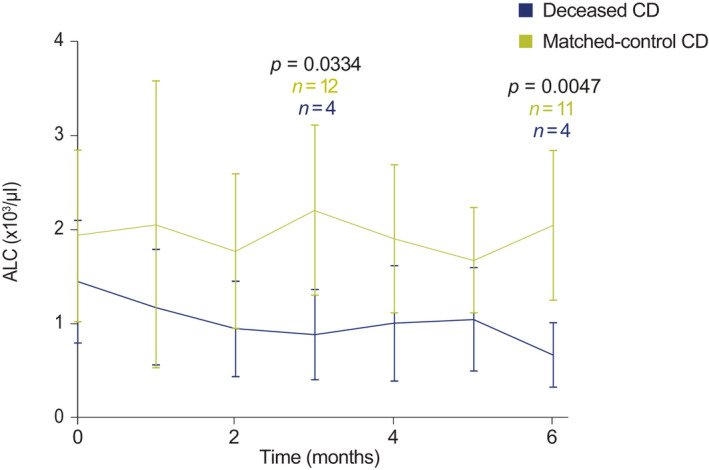
Repeated‐measures linear mixed model for monthly ALC in the deceased CD and matched‐control CD cohorts. Values in the graph represent mean ± SD. The values at month 0 show the ALC counts at the time of diagnosis. ALC, absolute lymphocyte count; CD, Castleman disease; SD, standard deviation. Note: p values changed over time, but only significant time points have been highlighted
iMCD cohort (non‐deceased iMCD cohort, n = 66; deceased iMCD cohort, n = 6)
Around the time of diagnosis, immunoglobulin M (IgM; t = 4.655, p = 0.0002), international normalised ratio (INR; t = 2.066, p = 0.0427) and platelet count (t = 3.144, p = 0.0114) were significantly decreased in the deceased iMCD group compared with the non‐deceased iMCD group (Figure 1 and Table 3). IgM was below the lower limit of normal (LLN) for three patients from the deceased iMCD group, and platelet count was below the LLN for four deceased iMCD patients. Unlike the other two parameters, the INR levels were closer to normal in the deceased iMCD group than in the non‐deceased iMCD group (Table 3).
FIGURE 1.
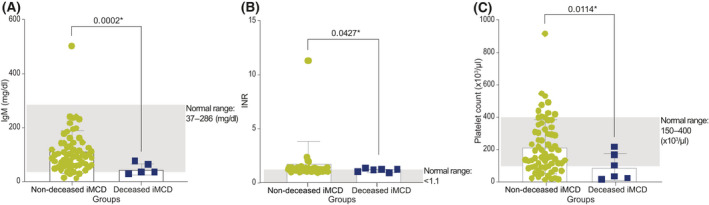
Patients with iMCD in the deceased group (n = 6) and non‐deceased iMCD group (n = 66). (A), (B) and (C) show the mean ± SD of IgM, INR and platelet count respectively, in the deceased iMCD and non‐deceased iMCD groups. IgM, immunoglobin M; iMCD, idiopathic multicentric Castleman disease; INR, international normalised ratio; SD, standard deviation. *, significance
TABLE 3.
Laboratory values at time of diagnosis (non‐deceased iMCD, n = 66 and deceased iMCD cohorts, n = 6)
| iMCD group | Non‐deceased iMCD | Deceased iMCD |
p value |
||||
|---|---|---|---|---|---|---|---|
| Laboratory measures | Mean | SD | n | Mean | SD | n | |
| Haematology | |||||||
| Absolute basophil count (×109/l) | 0.24 | 1.60 | 65 | 0.06 | 0.08 | 5 | 0.3851 |
| Absolute eosinophil count (×109/l) | 0.14 | 0.15 | 65 | 0.10 | 0.15 | 5 | 0.6791 |
| Absolute lymphocyte count (×109/l) | 2.23 | 3.02 | 65 | 1.61 | 0.66 | 5 | 0.2265 |
| Absolute monocyte count (×109/l) | 2.78 | 16.00 | 65 | 1.28 | 0.96 | 5 | 0.4671 |
| Absolute neutrophil count (×109/l) | 7.18 | 3.85 | 66 | 6.65 | 3.86 | 6 | 0.7810 |
| Haemoglobin (g/l) a | 104.8 | 25.1 | 66 | 85.0 | 28.4 | 6 | 0.1843 |
| International normalised ratio | 1.70 | 2.09 | 63 | 1.13 | 0.14 | 6 | 0.0427 |
| Platelet count (×109/l) | 217.97 | 166.80 | 66 | 92.67 | 76.17 | 6 | 0.0114 |
| Prothrombin time | 14.16 | 2.94 | 55 | 14.40 | 2.78 | 6 | 0.8612 |
| White blood cell count (×109/l) | 10.18 | 3.83 | 65 | 12.89 | 10.65 | 6 | 0.5944 |
| Chemistry | |||||||
| Albumin (g/dl) a | 31.4 | 14.8 | 66 | 28.8 | 0.99 | 6 | 0.6113 |
| Alkaline phosphatase (μkat/l) | 2.32 | 1.61 | 66 | 2.47 | 1.14 | 6 | 0.7898 |
| Alanine aminotransferase (μkat/l) | 0.35 | 0.30 | 66 | 0.51 | 0.38 | 6 | 0.3828 |
| Aspartate aminotransferase (μkat/l) | 0.39 | 0.25 | 66 | 0.50 | 0.19 | 6 | 0.2912 |
| Blood urea nitrogen (mmol/l) | 0.89 | 6.24 | 66 | 10.21 | 4.74 | 5 | 0.4231 |
| Creatinine (μmol/l) | 248.40 | 1155.39 | 66 | 91.94 | 20.33 | 6 | 0.2816 |
| C‐reactive protein (mg/l) a | 76.83 | 88.18 | 61 | 138.54 | 88.42 | 6 | 0.1855 |
| Estimated glomerular filtration (rate ml/min/1.73m2) | 1.63 | 0.48 | 60 | 1.20 | 0.40 | 5 | 0.0956 |
| Lactate dehydrogenase (μkat/l) | 4.20 | 3.14 | 64 | 8.65 | 9.52 | 6 | 0.3450 |
| Total bilirubin (μmol/l) | 20.87 | 31.3 | 65 | 33.69 | 52.51 | 6 | 0.6149 |
| Uric acid (mmol/l) | 0.40 | 0.18 | 53 | 0.34 | 0.13 | 5 | 0.4357 |
| Immunology | |||||||
| Immunoglobulin A (g/l) | 2.46 | 1.71 | 58 | 1.40 | 0.78 | 4 | 0.0934 |
| Immunoglobulin G (g/l) | 18.76 | 16.11 | 61 | 15.67 | 9.27 | 6 | 0.5246 |
| Immunoglobulin M (g/l) | 1.09 | 0.77 | 60 | 0.46 | 0.18 | 5 | 0.0002 |
p values in bold denote significance. iMCD, idiopathic multicentric Castleman disease; SD, standard deviation.
Patients were matched in part based on C‐reactive protein, haemoglobin and albumin (CHA), so the results of these differences in CHA would not be expected to be different.
DISCUSSION
This study provides new insights into differences between patients with CD who have fatal outcomes and those with non‐fatal outcomes. Preliminary analysis of this small cohort of deceased patients demonstrates that MCD can cause severe illness requiring frequent hospitalisation and urgent treatment upon diagnosis.
The exploratory analyses highlight potential factors for identifying CD patients at high risk of deterioration or poor outcomes. A longitudinal decrease in ALC levels was seen in the deceased CD cohort compared with matched controls and was significantly reduced at months 3 and 6 relative to matched controls. Lymphocytopenia increases the risk of recurrent infections and suggests immunodeficiency or homing of lymphocytes to secondary lymphoid organs. 35 It is unclear whether the decrease in ALC levels over time in deceased patients is due to underlying pathophysiological differences in disease or is a result of more intensive treatment, particularly with chemotherapy, or rituximab and cumulative steroid exposure. The deceased CD cohort was treated with significantly more regimens per year than the matched‐control CD cohort, although the total number of regimens administered was similar between the groups. Interestingly, ALC has been proposed as a prognostic marker in COVID‐19, in which cytokine storm and immune system over‐activity can cause severe illness. 36 , 37 , 38 Low ALC levels are suggestive of greater disease severity and worse outcome in COVID‐19, which is consistent with our findings. Thus, diligent evaluation of ALC could help assess risk of fatal outcome.
These analyses also reveal potential factors around the time of diagnosis that may be helpful for identifying patients with iMCD who are at high risk of deterioration or poor outcomes, including IgM, platelet count and INR. These preliminary findings indicate that the deceased iMCD group may be in a state of greater immune dysregulation (indicated by significantly lower IgM at time of diagnosis) and at increased risk of bleeding events (significantly lower platelet count at diagnosis) compared with patients who survive. The lower platelet count in the iMCD patients with a fatal outcome is likely reflective of the increased frequency of patients with the iMCD‐TAFRO clinical subtype among the deceased group, as thrombocytopenia is a key feature of TAFRO. The greater proportion of iMCD‐TAFRO cases in the deceased cohort also supports previous literature highlighting the high and early mortality observed in patients with this subtype. 18 , 39 , 40 It is also worth noting the relatively high proportion of iMCD‐TAFRO cases (54.2%) across all the iMCD patients in ACCELERATE, given that these are exclusively from the United States, and there was a sense early on that iMCD‐TAFRO was primarily found in Asia, where it was first described. 41 , 42
Interestingly, other groups have identified low platelet count as a risk factor for mortality: in a 2012 CD cohort analysis by Dispenzieri et al., age, sclerotic bone lesions, low platelet count and low serum albumin were suggested as risk factors for death in patients with CD. 26 Additionally, for patients with iMCD‐TAFRO, a drop in platelet count tends to reflect a flare in iMCD activity. 8 Unlike IgM and platelet count, around the time of diagnosis, INR levels were closer to normal in the deceased iMCD group than in the non‐deceased iMCD group. Additional research is needed into the use of these markers as predictors of mortality and the timing of changes. Although the sample size was too small to identify predictors of response to certain therapies in deceased and non‐deceased iMCD patients, previous work has identified laboratory tests and a proteomic panel capable of predicting response to siltuximab. 43 , 44 Notably, insufficient evidence existed to use the histopathological subtype to guide treatment approaches. 45 Patients in the deceased CD cohort had significantly more treatment regimens and hospitalisations per year than the matched controls. The observation that patients in the deceased CD cohort received many treatments within a short time suggests that a lack of treatment or undertreatment were not factors associated with mortality, but that there was a lack of clinical response to the treatments.
Substantial variability in time to diagnosis and time to treatment for patients with CD was also seen, reinforcing the need to improve diagnosis and time to treatment for patients.
The majority of iMCD patients were treated before the first‐ever iMCD consensus treatment guidelines were published in 2018. 20 Regimens were therefore likely selected based on evidence from other diseases or the experience of the treating physician.
Siltuximab is the only FDA‐ and EMA‐approved targeted treatment for iMCD and is recommended as the first‐choice treatment in iMCD consensus guidelines. 20 , 46 , 47 Our analysis revealed that many patients with iMCD were treated with siltuximab rather than with tocilizumab (25.0% received only tocilizumab, whereas 54.1% received only siltuximab as the IL6‐blocking therapy). In the deceased iMCD cohort, the most common therapies administered were cytotoxic chemotherapies, corticosteroids with other treatments and rituximab in combination with other treatments. In line with the broader iMCD population, half of the deceased cohort received siltuximab therapy. Of the six deceased iMCD patients, five were diagnosed after the approval of siltuximab (2014), but only one patient was diagnosed after the iMCD consensus guidelines were published (2018). 21 , 46 , 47 , 48 The reduced frequency of siltuximab use in the eligible iMCD population may have been due to a lack of familiarity with, or awareness of, siltuximab or the consensus‐based iMCD treatment guidelines, or a lack of local availability.
There are several limitations to our analyses. Although the dataset came from the largest registry of CD to date, the sample size of the deceased CD cohort was limited. As a retrospective observational study with a small sample size, the interpretation and generalisability of the results are limited, and we cannot exclude the possibility of unmeasured confounding factors. The limited sample size also affected the matched‐control CD group, as there were not enough patients to match two controls to each deceased patient. Laboratory measures such as haemoglobin, albumin and CRP may be important prognostic factors, but since we matched cases and controls on these measures, we were unable to assess their prognostic role and needed to find additional factors. Furthermore, these are real‐world data and therefore not collected according to a consistent schedule. Low sample size meant that it was not possible to capture and analyse the effects of various treatments on mortality. The repeated‐measures analysis of variance/linear mixed modelling results may not be robust, as the sample size was low (although changes in ALC had a large effect size). Since no correction for multiple comparisons was performed, the results are prone to type I error. The data presented are thus exploratory, and a future corollary study with a larger sample size is required to confirm our findings. Finally, variability in treatments used between patients presents another possible confounder. Understanding what is driving treatment choices and how to facilitate effective management with targeted therapy could improve outcomes in patients with CD.
Our study demonstrates that there may be differences between patients who have fatal and non‐fatal outcomes, and we provide preliminary suggestions for parameters to evaluate (platelet count, IgM and ALC) that may help to improve patient management. To improve our understanding of how to monitor and optimise treatment of patients with CD, further patient enrolment (www.CDCN.org/ACCELERATE), data collection and real‐world analyses through studies such as ACCELERATE are essential.
CONFLICT OF INTERESTS
David C. Fajgenbaum has received research funding for the ACCELERATE registry from EUSA Pharma and consulting fees from EUSA Pharma, as well as study drug from Pfizer for a clinical trial of sirolimus, and holds pending provisional patents for “Methods of treating idiopathic multicentric Castleman disease with JAK1/2 inhibition” and “Discovery and validation of a novel subgroup and therapeutic target in idiopathic multicentric Castleman disease.” Karan Kanhai is an employee of EUSA Pharma. Megan S. Lim received honoraria for participation in advisory board meetings for EUSA Pharma. Gordan Srkalovic reports speakers' bureau involvement with Takeda, Janssen Pharmaceuticals, Foundation Medicine and EUSA Pharma. Thomas S. Uldrick reports research support from Roche and Celgene, receives study drug for a clinical trial from Merck & Co, and is an employee of Regeneron. Frits van Rhee reports a consultancy relationship with Takeda, Sanofi Genzyme, EUSA Pharma, Adicet Bio, Kite Pharma and Karyopharm Therapeutics. All of the other authors report no competing interests.
AUTHOR CONTRIBUTIONS
All authors critically discussed and commented on the paper. David C. Fajgenbaum and Sheila K. Pierson defined the concept of the paper and supervised the ACCELERATE registry.
Supporting information
Table S1. Regimens and hospitalisations (deceased CD and matched‐control CD cohorts).
Table S2. Laboratory values at time of diagnosis (deceased CD and matched‐control CD cohorts).
ACKNOWLEDGEMENTS
The authors would like to thank all the patients and their families for their participation in the registry. ACCELERATE was established through a collaborative partnership between the Castleman Disease Collaborative Network (CDCN), Janssen Pharmaceuticals and UPenn. UPenn served as the regulatory study sponsor, managed the central database, retains ownership of all study data and supported the study infrastructure. Janssen served as the financial sponsor and provided advice regarding large‐scale study design, safety tracking, regulatory reports and site management before transferring this role to EUSA Pharma.
Medical writing support for the preparation of this manuscript was provided by William Shieu of TVF Communications, London, United Kingdom, and funded by EUSA Pharma Ltd.
EUSA Pharma is financially supporting the ACCELERATE study, as well as the development of the manuscript. David C. Fajgenbaum also receives funding support from the National Heart Lung & Blood Institute (R01HL141408).
Fajgenbaum DC, Pierson SK, Kanhai K, Bagg A, Alapat D, Lim MS, et al. The disease course of Castleman disease patients with fatal outcomes in the ACCELERATE registry. Br J Haematol. 2022;198:307–316. 10.1111/bjh.18214
Funding information
The funders of the study were involved in the data analysis, data interpretation and writing of the report. The corresponding authors had full access to all data in the study and had responsibility for the decision to submit for publication. EUSA Pharma provided funding for data collection and medical writing support.
REFERENCES
- 1. Flendrig J, Schillings P. Benign giant lymphoma: The clinical signs and symptoms and the morphological aspects. Folia Med (Plovdiv). 1969;12:119–20. [Google Scholar]
- 2. Keller AR, Hochholzer L, Castleman B. Hyaline‐vascular and plasma‐cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29(3):670–83. [DOI] [PubMed] [Google Scholar]
- 3. Oksenhendler E, Boutboul D, Fajgenbaum DC, Mirouse A, Fieschi C, Malphettes M, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol. 2018;180(2):206–16. [DOI] [PubMed] [Google Scholar]
- 4. Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135(16):1353–64. [DOI] [PubMed] [Google Scholar]
- 5. Wong RSM. Unicentric Castleman Disease. Hematol Oncol Clin North Am. 2018;32(1):65–73. [DOI] [PubMed] [Google Scholar]
- 6. Fajgenbaum DC, van Rhee F, Nabel CS. HHV‐8‐negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924–33. [DOI] [PubMed] [Google Scholar]
- 7. Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK, et al. POEMS syndrome and Castleman's disease. Biology and Management of Unusual Plasma Cell Dyscrasias. New York: Springer; 2016. p. 41–69. [Google Scholar]
- 8. Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence‐based consensus diagnostic criteria for HHV‐8‐negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dupin N, Diss TL, Kellam P, Tulliez M, Du M‐Q, Sicard D, et al. HHV‐8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV‐8–positive plasmablastic lymphoma. Blood. 2000;95(4):1406–12. [PubMed] [Google Scholar]
- 10. Fajgenbaum DC. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood. 2018;132(22):2323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Koga T, Sumiyoshi R, Kawakami A, Yoshizaki K. A benefit and the prospects of IL‐6 inhibitors in idiopathic multicentric Castleman's disease. Mod Rheumatol. 2019;29(2):302–5. [DOI] [PubMed] [Google Scholar]
- 12. Pierson SK, Stonestrom AJ, Shilling D, Ruth J, Nabel CS, Singh A, et al. Plasma proteomics identifies a ‘chemokine storm’ in idiopathic multicentric Castleman disease. Am J Hematol. 2018;93(7):902–12. [DOI] [PubMed] [Google Scholar]
- 13. Fajgenbaum DC, Langan R‐A, Japp AS, Partridge HL, Pierson SK, Singh A, et al. Identifying and targeting pathogenic PI3K/AKT/mTOR signaling in IL‐6‐blockade‐refractory idiopathic multicentric Castleman disease. J Clin Invest. 2019;129(10):4451–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshimi A, Trippett TM, Zhang N, Chen X, Penson AV, Arcila ME, et al. Genetic basis for iMCD‐TAFRO. Oncogene. 2020;39(15):3218–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arenas DJ, Floess K, Kobrin D, Pai R‐AL, Srkalovic MB, Tamakloe M‐A, et al. Increased mTOR activation in idiopathic multicentric Castleman disease. Blood. 2020;135(19):1673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pai R‐AL, Japp AS, Gonzalez M, Rasheed RF, Okumura M, Arenas D, et al. Type I IFN response associated with mTOR activation in the TAFRO subtype of idiopathic multicentric Castleman disease. JCI Insight. 2020;5(9):135031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nabel CS, Sameroff S, Shilling D, Alapat D, Ruth JR, Kawano M, et al. Virome capture sequencing does not identify active viral infection in unicentric and idiopathic multicentric Castleman disease. PLoS One. 2019;14(6):e0218660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fujimoto S, Sakai T, Kawabata H, Kurose N, Yamada S, Takai K, et al. Is TAFRO syndrome a subtype of idiopathic multicentric Castleman disease? Am J Hematol. 2019;94(9):975–83. [DOI] [PubMed] [Google Scholar]
- 19. Nishimura Y, Fajgenbaum DC, Pierson SK, Iwaki N, Nishikori A, Kawano M, et al. Validated international definition of the thrombocytopenia, anasarca, fever, reticulin fibrosis, renal insufficiency, and organomegaly clinical subtype (TAFRO) of idiopathic multicentric Castleman disease. Am J Hematol. 2021;96(10):1241–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Rhee F, Voorhees P, Dispenzieri A, Fosså A, Srkalovic G, Ide M, et al. International, evidence‐based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deisseroth A, Ko C‐W, Nie L, Zirkelbach JF, Zhao L, Bullock J, et al. FDA Approval: siltuximab for the treatment of patients with multicentric Castleman disease. Clin Cancer Res. 2015;21(5):950–4. [DOI] [PubMed] [Google Scholar]
- 22. van Rhee F, Wong RS, Munshi N, Rossi JF, Ke XY, Fosså A, et al. Siltuximab for multicentric Castleman's disease: a randomised, double‐blind, placebo‐controlled trial. Lancet Oncol. 2014;15(9):966–74. [DOI] [PubMed] [Google Scholar]
- 23. Ramasamy K, Gandhi S, Tenant‐Flowers M, Ceesay M, Corderoy S, Marcus R, et al. Rituximab and thalidomide combination therapy for Castleman disease. Br J Haematol. 2012;158(3):421–3. [DOI] [PubMed] [Google Scholar]
- 24. Starkey CR, Joste NE, Lee F‐C. Near‐total resolution of multicentric Castleman disease by prolonged treatment with thalidomide. Am J Hematol. 2006;81(4):303–4. [DOI] [PubMed] [Google Scholar]
- 25. Zhu S‐H, Yu Y‐H, Zhang Y, Sun J‐J, Han D‐L, Li J. Clinical features and outcome of patients with HIV‐negative multicentric Castleman's disease treated with combination chemotherapy: a report on 10 patients. Med Oncol Northwood Lond Engl. 2013;30(1):492. [DOI] [PubMed] [Google Scholar]
- 26. Dispenzieri A, Armitage JO, Loe MJ, Geyer SM, Allred J, Camoriano JK, et al. The clinical spectrum of Castleman's disease. Am J Hematol. 2012;87(11):997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pierson SK, Khor JS, Ziglar J, Liu A, Floess K, NaPier E, et al. ACCELERATE: A patient‐powered natural history study design enabling clinical and therapeutic discoveries in a rare disorder. Cell Rep Med. 2020;1(9):100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shin S, Kim J‐A, Lee B, Min CK, Kwak J‐Y, Yhim H‐Y. Achievement of complete remission after high‐dose melphalan and autologous stem cell transplantation Is the only important prognostic factor in patients with multiple myeloma. Blood. 2011;118(21):2018. [Google Scholar]
- 29. Melikyan AL, Egorova EK, Kovrigina АМ, Subortseva IN, Gilyazitdinova EA, Karagyulyan SR, et al. Clinical and morphological features of different types of Castleman's disease. Ter Arkh. 2015;87(7):64–71. [DOI] [PubMed] [Google Scholar]
- 30. Seo S, Yoo C, Yoon DH, Kim S, Park JS, Park C‐S, et al. Clinical features and outcomes in patients with human immunodeficiency virus‐negative, multicentric Castleman's disease: a single medical center experience. Blood Res. 2014;49(4):253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang X, Rao H, Xu X, Li Z, Liao B, Wu H, et al. Clinical characteristics and outcomes of Castleman disease: a multicenter study of 185 Chinese patients. Cancer Sci. 2018;109(1):199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dong Y, Wang M, Nong L, Wang L, Cen X, Liu W, et al. Clinical and laboratory characterization of 114 cases of Castleman disease patients from a single centre: paraneoplastic pemphigus is an unfavourable prognostic factor. Br J Haematol. 2015;169(6):834–42. [DOI] [PubMed] [Google Scholar]
- 33. Cohen AB, Swaminathan A, Wang X, Zamora S, Repucci MA, Kelly J, et al. Clinical characteristics, treatment patterns, and overall survival of real‐world patients with idiopathic multicentric Castleman disease. J Clin Oncol. 2021;39(15_suppl):7048. [Google Scholar]
- 34. Fujimoto S, Koga T, Kawakami A, Kawabata H, Okamoto S, Mizuki M, et al. Tentative diagnostic criteria and disease severity classification for Castleman disease: A report of the research group on Castleman disease in Japan. Mod Rheumatol. 2018;28(1):161–7. [DOI] [PubMed] [Google Scholar]
- 35. Warny M, Helby J, Nordestgaard BG, Birgens H, Bojesen SE. Lymphopenia and risk of infection and infection‐related death in 98,344 individuals from a prospective Danish population‐based study. PLoS Med. 2018;15(11):e1002685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wagner J, DuPont A, Larson S, Cash B, Farooq A. Absolute lymphocyte count is a prognostic marker in Covid‐19: a retrospective cohort review. Int J Lab Hematol. 2020;42(6):761–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang H‐J, Qi G‐Q, Gu X, Zhang X‐Y, Fang Y‐F, Jiang H, et al. Lymphocyte blood levels that remain low can predict the death of patients with COVID‐19. Medicine (Baltimore). 2021;100(28):e26503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020;383(23):2255–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sakashita K, Murata K, Takamori M. TAFRO syndrome: current perspectives. J Blood Med. 2018;9:15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ma W‐L, Zhang L, Zhu T‐N, Zhou D‐B, Li J, Sun J, et al. TAFRO Syndrome – a specific subtype of Castleman's disease in China. Chin Med J. 2018;131(15):1868–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iwaki N, Fajgenbaum DC, Nabel CS, Gion Y, Kondo E, Kawano M, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV‐8‐negative multicentric Castleman disease. Am J Hematol. 2016;91(2):220–6. [DOI] [PubMed] [Google Scholar]
- 42. Louis C, Vijgen S, Samii K, Chalandon Y, Terriou L, Launay D, et al. TAFRO syndrome in Caucasians: a case report and review of the literature. Front Med [Internet]. 2017;4(149):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morra DE, Pierson SK, Shilling D, Nemat S, Appiani C, Guilfoyle M, et al. Predictors of response to anti‐IL6 monoclonal antibody therapy (siltuximab) in idiopathic multicentric Castleman disease: secondary analyses of phase II clinical trial data. Br J Haematol. 2019;184(2):232–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pierson SK, Shenoy S, Oromendia AB, Gorzewski AM, Langan Pai R‐A, Nabel CS, et al. Discovery and validation of a novel subgroup and therapeutic target in idiopathic multicentric Castleman disease. Blood Adv. 2021;5(17):3445–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fajgenbaum DC, Wu D, Goodman A, Wong R, Chadburn A, Nasta S, et al. Insufficient evidence exists to use histopathologic subtype to guide treatment of idiopathic multicentric Castleman disease. Am J Hematol. 2020;95 (12):1553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. EMA. Sylvant (siltuximab) Summary of Product Characteristics. Available from: https://www.ema.europa.eu/en/documents/product‐information/sylvant‐epar‐product‐information_en.pdf
- 47. FDA. Sylvant (siltuximab) Prescribing Information. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125496s000lbl.pdf
- 48. van Rhee F, Oksenhendler E, Srkalovic G, Voorhees P, Lim M, Dispenzieri A, et al. International evidence‐based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv. 2020;4(23):6039–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Regimens and hospitalisations (deceased CD and matched‐control CD cohorts).
Table S2. Laboratory values at time of diagnosis (deceased CD and matched‐control CD cohorts).