

Abstract
Background
Hemoglobinopathies, inherited disorders of hemoglobin (Hb), are the most common hereditary monogenic diseases of the red cell in the world. Few studies have been conducted on hemoglobinopathies in Mauritania. Therefore, the aim of this work is to establish the molecular and epidemiological basis of hemoglobinopathies in a cohort of Mauritanian patients and to determine the haplotype of the β‐globin gene cluster in sickle cell subjects.
Methods
The molecular screening of Hb disorders in 40 Mauritanian patients was done by a polymerase‐restriction fragment length polymorphism (RFLP) for the sickle cell disease (SCD) mutation, a PCR/sequencing method for β‐thalassemia mutations, and by the multiplex polymerase chain reaction method for the α‐thalassemia. The exploration of eight polymorphic sites (SNPs) within the β‐globin gene cluster was conducted by PCR/RFLP method, to identify the HbS haplotypes from the sickle cell subjects.
Results
The epidemiological study of our patients showed a high incidence in the Senegal River area (52.5%) and a high ethnic prevalence for the Heratin (47.5%) and the Pular (35%). Molecular study allowed us to identify eight different mutations in our sample analyzed. They are respectively: HbS (HBB:c.20A>T) (68.75%), Cd44 ‐C (HBB:c.135delC) (8.75%), −29A>G (HBB:c.‐79A>G) (4.8%), −α‐3.7 (g.34164_37967del3804) (3.75%), IVS‐II‐849A>G (HBB:c.316‐2A>G) (2.25%) and Cd24T>A (HBB:c.75T>A), Hb Siirt (HBB:c.83C>G) and HbC (HBB:c.19G>A) each with (1.25%). Six different haplotypes are being explored among the SCD subjects with the Senegal haplotype as the most prevalent (66.7%), followed by Benin (10%), Arab‐Indians (6.7%), Bantu (3.3%), and two atypical haplotypes.
Conclusion
Our findings enrich the epidemiological data in our population and could contribute to the establishment of a strategy of prevention and management through screening, genetic counseling, and prenatal diagnosis of Hemoglobinopathies in the Mauritanian population.
Keywords: haplotypes, hemoglobin (Hb) disorders, hemoglobinopathies, sickle cell disease (SCD), α‐thalassemia, β‐thalassemia
Hemoglobinopathies, inherited disorders of hemoglobin (Hb), are the most common hereditary monogenic diseases of the red cell in the world. Few studies have been conducted on Hemoglobinopathies in Mauritania. Our results showed a high prevalence of hemoglobinopathies patients in Bank Senegal River in the South‐West of the country. Two Mauritanian ethnic groups, the Heratin and the Pular, were the most affected.

1. INTRODUCTION
Hemoglobinopathies are the most common hereditary monogenic diseases of the red blood cell in the world (Weatherall, 2007). They are a group of inherited disorders of hemoglobin (Hb) that are mostly transmitted in an autosomal recessive mode. These inherited disorders are characterized by either abnormal globin chain variants like sickle cell anemia or deficit, total or partial, globin chain synthesis in erythroid cells during hematopoiesis at the origin of thalassemia syndromes (Rees et al., 2010).
The most known forms are the β‐thalassemia which prevails primarily in the carrier of the Mediterranean basin, and the sickle cell disease (SCD) which is more frequent in the countries of sub‐Saharan Africa (Weatherall & Clegg, 2001).
The heterogeneous ethnic and epidemiological distribution of these diseases and the presence of wide variability in the clinical expression linked to the genetic and environmental factors constitute an obstacle for the development of hemoglobinopathies control programs (Zohreh, 2013).
The Mauritanian geographic situation constitutes a link between the population of West Africa and those from the Maghreb. The Mauritanian population consists of two big groups: the Arabo‐Berber whose members are commonly named Moor (Heratin and Bidan) and the origin African composed of Pular, Soninke, Wolof, and a minority of Bambara (Merchesin, 1992). Few studies have been carried out on hemoglobinopathies in Mauritania. The first study evaluated the frequency of the βS at around 8.71% (Deyde et al., 2002), however, the second study shows a frequency of 5.71% (Veten et al., 2012).
With regard to the genetic study of Hemoglobinopathies, nearly a thousand mutant alleles have now been reported (https://globin.bx.psu.edu/hbvar/menu.html). The mutations are regionally specific and in most cases, the geographical and ethnic distributions have been determined providing the foundation for a program of control through screening, genetic counseling, and prenatal diagnosis (Giardine et al., 2021; Hardison et al., 2002; Swee Lay, 2013).
It should be noted that the HbS mutation has been described on five distinct haplotypes based on the presence or absence of the eight different restriction enzyme sites in the genomic regions retching from the 5′ε‐globin to the 3′β‐globin gene on chromosome 11 (~60 kb) (Kamel, 1979; Sutton et al., 1989). These haplotypes are known as Benin, Bantu or Central African Republic (CAR), Senegal, Cameroon, and Asian (Arabo‐Indian) according to their ethnic and geographical origins. In fact, the first four are African haplotypes (Pagnier et al., 1984), while the last one was described in Central India and in Saudi Arabia (Nagel & Fleming, 1992). It has been previously reported that the CAR haplotype is usually associated with a more severe disease when compared with the intermediate phenotype of the Benin haplotype and to the milder conditions associated with the Senegal and the Arabo‐Indian (Padmos et al., 1991). Analysis of the polymorphic sites of the β genes cluster is of genetic, anthropologic, and clinical interest, and it can also be used to predict disease prognosis and to plan appropriate treatment.
In this study, we aim to determine the molecular and epidemiological basis of hemoglobinopathies in a group of Mauritanian patients. To do this, we have explored the β‐ and α‐ globin genes to identify mutations responsible for hemoglobinopathies. Then, we focused on the exploration of eight polymorphic sites (SNPs) within the β‐globin gene cluster to establish the HbS haplotype in the sickle cell subjects. Also, the epidemiological study and the correlation phenotype/genotype study were carried out in order to better understand the clinical and hematological characteristics of hemoglobinopathies in our sample study.
2. MATERIALS AND METHODS
2.1. Patients
This study included 40 Mauritanian patients that were affected by hereditary hemoglobin diseases, mainly SCD, β‐thalassemia, and α‐thalassemia. They are originated from different Mauritania cities and are regularly followed in the Pediatric Hemato‐Oncology department of the National Oncology Center (CNO) and at the MaurLab laboratory in Nouakchott, Mauritania during 2018 to 2021. Information about blood transfusion, age of diagnosis, splenomegaly, vaso‐occlusive crises, and the hematological parameters were obtained by retrospective clinical data. Our subjects are aged between 1 and 29 years old with an average age of 10 years. Our cohort consists of 25 males (60.97%) and 15 females (39.02%) with 1.6 as gender ratio. Our demographic data indicates that 29.3% of patients were born from consanguineous couples.
After obtaining authorization, 3–5 ml of whole blood from each subject were collected on an EDTA tube and then referred to the Biochemistry and Molecular Laboratory at Children's Hospital of Tunisia for molecular study. Written consent was obtained from all patients and parents of minor patients before enrollment in the study.
This study was approved by the ethics committee of Pediatric Hemato‐Oncology department of the National Oncology Center in Mauritania and the Children's Hospital of Tunis.
2.2. Hematological data and hemoglobin analysis
Hematological parameters were determined by cytometry on an automated Beckman LH750 TM Hematology analyzer (Beckman). Hb identification was carried out by a cation exchange high‐performance liquid chromatography (HPLC), using Variant TM II System (Bio‐Rad Laboratories). Hb fractions were eluted from the cartridge based on their ionic interaction with the cartridge material, and the different Hbs were eluted in the order: HbF, glycated HbA, HbA, HbA 2, and HbS.
2.3. DNA analysis and molecular study
Genomic DNA was isolated from white blood cells by the salting‐out procedure (Miller et al., 1988) or by the PureLink® Genomic DNA Kit (Invitrogen, ThermoFiher Scientific). The research on βS mutation [β6, GAG>GTG, Glu>Val] was carried out by the PCR‐RFLP (Polymerase‐Restriction Fragment Length Polymorphism) with the Bsu36I restriction enzyme (Livingstone, 1967).
Molecular screening of the β‐thalassemia mutations was done by PCR/Sequencing, using the specific primers (Chaima et al., 2013) and the SeqStudio™ Genetic Analyzer System (Thermo Fisher Scientific) for the systematic sequencing of β‐globin gene (HBB ) (Sanger et al., 1977).
Common α‐thalassemia mutation –α3.7deletion was screened among patients, by multiplex polymerase chain reaction method (Arnold et al., 2001), using the Go Taq® Long Mater Mix kit (Promega).
The β‐globin gene cluster haplotype was characterized by the PCR‐RFLP method (Orkin et al., 1982; Saleh‐Gohari & Mohammadi‐Anaie, 2012). The common haplotype of the β‐globin gene cluster was determined according to the presence (+) or absence (−) of a composition of SNPs (Single Nucleotide Polymorphism) corresponding to the traditional eight restriction sites were analyzed including the HincII (5′ of ε‐globin gene), XmnI (5′ of the Gγgene), HindIII (intron 2 of the Gγgene), HindIII (Aγ‐globingene), HincII (5′ of the ψβ‐globin gene), HincII (3′ of the ψβ‐globin gene), AvaII (intron 2 of β‐globin gene) and BamHI (3′ of the β‐globin gene).
The statistical analysis was performed using version 20.0 of the statistical package for the social sciences software SPSS (SPSS). The SNPs analysis and the haplotypes determination were performed by the haploview 4.2 software.
3. RESULTS
The epidemiological data analysis showed a wide distribution of hemoglobinopathies throughout the Mauritanian territory with a high incidence on the area of the Senegal River bank (52.5%), located in the South‐West, which included the cities of Gorgol, Guidimakha, Trarza, and Brakna. The Mauritanian population is made up of two big groups: the Negro‐Africans (Pular, Wolof, and Soninke) and the Arabo‐Berbers (Heratin and Bidan). Indeed, the ethnic distribution of our sample showed a high prevalence of Hemoglobinopathies in favor of Heratin (47.5%) and Pular (35%) (Figures 1 and 2).
FIGURE 1.
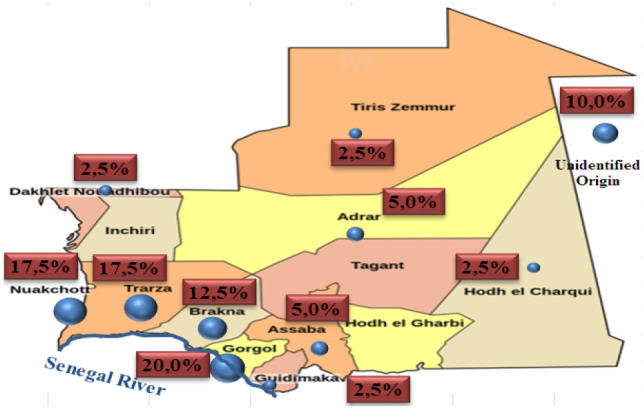
Geographical distribution of our patients in Mauritania
FIGURE 2.
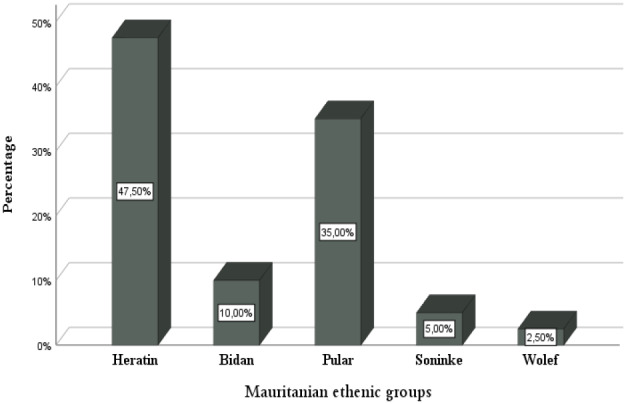
Ethnic distribution of our Mauritanian patients
The molecular study allowed us to identify five types of Hb disorders (Hb) in overall samples: the HbS variant (βS) (c.20A>T) is the most frequent accounting 68, 75% followed by β‐Thal alleles (15%), α‐Thal alleles (3.75%), HbC (c.19G>A) (1.25%) and Hb Siirt (c.83C>G) (1.25%) (Table 1). It should be noted that the last variant is found for the first time in the Mauritanian population in a heterozygous state.
TABLE 1.
Allelic frequency of hemoglobin defects found in our sample study
| Hb defects | Number of alleles (%) |
|---|---|
| β−S | 55 (68.75) |
| β−Thal | 12 (15.0) |
| α−Thal | 3 (3.75) |
| β−A | 8 (10.0) |
| HbC | 1 (1.25) |
| Hb Siirt | 1 (1.25) |
Eight different mutations were detected in our study (Table 2). The β‐thal point mutations were respectively the Cd44 ‐C (c.135delC) with 8.75%, the −29 A>G (c.‐79A>G) with 4.8%, the IVS‐II‐849 A>G (c.316‐2A>G) with 2.25% and the Cd24 T>A (c.75T>A) with 1.25%. –α‐3.7deletion (g.34164_37967del3804) was detected on the α‐globin gene with 3.75%. Three SNP were also reported: rs10768683 or IVS2‐16 C>G (15%), rs713040 or cd2 C>T (7.5%) and rs16098012 or IVS2‐666 (10%).
TABLE 2.
Different mutations identified in our sample study and their ethnic origin (https://globin.bx.psu.edu/hbvar/menu.html)
| Mutations | Type | Number of alleles (%) | Ethnic origin |
|---|---|---|---|
| HBB:c.20A>T | Hb Variant | 55 (68.75) | Black American and Indian |
| HBB: c.135delC | β0 | 7 (8.75) | Oman, Tunisian, Bahraini… |
| HBB:c.‐79A>G | β+ | 4 (4.8) | Black & Chinese |
| NG_000006.1:g.34164_37967del3804 | α−thal−2 | 3 (3.75) | Indian, Far Eastern; African, Mediterranean |
| HBB:c.316‐2A>G | β0 | 2 (2.25) | African black |
| HBB:c.75T>A | β+ | 1 (1.25) | American black, Japanese |
| HBB:c.83C>G | Hb variant | 1 (1.25) | Kurdish |
| HBB:c.19G>A | Hb variant | 1 (1,25) | African black |
| rs10768683 (G>C) | SNP | 12 (15) | Indian |
| rs1609812 (C>T) | SNP | 8 (10) | Indian |
| rs713040 (T>C) | SNP | 6 (7.5) | Indian |
Abbreviations: Hb variant, hemoglobin variant; β+, a partial deficit of β‐globin chains synthesis; β0, a total deficit of β‐globin chains synthesis; SNP, Single Nucleotide Polymorphism; α−thal−2, alpha‐thalassemia type 2.
The Genotypic analysis showed the presence of eight different genotypes in our subjects. The β S /β S (55%) and β A /β −Thal (17.5%) were the most common genotypes. While, the β S /β C was the less frequent one (Table 3). Moreover, our hematological data showed that the βS/βthal genotype increases levels of hematocrite and HbA1, and decreases levels of HbF and HbS as compared to the β S /β S . While, β S /β S , αα/−− genotype decreases MCV, MCH, hematocryte, and HbA2 more than the β S /β S genotype (Table 3).
TABLE 3.
The hematological parameters of the different genotypes identified in our study
| Genotypes | Hb (g/dl) | MCH (pg) | MCV (fl) | Ht (%) | HbA (%) | HbA2 (%) | HbF (%) | HbS (%) | HbC (%) | N (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| βA/βS | 9.15 | 23.63 | 72.30 | 27.50 | 54.50 | 2.56 | 4.2 | 40.13 | 3 (7.5) | |
| βS/βS | 7.44 | 27.80 | 83.90 | 21.50 | 2.78 | 3.18 | 12.64 | 79.43 | 22 (55) | |
| βS/βC | 11.3 | 25 | 75 | 33 | 2.5 | 1.9 | 3.1 | 41.7 | 40.8 | 1 (2.5) |
| β−Thal/β−Thal | 6.50 | 24.5 | 69.30 | 15.4 | 36.65 | 5.95 | 40.8 | 2 (5) | ||
| βA/β−Thal | 9.38 | 21.23 | 74.4 | 12.33 | 70.08 | 5.51 | 18.00 | 0 | 7 (17.5) | |
| βS/β−Thal | 10.25 | 21.3 | 67.5 | 32.5 | 28.65 | 2.55 | 3.15 | 58.15 | 2 (5) | |
| βS/βS, αα/−− | 6.4 | 26.75 | 72.50 | 15 | 1.1 | 1.8 | 8.4 | 86.5 | 2(5) | |
| βA/βS, αα/−− | 10.30 | 19 | 61 | 33.0 | 47.10 | 1.2 | 0.70 | 33.80 | 1 (2.5) | |
| βA/βS | 9.15 | 23.63 | 72.30 | 27.50 | 54.50 | 2.56 | 4.2 | 40.13 | 3 (7.5) | |
| βS/βS | 7.44 | 27.80 | 83.90 | 21.50 | 2.78 | 3.18 | 12.64 | 79.43 | 22 (55) | |
| βS/βC | 11.3 | 25 | 75 | 33 | 2.5 | 1.9 | 3.1 | 41.7 | 40.8 | 1 (2.5) |
| β−Thal/β−Thal | 6.50 | 24.5 | 69.30 | 15.4 | 36.65 | 5.95 | 40.8 | 2 (5) | ||
| βA/β−Thal | 9.38 | 21.23 | 74.4 | 12.33 | 70.08 | 5.51 | 18.00 | 0 | 7 (17.5) | |
| βS/β−Thal | 10.25 | 21.3 | 67.5 | 32.5 | 28.65 | 2.55 | 3.15 | 58.15 | 2 (5) | |
| βS/βS, αα/−− | 6.4 | 26.75 | 72.50 | 15 | 1.1 | 1.8 | 8.4 | 86.5 | 2 (5) | |
| βA/βS, αα/−− | 10.30 | 19 | 61 | 33.0 | 47.10 | 1.2 | 0.70 | 33.80 | 1 (2,5) |
Abbreviations: Hb, hemoglobin; MCH, mean corpuscular hemoglobin; MCV, mean cell volume; Ht, hematocrite; HbA, hemoglobin A; HbA2, hemoglobina2; HbF, fetal hemoglobin; HbS, sickle hemoglobin; HbC, hemoglobin C.
We were also carried out a molecular exploration of eight Single Nucleotide Polymorphisms (SNPs) within the β‐globin gene cluster in order to establish the haplotypes from 30 sickle cell subjects in our cohort. This study showed that there were six different haplotypes in our cohort, revealing the Senegalese haplotype as the most frequent (66.66%), followed by Benin (10%), Arabo‐Indian (6.66%), and Bantu (3.33%). In our cohort, we noted Two atypical haplotypes (different combinations), termed here as Aty1 and Aty2 with 6.66% each other (Table 4, Figure 3).
TABLE 4.
Frequencies of restriction haplotypes established in sickle cell subjects
| Haplotypes | 3′‐ɛ Hinc II rs3834466 | 5′‐Gγ XmnI rs7482144 | Gγ HindIII rs2070972 | Aγ HindIII rs28440105 | Ψβ HincII rs10128556 | 3′ ψβ HincII rs968857 | β AvaII rs1076863 | 3′β BamHI no RSID | n (%) |
|---|---|---|---|---|---|---|---|---|---|
| Senegal | − | + | + | − | + | + | + | + | 40 (66.7) |
| Benin | − | − | − | − | − | + | + | + | 6 (10) |
| Arab‐Indian | + | + | + | − | + | + | + | − | 4 (6.7) |
| Bantu | − | − | + | − | − | − | + | + | 2 (3.3) |
| Atypical 1 | − | + | + | − | + | − | + | + | 4 (6.7) |
| Atypical 2 | − | + | + | − | − | + | + | + | 4 (6.7) |
Abbreviations: +, presence of the SNP; −, absence of the SNP.
FIGURE 3.

Different haplotypes found in our cohort
In order to understand the clinical heterogeneity observed from our SCD subjects, we have analyzed the clinical and hematological data related to each haplotypes. We noted that patients carrying the haplotype the Senegalese haplotype present a less severe phenotype compared to Benin, Bantu, and Arabo‐Indian haplotypes. In fact, among the subjects who carried the Senegalese haplotype only 6.66% present splenomegaly, 46.6% present a vaso‐occlusive crisis and 13.3% require a regular blood transfusion (Table 5).
TABLE 5.
The clinical and hematological data associated with different haplotypes established from sickle cell subjects
| Haplotypes | Hb (g/dl) p = 0.58 | HbF (%) p = 0.35 | HbS (%) p = 0.87 | BT (%) p = 0.59 | VOC (%) p = 1.00 | SHM (%) p = 0.33 |
|---|---|---|---|---|---|---|
| Senegal | 7.47 | 11.09 | 74.82 | 13.3 | 46.6 | 6.6 |
| Benin | 7.36 | 9.15 | 71.31 | 33.3 | 50.0 | 33.3 |
| Arabo‐Indian | 10 | 6.8 | 69.05 | 50.0 | 50.0 | 50.0 |
| Bantu | 7.10 | 20.33 | 76.7 | 0.0 | 50.0 | 0.0 |
| Atypical 1 | 8.10 | 5.45 | 56.35 | 0.0 | 50.0 | 0.0 |
| Atypical 2 | 9.50 | 19.00 | 69.6 | 0.0 | 50.0 | 0.0 |
Abbreviations: Hb, mean Hemoglobin level; HbF, mean level of fetal hemoglobin; HbS, mean level of hemoglobin S; BT, blood transfusion frequency; VOC, vaso‐occlusive crises frequency; SHM, spleno‐hepato‐megaly frequency.
4. DISCUSSION
Hemoglobinopathies are widely spread throughout North Africa (Fattoum, 2009). They represent a public health problem in Mauritania due to lack of knowledge and diagnostic errors. In order to establish a molecular basis for these genetic disorders, a cohort of 40 Mauritanian patients was studied (Appendix A, Table A1).
The epidemiological data analysis showed an important distribution of hemoglobin disorders throughout the Mauritanian territory with a high incidence in the Senegal River bank (52.5%) located in the South‐West of the country. This high prevalence of hemoglobinopathies in the area of Senegal River bank could be due to speared of hemoglobin defects by the gene flow from Senegal to Mauritania, especially the SCD and α‐thalassemia, which have been reported as very common in the Senegalese population (Gueye Tall et al., 2017; OMS, 2006). The ethnic distribution in our group of patients showed a high incidence in favor of Heratin and Pular with 47.5% and 35% respectively (Figure 2). These results are in contradiction with those found by Deyde et al. (2002). This difference can be explained by the number of analyzed patients and the location of the hospitals study centers. Our finding indicates that these two Mauritanian groups ethnic (Heratin and Pular) may constitute a high‐risk population which implies the establishment of systematic screening program among their members.
Concerning the molecular screening of α‐ and β‐globin genes, we detected eight different mutations and three SNPs in the 82 alleles studied. In fact, the βS mutation, was found to be the most common with 68.75%, followed by Cd44 ‐C (c.135delC) (8.75%), −29 A>G (c.‐79A>G) (4.8%), −α‐3.7 (g.34164_37967del3804) (3.75%), and IVS‐II‐849 A>G (c.316‐2A>G) (2.25%) mutation.
Most of these mutations have been reported previously in the African, Mediterranean, and Asian population (Amselem et al., 1988; Antonarakis & Cheng, 1984; Schilirò et al., 1995). This finding is in line with the history of the Mauritanian population that was a link between the Black population of West Africa and those from the Maghreb (Merchesin, 1992).
The Hb Siirt variant was found in a 16‐year‐old patient who presented clinical feature of β‐thalassemia intermediate with mild anemia and splenomegaly (Hb: 7.4 g/dl; A2:4.2% and HbF: 51%). The Hb Siirt variant has been previously described from Kurdish patient as a silent Hb variant (Bianco et al., 1997). A recent study reported a possible small contribution to the instability of helix β of hemoglobin β (Cappabianca et al., 2017).
We compared the frequency of mutations detected in this study with other data reported for the neighboring countries such as Algeria, Tunisia, and Senegal. The βS (HBB:c.20A>T), and –α‐3.7 (g.34164_37967del3804) have been previously reported in these three countries with a high frequency in Senegal with 8%–10% for the βS and 21% for –α‐3.7 (Gueye Tall et al., 2017; OMS, 2006). Two previously study reported the high frequency of βS in Mauritanian population with 5.71% (Veten et al., 2012) and 8.71% (Deyde et al., 2002). In Tunisia and in Algeria, the frequency of βS was, respectively, 1.89% (Fattoum, 2006; Fattoum et al., 2004) and 0.8%–3.5% (Boudrahem‐Addour et al., 2009; Mesbah‐Amroun et al., 2008). We noted that cd24 T>A (HBB:c.75T>A) and Hb Siirt (HBB:c.83C>G) seem to be specific of Mauritanian population.
The genotypic study showed eight different genotypes in our cohort. The analysis of the hematological indices associated with these genotypes indicates that the combination of βS/β−thal increases levels of hematocrite and HbA1, and decreases levels of HbF and HbS as compared to the β S /β S . Whereas, β S /β S , αα/−− genotype decreases MCV, hematocryte, and HbA2 more than the β S /β S genotype. These findings are in agreement with the fact that coinheritance of βS/βThal and βS/α−thal modulates the severity of the disease (Saleh‐Gohari & Mohammadi‐Anaie, 2012; Valavi et al., 2010).
The molecular study of eight SNP within the β‐globin gene cluster showed that six different haplotypes among the SCD subjects with the Senegalese haplotype as the most prevalent (66.7%), followed by Benin, Arabo‐Indian, Bantu and two atypical haplotypes. These results are agreement with those found in a previous Mauritanian study carried (Veten et al., 2012) and in disagreement with those found in the Tunisian population whose the most predominant haplotype is the Benin (Moumni et al., 2011). These different haplotypes observed here reveal that was a multiple origin of βS mutation in the Mauritanian population as well as the high prevalence of the Senegalese haplotype appears to be due to the gene flow of βS‐gene from the Senegal to the Mauritanian population.
The analysis of clinical and hematological data showed that the Senegalese haplotype presented a less severe phenotype compared to Benin, Bantu, and Arabo‐Indian haplotypes. This finding would indicate the existence of clinical variability associated with βS‐globin haplotypes described in our cohort. However, this association was not significant. Further studies, including a large sample size, are required to confirm our findings.
In fact, it has been previously reported that the Senegal and Arabo–Indian haplotypes are generally associated with a mild course in SCD and the individuals with the Bantu haplotype appear to have a more severe disease (Nagel & Fleming, 1992; Padmos et al., 1991).
5. CONCLUSION
Our results showed a high prevalence of Hemoglobinopathies patients in Bank Senegal River in the South‐West of the country. Two Mauritanian ethnic groups, the Heratin and the Pular, were the most affected.
The molecular study showed the presence of different Hemoglobinopathies in Mauritania, with SCD and β‐thalassemia as the most common hemoglobin disorders, and with an admixture of the African, Mediterranean, and Asian mutations on the Mauritanian population. The βS mutation was the most common one 68.75%. Six different haplotypes have been described among the SCD subjects with the Senegal haplotype as the most prevalent. This genetic variability could be due to the geographical origin and to the multi‐ethnicity of the Mauritanian population.
Our results enrich the epidemiological data in our population and help to provide an efficient genetic counseling among families. Unfortunately, our study is not without limitations and these should be mentioned: First, the sample size is limited. Second, our patients only come from two hospital centers in Nouakchott, a multicenter study is necessary to increase the number of patients and reach all Mauritanian cities.
AUTHOR CONTRIBUTIONS
Mr. Taher Mahmoud: The data collection, practice, data analysis, and manuscript preparation. Dr. Chaima Sahli: Experimental protocol, data analysis, and manuscript preparation. Dr. Sondess Hadj Fredj: manuscript revision Dr. Yessine Amri: Experimental protocol and data analysis. Mrs. Rim Othmani: Experimental protocol and data analysis. Pr. Taieb Messaoud: Planning, conduct, and data analysis. Dr. Sidi M. Ghaber: Conceptualization and design analysis. Dr.Ekhtelbenina Zein. The doctor providing the clinical data of patients.
CONFLICT OF INTEREST
The authors report no conflict of interest.
ACKNOWLEDGMENTS
We would like to thank the staff of the Pediatric Hemato‐Oncology Department of the National Oncology Center (Nouakchott, Mauritania) for providing the clinical data of patients and the Biochemistry and Molecular Biology Laboratory LR00SP03 of Children's Hospital Bechir Hamza (Tunis, Tunisia).
APPENDIX A.
TABLE A1 Demographic, hematological, and clinical data of the sample study
| Patients | Age (an) | Sex | Ethnic group | Origin | Hb g/dl | MCV (fl) | MCH Pg | Ht (%) | A (%) | (A2%) | F (%) | S (%) | C (%) | Clinical signs | Genotypes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 13 | F | Heratin | Gorgol | 8.1 | 62.4 | 21.5 | 23.49 | 45 | 4.4 | 17.4 | MA and BT | β−Thal/β−Thal | ||
| 2 | 3 | F | Heratin | Gorgol | 10.2 | 68.8 | 15 | 19.10 | 77.9 | 5.7 | 5.4 | MA | βA/β−Thal | ||
| 3 | 4 | M | Heratin | Gorgol | 5.4 | 68.8 | 23.8 | 15.64 | 68.4 | 3.3 | 11 | SA and SHM | βA/β−Thal | ||
| 4 | 14 | M | Heratin | Noukchott | 4.9 | 76.2 | 27.5 | 7.30 | 28.3 | 7.5 | 64.2 | AS, BT, and SHM | β−Thal/β−Thal | ||
| 5 | 6 | M | Pular | Trarza | 9.5 | 67 | 21.9 | 29 | 35.6 | 2 | 4.8 | 45.9 | MA and VOC | βS/β−Thal | |
| 6 | 16 | M | Heratin | – | 7.4 | 103 | 32.9 | 22.6 | 26 | 4.2 | 51.1 | MA and SHM | βA/β−Thal (Hb Siirt) | ||
| 7 | 15 | M | Heratin | Assaba | 11 | 68 | 20.7 | 36 | 21.7 | 3.1 | 1.5 | 70.4 | MA and BT | βS/β−Thal | |
| 8 | 7 | F | – | 12 | 68 | 20.3 | – | 94.3 | 5.7 | MA | βA/β−Thal | ||||
| 9 | 29 | M | Soninke | – | 13.4 | 67.8 | 21.4 | – | 89 | 7.4 | 3.6 | βA/β−Thal | |||
| 10 | 19 | M | Pular | – | 8.3 | 70 | 14 | – | 61.9 | 6 | 32.9 | MA | βA/β−Thal | ||
| 11 | 5 | M | Soninke | GuidiMaka | 5.4 | 84 | 29.8 | 15 | 2.2 | 1.7 | 16.8 | 75 | SA and SHM | βS/βS, αα/α‐Thal | |
| 12 | 3 | F | Pular | Gorgol | 8.4 | 83 | 27.1 | 26 | 0 | 3 | 26.7 | 70.3 | AM, BT, and VOC | βS/βS | |
| 13 | 10 | F | Pular | Brakna | 8.3 | 61.2 | 21.7 | 11 | 2.3 | 1.6 | 8.3 | 83.6 | AM, BT, VOC, and SHM | βS/βS | |
| 14 | 13 | F | Pular | Gorgol | 8.6 | 86.8 | 29.7 | 25 | 21.8 | 2.7 | 6 | 62.5 | MA and BT | βS/βS | |
| 15 | 11 | F | Heratin | Gorgol | 5.8 | 88.7 | 28.2 | 18.20 | 0 | 3 | 23.4 | 73.6 | SA, BT, and VOC | βS/βS | |
| 16 | 5 | F | Pular | H.Chargui | 7.3 | 72.8 | 23.2 | 23.02 | 53.3 | 2.6 | 8.4 | 35.7 | MA and SHM | βA/βS | |
| 17 | 7 | M | Heratin | Nouakchott | 7.4 | 61 | 23.7 | – | 0 | 2 | 0 | 98 | MA | βS/βS, αα/αThal | |
| 18 | 8 | M | Pular | Nouakchott | 7.7 | 78 | 26 | 14 | 0 | 2.4 | 30.8 | 66.8 | MA and VOC | βS/βS | |
| 19 | 10 | M | Pular | Trarza | 10.3 | 33.4 | – | 0 | 3.2 | 12.1 | 84.7 | MA | βS/βS | ||
| 20 | 15 | M | Heratin | Brakna | 8.1 | 96 | 31.2 | 25 | 3.1 | 1 | 10.9 | 76 | MA and VOC | βS/βS | |
| 21 | 15 | M | Bidan | Nouakchott | 11 | 71.3 | 23.7 | – | 50 | 2 | 48 | MA | βA/βS | ||
| 22 | 14 | M | Heratin | Nouakchott | 8.2 | 93 | 33 | 23 | 0 | 3.4 | 6.8 | 89.8 | MA and VOC | βS/βS | |
| 23 | 9 | M | Heratin | Nouakchott | 3.3 | 70 | 18.9 | 12 | 4.9 | 5.2 | 0 | 89.9 | SA | βS/βS | |
| 24 | 9 | M | Heratin | Trarza | 4.3 | 76 | 19.6 | – | 0 | 4.3 | 3.4 | 92.3 | SA, BT, and SHM | βS/βS | |
| 25 | 4 | M | Heratin | Gorgol | 9 | 82 | 30 | – | 0 | 2.9 | 20.9 | 76.2 | MA, BT, and VOC | βS/βS | |
| 26 | 15 | F | Pular | Trarza | – | 0 | 3.1 | 6.8 | 90.1 | MA, BT, and VOC | βS/βS | ||||
| 27 | 6 | F | Pular | Gorgol | 7.3 | 88.6 | 31 | 20.80 | 13.8 | 1.3 | 5.5 | 65.9 | MA, BT, and VOC | βS/βS | |
| 28 | 8 | M | Pular | Brakna | 10.3 | 61 | 19.1 | 23 | 47.1 | 1.2 | 0.7 | 33.8 | MA | βA/βS, αα/α‐Thal | |
| 29 | 11 | M | Heratin | Assaba | 11.3 | 75 | 25 | 33 | 2.5 | 1.9 | 3.1 | 41.7 | 40.8 | MA | βS/βC |
| 30 | 8 | M | Bidan | Tris‐Zemour | 10 | 102 | 33.2 | 30.10 | 2.7 | 0.3 | 26.2 | 62 | MA and VOC. | βS/βS | |
| 31 | 5 | F | Bidan | Adrar | 9.5 | 96.6 | 33.6 | 27 | 0 | 2.4 | 18.4 | 79.2 | MA and VOC. | βS/βS | |
| 32 | Heratin | Nouadhibou | – | 84 | 2.8 | 0.8 | βA/β‐Thal | ||||||||
| 33 | 12 | F | Pular | Brakna | 2.9 | 73 | 22.5 | – | 0.3 | 11 | 19 | 69.6 | SA and BT | βS/βS | |
| 34 | 13 | M | Pular | Trarza | 6.8 | 89 | 29.5 | 18.90 | 12.3 | 3.2 | 5.1 | 79.4 | SA, BT, and VOC | βS/βS | |
| 35 | 1 | M | Pular | Brakna | 5.4 | 92 | 28.5 | 17 | 0 | 2.8 | 14.3 | 82 | SA, BT, and VOC | βS/βS | |
| 36 | 22 | F | Wolef | Nouakchott | 72.8 | 24 | 32 | 60.2 | 3.1 | 0 | 36.7 | βA/βS | |||
| 37 | 11 | F | Heratin | Gorgol | 6.8 | 89 | 28.5 | 21 | 0 | 3.9 | 5.7 | 90.4 | SA, BT, and VOC | βS/βS | |
| 38 | 11 | M | Bidan | Adrar | 6.4 | 96 | 31.4 | – | 0 | 3 | 7.9 | 89.1 | SA, BT, andVOC | βS/βS | |
| 39 | 8 | F | Heratin | Trarza | 7.7 | 82.3 | 26 | – | 0 | 3.1 | 12.2 | 84.7 | AM | βS/βS | |
| 40 | 14 | M | Heratin | Trarza | 8.3 | 79.4 | 29.7 | – | 0 | 3.2 | 7.8 | 89 | AM | βS/βS |
Abbreviations: F, female; M, male; Origin, Mauritanian cities; Hb, hemoglobin; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; Ht, hematocrite; A, A2, F, S, and C, hemoglobin fractions; MA, moderate anemia; SA, severe anemia; BT, blood transfusion. VOC, vaso‐occlusive crises, SHM, Spleno‐Hepato‐Megaly; −, unidentified origin.
Mahmoud, T. , Sahli, C. , Hadj Fredj, S. , Amri, Y. , Othmani, R. , Mohamed, G. S. , Zein, E. , & Messaoud, T. (2022). Epidemiological and molecular study of hemoglobinopathies in Mauritanian patients. Molecular Genetics & Genomic Medicine, 10, e2048. 10.1002/mgg3.2048
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- Amselem, S. , Nunes, V. , Vidaud, M. , Estivill, X. , Wong, C. , d'Auriol, L. , Vidaud, D. , Galibert, F. , Baiget, M. , Goossens, M. , Amselem, S. N. V. , & Vidaud, M. (1988). Determination of the spectrum of β thalassemia genes in Spain by use of dot blot analysis of amplified β globin DNA. American Journal of Human Genetics, 43, 95–100. [PMC free article] [PubMed] [Google Scholar]
- Antonarakis, S. E. , Irkin, S. H. , Cheng, T. C. , Scott, A. F. , Sexton, J. P. , Trusko, S. P. , Charache, S. , & Kazazian, H. H., Jr. (1984). β‐Thalassemia in American blacks: Novel mutations in the “TATA” box and an acceptor splice sites. Proceedings of the National Academy of Sciences of the United States of America, 81(4), 1154–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold, S. C. , Quah, T. C. , Low, P. S. , & Chong, S. S. (2001). A rapid and reliable 7‐deletion multiplex polymerase chain reaction assay for a‐thalassemia. Blood, 98(1), 250–251. [DOI] [PubMed] [Google Scholar]
- Bianco, M. C. P. , Lerone, M. , Morlupi, L. , & Rinaldi, S. (1997). HB Siirt [β27(β9)ALA→GLY]: A new, Electrophoretically silent, hemoglobin variant. Hemoglobin, 21(6), 495–497. 10.3109/03630269708999180 [DOI] [PubMed] [Google Scholar]
- Boudrahem‐Addour, N. , Zidani, N. , Carion, N. , Labie, D. , Belhani, M. , & Beldjord, C. (2009). Molecular heterogeneity of beta‐thalassemia in Algeria: How to face up to a major health problem. Hemoglobin, 33, 34–36. [DOI] [PubMed] [Google Scholar]
- Cappabianca, M. P. , Colosimo, A. , Sabatucci, A. , Dainese, E. , Di Biagio, P. , Piscitelli, R. , Sarra, O. , Zei, D. , & Amato, A. (2017). A clinical update of the Hb Siirt [β27(B9)ala→Gly; HBB: C.83C>G] hemoglobin variant. Hemoglobin, 41(1), 53–55. 10.1080/03630269.2017.1302469 [DOI] [PubMed] [Google Scholar]
- Chaima, C. A. , Bibi, A. , Ouali, F. , Fredj, S. H. , Dakhlaoui, B. , Othmani, R. , Laaouini, N. , Jouini, L. , Ouenniche, F. , Siala, H. , Tohami, I. , Becher, M. , Fattoum, S. , El Houda Toumi, N. , & Messaoud, T. (2013). Indices érythrocytaires : différenciation entre trait β‐thalassémique et anémie ferriprive et application à la drépanocytose et à la thalassémie drépanocytaire. Clinical Chemistry and Laboratory Medicine, 51(8), 1595–1603. [DOI] [PubMed] [Google Scholar]
- Deyde, V. M. , Lo, B. B. , Khalifa, I. O. , Ly, B. , Ball, A. , & Fattoum, S. (2002). Epidemiological profile of hemoglobinopathies in the Mauritanian population. Annals of Hematology, 81(6), 320–321. 10.1007/s00277-002-0471-6 [DOI] [PubMed] [Google Scholar]
- Fattoum, S. (2006). Hemoglobinopathies in Tunisia. An updated review of the epidemiologic and molecular data. La Tunisie Médicale, 84(11), 687–696. [PubMed] [Google Scholar]
- Fattoum, S. (2009). Evolution of hemoglobinopathy prevention in Africa: Results, problems and prospect. Mediterranean Journal of Hematology and Infectious Diseases, 1(1), e2009005. 10.4084/MJHID.2009.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattoum, S. , Messaoud, T. , & Bibi, A. J. H. (2004). Molecular basis of β‐thalassemia in the population of Tunisia. Hemoglobin, 28(3), 177–187. [DOI] [PubMed] [Google Scholar]
- Giardine, B. , Joly, P. , Pissard, S. , Wajcman, H. , Chui, D. H. K. , Hardison, R. C. , & Patrinos, G. P. (2021). Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Research, 49(D1), D1192–D1196. 10.1093/nar/gkaa959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueye Tall, F. , Ndour, E. H. M. , Ly, I. D. , Ndiaye, R. , Cisse, A. , Djite, M. , Guéye, P. M. , & Sall, P. L. (2017). Prévalence de l'alPha‐thalassémie au sein d'une PoPulation dréPanocytaire sénégalaise. Revue Africaine et Malgache de Recherche Scientifique/Sciences de la Santé, 5(2). [Google Scholar]
- Hardison, R. C. , Chui, D. H. , Giardine, B. , Riemer, C. , Patrinos, G. P. , Anagnou, N. , Miller, W. , & Wajcman, H. (2002). HbVar: A relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Human Mutation, 19(3), 225–233. [DOI] [PubMed] [Google Scholar]
- Kamel, K. (1979). Heterogeneity of sickle cellanaemia in Arabs: Review of cases with various amounts of fetal haemoglobin. Journal of Medical Genetics, 16, 428–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingstone, F. (1967). Abnormal Hemoglobins in human populations; a summary and interpretation (pp. 23–25). Aldine Publishing Company. [Google Scholar]
- Merchesin, P. (1992). Tribus, Ethnies et pouvoir en Mauritanie (pp. 11–57). Kharthala. [Google Scholar]
- Mesbah‐Amroun, H. , Rouabhi, F. , Ducrocq, R. , & Elion, J. (2008). Molecular basis of alpha‐thalassemia in Algeria. Hemoglobin, 32, 273–278. [DOI] [PubMed] [Google Scholar]
- Miller, S. A. , Dykes, D. D. , & Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cell. Nucleic Acids Research, 16(3), 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moumni, I. , Ikbel, B. M. M. , Leila, C. , Fethi, M. , Amine, Z. , Mohamed, B. , & Salem, A. (2011). Restriction mapping of bS locus among Tunisian sickle‐cell patients. American Journal of Human Biology, 23, 815–819. [DOI] [PubMed] [Google Scholar]
- Nagel, R. L. , & Fleming, A. F. (1992). Genetic epidemiology of the βs gene. Baillière's Clinical Haematology, 5, 33–165. [DOI] [PubMed] [Google Scholar]
- OMS . (2006). Rapport du secrétariat: Prévalence de la drépanocytose. Cinquante neuvieme assemblée mondiale de la santé. https://apps.who.int/iris/bitstream/handle/10665/21941/A59_9‐fr.pdf?sequence=1&isAllowed=y [Google Scholar]
- Orkin, S. H. , Kazazian, H. H. , Antonarakis, S. E. , Goff, S. C. , Boehm, C. D. , Sexton, J. P. , Waber, P. G. , & Giardina, P. J. (1982). Linkage of β thalasaemic mutation and β globin gene polymorphisms with DNA polymorphisms in the human β globin gene cluster. Nature, 296(5858), 627–631. [DOI] [PubMed] [Google Scholar]
- Padmos, M. A. , Sackey, K. , Roberts, G. T. , Kulozik, A. , Bail, S. , Morris, J. S. , Serjeant, B. E. , & Serjeant, G. R. (1991). Two different forms of homozygous sickle cell disease occur in Saudi Arabia. British Journal of Haematology, 79, 93–98. [DOI] [PubMed] [Google Scholar]
- Pagnier, J. , Mears, J. G. , Dunda‐Belkhodja, O. , Schaefer‐Rego, K. E. , Beldjord, C. , Nagel, R. L. , & Labie, D. (1984). Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proceedings of the National Academy of Sciences of the United States of America, 81, 1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees, D. C. , Williams, T. N. , & Gladwin, M. T. (2010). Sickle‐cell disease. Lancet, 376, 2018–2031. [DOI] [PubMed] [Google Scholar]
- Saleh‐Gohari, N. , & Mohammadi‐Anaie, M. (2012). Co‐inheritance of sickle cell trait and thalassemia mutations in south Central Iran. Iranian Journal of Public Health, 41, 81–86. [PMC free article] [PubMed] [Google Scholar]
- Sanger, F. , Nicklen, S. , & Coulson, A. R. (1977). DNA sequencing with chain‐terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 74(12), 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilirò, G. , Di Gregorio, F. , Samperi, P. , Mirabile, E. , Liang, R. , Cürük, M. A. , Ye, Z. , & Huisman, T. H. J. (1995). Genetic heterogeneity of β thalassemia in Southeast Sicily. American Journal of Hematology, 48, 5–11. [DOI] [PubMed] [Google Scholar]
- Sutton, M. , Bouhassira, E. E. , & Nagel, R. L. (1989). Polymerase chain reaction amplification applied to the determination of beta‐like globin gene cluster haplotypes. American Journal of Hematology, 32, 66–69. [DOI] [PubMed] [Google Scholar]
- Swee Lay, T. (2013). The molecular basis of β‐thalassemia. Cold Spring Harbor Perspectives in Medicine, 3, a011700. 10.1101/cshperspect.a011700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valavi, E. , Ansari, M. J. A. , & Zandian, K. (2010). How to reach rapid diagnosis in sickle cell disease. Iranian Journal of Pediatrics, 20(1), 69–74. [PMC free article] [PubMed] [Google Scholar]
- Veten, F. M. , Abdelhamid, I. O. , Meiloud, G. M. , Ghaber, S. M. , Salem, M. L. , Abbes, S. , & Houmeida, A. O. (2012). Hb S [β6 (A3) Glu→ Val, GA G> GTG] and β‐Globin Gene Cluster Haplotype Distribution in Mauritania. Hemoglobin, 36(4), 311–315. [DOI] [PubMed] [Google Scholar]
- Weatherall, D. J. (2007). Current trends in the diagnosis and management of haemoglobinopathies. Scandinavian Journal of Clinical and Laboratory Investigation, 67(1), 1–2. [DOI] [PubMed] [Google Scholar]
- Weatherall, D. J. , & Clegg, J. B. (2001). The thalassemia syndromes (4th ed.). Oxfored Scientific Publications. [Google Scholar]
- Zohreh, R. (2013). Genetic epidemiology, hematological and clinical features of hemoglobinopathies in Iran. BioMed Research Internationa., 2013, 1–10. 10.1155/2013/803487 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.