

Abstract
Background
Typical patients with KCNQ2 (OMIM# 602235) epileptic encephalopathy present early neonatal‐onset intractable seizures with a burst suppression EEG pattern and severe developmental delay or regression, and those patients always fail first‐line treatment with sodium channel blockers. Vitamin B6, either pyridoxine or pyridoxal 50‐phosphate, has been demonstrated to improve seizure control in intractable epilepsy.
Methods
Here, we collected and summarized the clinical data for four independent cases diagnosed with pyridoxine‐responsive epileptic encephalopathy, and their exome sequencing data. Moreover, we reviewed all published cases and summarized the clinical features, genetic variants, and treatment of pyridoxine‐responsive KCNQ2 epileptic encephalopathy.
Results
All four cases showed refractory seizures during the neonatal period or infancy, accompanied by global development delay. Four pathogenetic variants of KCNQ2 were uncovered and confirmed by Sanger sequencing: KCNQ2 [NM_172107.4: c.2312C > T (p.Thr771Ile), c.873G > C (p.Arg291Ser), c.652 T > A (p.Trp218Arg) and c.913‐915del (p. Phe305del)]. Sodium channel blockers and other anti‐seizure medications failed to control their seizures. The frequency of seizures gradually decreased after treatment with high‐dose pyridoxine. In case 1, case 2, and case 4, clinical seizures relapsed when pyridoxine was withdrawn, and seizures were controlled again when pyridoxine treatment was resumed.
Conclusion
Our study suggests that pyridoxine may be a promising adjunctive treatment option for patients with KCNQ2 epileptic encephalopathy.
Keywords: epileptic encephalopathy, gene mutation, KCNQ2, pyridoxine, pyridoxine‐responsive
We introduce another four cases with new variants, after combined all reported B6‐responsive cases, we found refractory epileptic encephalopathy patients may be responsive to pyridoxine with the variants located in ion transport domain of KCNQ2.

1. INTRODUCTION
The Kv7 family of voltage‐gated potassium (K+) channels consists of five members (from Kv7.1 to Kv7.5) encoded by KCNQ genes (KCNQ1‐5). Subunit Kv7.2 encoded by KCNQ2 is expressed in various brain areas, including the cerebral cortex, hippocampal formation, amygdala, basal ganglia, and hypothalamus. It has been demonstrated to be involved in the intrinsic excitability of neurons and plasticity synaptic transmission and play a role in hippocampus‐dependent learning and memory. More importantly, KCNQ2 has been found to have a close relationship with neonatal‐onset epilepsy. KCNQ2 and its homolog KCNQ3 form homotetrameric or heterotetrameric ion channels on the neuron plasma membrane and are responsible for a repolarizing M‐current that reduces neuronal excitability. Additionally, KCNQ2 can increase voltage‐gated Na + channel (Nav) availability and stabilize the resting membrane potential in nodal domains. The pathogenic variants of KCNQ2 caused a reduction in the M‐current, resulting in the recurrence of seizures and mild to severe intellectual retardation.
To date, sodium channel blocker (SCB) is considered the first‐line drug for the treatment of KCNQ2‐related epilepsy, probably through the modulation of the channel complex. Carbamazepine can stabilize the inactive state of voltage‐gated sodium channels, which colocalize with KCNQ potassium channels in neuronal membranes. Furthermore, sodium valproate (VPA), phenytoin sodium (PHT), levetiracetam (LEV), high‐dose steroids, and a ketogenic diet have also been effective in some patients (Kuersten et al., 2020). Retigabine (RTG, or ezogabine), specifically targeting KCNQ channels, was the first approved antiepileptic drug that promotes the activation of potassium channels. However, the clinical use of retigabine has been discontinued due to its side effects.
The application of pyridoxine in the adjuvant treatment of epilepsy was first reported in 1940. To date, pyridoxine has been widely used in various epilepsy and epilepsy syndromes. Here, we present the clinical and molecular genetic findings of four patients with KCNQ2 epileptic encephalopathy who were responsive to pyridoxine supplementation. Our results provide a promising adjunct treatment option for patients with intractable KCNQ2 epileptic encephalopathy.
2. MATERIALS AND METHODS
2.1. Ethical compliance
This study was approved by the institutional review board of the West China Second University Hospital (No. 138, Year: 2021). Informed consent was obtained from their families. Clinical manifestations, electroencephalogram (EEG), brain magnetic resonance imaging (MRI), malformations, investigations of other organs, and gene variations were analyzed.
2.2. Cases
In our center, the treatment protocol for patients with developmental epileptic encephalopathy is trying high‐dose pyridoxine first, regardless of the results of genetic testing. In this study, we retrospectively reviewed 64 patients with KCNQ2‐associated epileptic encephalopathy (16 pathogenic, 29 likely pathogenic, and 19 variants of uncertain significance) and found 4 patients with pyridoxine‐responsive KCNQ2 (NM_172107.4) epileptic encephalopathy. We also pooled the 8 pyridoxine‐responsive KCNQ2 (NM_172107.4) variant‐related cases reported previously in our analysis. Additional phenotype data and genetic findings for individuals are summarized in Table S1.
2.3. Whole‐exome sequencing and bioinformatics
Peripheral blood samples were collected from the probands and their families. A total of 1.0 μg genomic DNA per sample was utilized for sequencing. The exom capture libraries were generated using xGen Exome Research Panel probes (IDT, USA) following the manufacturer's recommendations. Sequencing was performed on the Illumina NovaSeq 6000 platform.
Burrows–Wheeler Aligner (BWA) was performed to map the paired‐end clean reads to the human reference genome (hg38). GATK was utilized for SNP, and short indel calling. ANNOVAR was utilized for variant annotation. Variants were picked up in exonic and splicing regions with a minor allele frequency of ≤0.005 in the SNP database (ExAC, 1000Genomes, gnomAD). Candidate variants were further validated by Sanger sequencing. The ACMG guidelines were used to evaluate the pathogenicity of candidate variants.
To understand the location information of B6‐positive responding variants of KCNQ2 (NM_172107.4), the online tool SWISS‐MODEL (https://swissmodel.expasy.org) was utilized for domain information.
3. RESULTS
3.1. Case report
Case 1 was a full‐term, seven‐month‐old girl with uncomplicated prenatal and neonatal courses. Her other family members were all healthy. This girl had seizure onset at six days after birth, with focal to generalized tonic–clonic seizures. Her head circumference and facial features were normal, with normal investigations of other organs (heart, lung, liver, kidney, etc.). She had a failure to thrive, and her development was delayed and stagnant. Upon admission at seven months old, she could not lift her neck or roll over and had a poor reaction to human faces, lights, and sound. The EEG showed multifocal sharp/spike slow waves in bilateral occipital and temporal regions, with normal brain MRI (Figure S1). Cerebrospinal fluid examination and blood and urine metabolic screening were normal. Phenobarbital, levetiracetam, oxcarbazepine, sodium valproate, and topiramate were tried before admission. However, her seizures were not effectively controlled, often lasting for 10–20 s 3–10 times per day. After admission, we administered high‐dose pyridoxine (50 mg/kg.d) via intravenous treatment for 1 week, and seizures were absent. After discharge, the dosage of pyridoxine was gradually tapered down to 30 mg per day, and seizures reoccurred. Then, the dosage of pyridoxine was increased to 60 mg per day for oral treatment, and the seizures were quickly controlled again. At her latest follow‐up assessment at 2 years old, she was seizure‐free with the combination treatment of oxcarbazepine (30 mg/kg.d) and pyridoxine (60 mg/d). Repeated EEG monitoring showed improvement in the epileptiform discharge. The development milestone was slightly improved: she could lift her neck and roll over, but she could still not sit alone or say “father” or “mother”; the Gesell Developmental Scale score was 40. Whole‐exome sequencing analysis found a de novo mutation in KCNQ2: c.873G > C (p.Arg291Ser) (Figure 1a).
FIGURE 1.
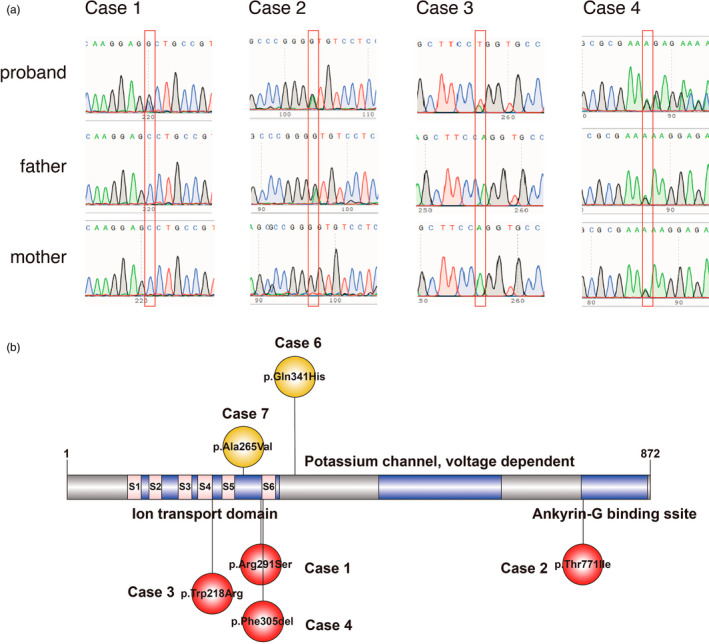
KCNQ2 (NM_172107.4) variants detected in our four cases and public database responding to B6. Sanger sequencing confirmed our cases (a). All variants positively responded to B6 (b). #: Case5 was a heterozygous 1.5‐mb deletion (not showed in the figure).
Case 2 was a ten‐month‐old boy with an uneventful birth. His father had a history of convulsions in infancy, and his mother was healthy. This boy had seizure onset at seven months after birth, with focal to generalized tonic–clonic seizures. He is prone to diarrhea and respiratory tract infection. His head circumference was normal, and no malformations or other organs were identified; however, development was delayed. Upon admission at ten months old, he could still not roll over and sit alone or say “father” or “mother”. The EEG showed multifocal epileptiform discharges in the right side, while the brain MRI showed that the extracerebral space of the left temporal pole was slightly wider than the right side, and the left side ventricle was slightly enlarged compared with the right side (Figure S1). Cerebrospinal fluid examination and blood and urine metabolic screening were normal. He was treated with oxcarbazepine before admission with poor seizure control. He still had seizures 8–9 times per day, and they often lasted for 2–3 min. After admission, he was treated with midazolam (2 ug/kg/min) continuous intravenous infusion, oral clonazepam (0.5 mg QN), levetiracetam (50 mg/kg.d), sodium valproate (30 mg/kg.d), and topiramate (8 mg/kg.d) for antiepileptic treatment, but the frequency of seizures gradually increased (10–12 times per day). Then, we administered a pyridoxine (50 mg/kg.d) intravenous injection for three days, and the frequency of seizures gradually decreased. After discharge, the dosage of pyridoxine was gradually reduced to 20 mg/d orally, and seizures occurred again. Then, the dosage of pyridoxine was increased to 40 mg/d for oral treatment, and the seizures were quickly controlled. At her latest follow‐up assessment at 2 years old, she was seizure‐free with pyridoxine (20 mg/d) plus levetiracetam (40 mg/kg.d) treatment. Repeated EEG monitoring showed improvement in the epileptiform discharge with sharp/spike and slow wave discharge several times. The development milestone was slightly improved, and the Gesell Developmental Scale score was 35. Whole‐exome sequencing analysis found a KCNQ2 gene mutation c.2312C > T (p.Thr771Ile) (Figure 1a).
Case 3 was a three‐month‐old girl, delivered by cesarean section at 35 + 2 weeks of gestation for fetal intrauterine distress. The Apgar score of 1‐5‐10 min was 9‐10‐10 points. Her other family members were all healthy. This patient had seizure onset at three days after birth, with generalized tonic–clonic seizures. She could not lift her neck and had a poor reaction to light, and sound. The EEG showed frequent multifocal discharges of sharp slow waves and spike slow waves in the right parietal, occipital and posterior regions, while the brain MRI was normal (Figure S1). Cerebrospinal fluid examination and blood and urine metabolic screening were normal. Phenobarbital, levetiracetam, and sodium valproate were tried to control seizures before admission with poor outcomes, with seizures often lasting for 30 seconds 10–15 times per day. After admission, she was treated with topiramate which was still ineffective. Then, pyridoxine (50 mg/kg.d) intravenous treatment was administered for 1 week, and the patient was seizure‐free. After discharge, she continued taking levetiracetam, sodium valproate, topiramate and pyridoxine, and no seizures occurred during the treatment. When she was 1 year old, the development milestone was slightly improved, and the Gesell Developmental Scale score was 33. Repeated EEG monitoring showed improvement in the epileptiform discharge with sharp and slow wave discharge several times. Whole‐exome sequencing analysis found a de novo mutation in KCNQ2: c.652 T > A(p.Trp218Arg) (Figure 1a).
Case 4 was a two‐month‐old girl with uncomplicated prenatal and neonatal courses. Her other family members were all healthy. This patient had seizure onset at 20 days after birth, with generalized tonic–clonic seizures. She could not lift her neck and had a poor reaction to light, and sound. The EEG showed multifocal frequent discharges of sharp slow waves and spike slow waves, while the brain MRI was normal (Figure S1). Cerebrospinal fluid examination and blood and urine metabolic screening were normal. Phenobarbital, midazolam, levetiracetam, and topamax were tried in other hospitals, but her seizures were not effectively controlled, often lasting for a few seconds to 1 min 12–15 times per day. Whole‐exome sequencing analysis found a KCNQ2 gene mutation: c.913915del (p. Phe305del) (Figure 1a). After admission, we administered high‐dose pyridoxine (50 mg/kg.d) orally for 1 week, and seizures were absent. After discharge, she continued taking Phenobarbital (15 mg/kg.d), levetiracetam (50 mg/kg.d), topiramate (3 mg/kg.d) and pyridoxine. The dosage of pyridoxine was reduced to 10 mg/kg.d by oral administration, and seizures occurred again. Then, the dosage of pyridoxine was increased back to 20 mg/kg.d orally, and the seizures were quickly controlled.
3.2. Variations and interpretation
Four variants in the KCNQ2 gene were identified: the de novo variant [c.873G > C (p.Arg291Ser)] was detected in the first family; it was a nonsynonymous variant and resulted in a Arg‐to‐Ser substitution at the 291th codon of KCNQ2. It was absent in public databases (gnomAD, ExAC), predicted as damaging by multiple software, and finally classified as likely pathogenic according to the AMCG guide. The second variant [c.2312C > T (p.Thr771Ile)] was also a nonsynonymous variant and resulted in a Thr‐to‐Ile substitution at the 771th residue of KCNQ2. Its frequency in gnomAD is extremely rare (0.00005), and it was classified as having uncertain significance according to the AMCG guide. Moreover, it was collected in the ClinVar database from another report and classified as having uncertain significance. The third de novo variant [c.652 T > A(p.Trp218Arg)] was also a nonsynonymous variant and resulted in a Trp‐to‐Arg substitution at the 218th codon of KCNQ2. It was absent in public databases (gnomAD, ExAC), predicted as damaging by multiple software, and finally classified as likely pathogenic according to the AMCG guide. The fourth de novo variant [c.913‐915del (p. Phe305del)] was a frameshift deletion; it was collected in the ClinVar database and classified as pathogenic according to the ACMG guidelines.
3.3. Literature review
SCB like phenytoin and carbamazepine have been particularly effective in KCNQ2 encephalopathy and BFNE in clinical observations (11 patients or 73% of all the patients had remission of epilepsy) (Pisano et al., 2015). We retrieved KCNQ2‐related cases in PubMed and found that 7 reports detailing 8 patients with variants in KCNQ2 were reported to fail in the treatment of SCB but have a varying clinical response to pyridoxine or pyridoxal phosphate with the combination of other antiepileptic drugs (Allen et al., 2015; Klunker, 2017; Kwong et al., 2020; Mefford et al., 2012; Reid et al., 2015; Weckhuysen et al., 2012; Wilson, 2019). Importantly, three of them were treated with a high dose of pyridoxine (30–50 mg/kg.d) (Table S1); including with our cases, all missense or frameshift variants were found to be located in or near the ion transport domain (Figure 1b). These results indicate that refractory epileptic encephalopathy patients may be responsive to pyridoxine with variants located in the ion transport domain of KCNQ2. Futhermore, these results indicate the possible mechanism between B6‐positive responses and variant locations. All refractory epileptic encephalopathy patients with variants located in the ion transport domain of KCNQ2 might be treated with a high dose of pyridoxine.
4. DISCUSSION
Phenotypes of KCNQ2 variants vary from BFNE to developmental and epileptic encephalopathy (DEE), related to missense variants in the intracellular domain of S2 as well as S3 and variants in S6 with its adjacent regions respectively (Goto et al., 2019). KCNQ2 epileptic encephalopathy presents early neonatal‐onset intractable seizures, usually tonic seizures with a suppression burst EEG pattern and severe developmental delay (Orhan et al., 2014). In this paper, four independent children carrying KCNQ2 variants, presented with early onset intractable seizures with severe global development delay and multifocal epileptiform discharges, which were consistent with the clinical manifestations of KCNQ2 epileptic encephalopathy. Ezogabine (EZO, or Retigabine) can acts directly on KCNQ2 channels, increasing their opening. Therefore, EZO use in cases where KCNQ2 variants diminish activity could be beneficial (Millichap et al., 2016). However, clinical trials with RTG could result in retinal pigmentation and discoloration of the skin after long‐term usage (Garin Shkolnik et al., 2014). SCB can efficacy against KCNQ2 epileptic encephalopathy by reducing neuronal hyperactivity arising from KCNQ2 channel deficiency (Pisano et al., 2015). However, seizures were not effectively controlled in some patients with KCNQ2 epileptic encephalopathy who received SCB treatment.
Pyridoxine plays a key supporting role in regulating many biochemical reactions in cell metabolism, including protein folding, amino acid biosynthesis, cellular storage compounds, tetrapyrrole biosynthesis, and neurotransmitter biosynthesis (Parra et al., 2018). Pyridoxal phosphate (PLP) as the biologically active form of pyridoxine, can promote the production of the inhibitory neurotransmitter gamma‐aminobutyric acid (GABA) and has a stabilizing effect on brain cells (Salvo et al., 2012). Both the detailed mechanisms of action of pyridoxal and the characteristics of responders to pyridoxine are not fully defined and may proceed through a variety of mechanisms: (1) Direct antagonist action on ion channels: PLP has recently been shown to inhibit P2X receptors in vitro (Jimenez‐Pacheco et al., 2013; Theriault et al., 2014). Certain P2X receptors, particularly P2X7R, have been shown to be activated during pathologic brain activity including neuronal necrosis due to excitotoxicity and prolonged or repeated brief seizures (Henshall et al., 2013). Therefore, PLP is an antagonist of the P2X receptor and has potent anticonvulsant effects. (2) Antioxidant effect on excessive reactive oxygen species (ROS) generated by increased neuronal firing: repeated seizure activity results in increased oxidation of cellular macromolecules such as proteins, lipids and nucleotides, which in turn can lead to neuronal death. Pyridoxine can also be attacked by oxygen‐derived free radicals thereby depleting PLP in the CSF (Footitt et al., 2011). Treatment with PN/PLP may therefore prevent secondary seizures due to oxidative stress‐induced PLP depletion as well as curtail the cycle of mitochondrial and neuronal dysfunction. (3) Replenishing the pool of PLP needed for the synthesis of some inhibitory neurotransmitters: a high dose of PLP can reduce the recurrent symptoms in children with PDE. This is because the PLP‐dependent biosynthesis of the inhibitory neurotransmitter GABA is reduced, which causes excessive consumption of PLP in the brain, resulting in excessive neuronal excitation and seizures (Clayton, 2006). To date, many reports suggest that a variety of children with epilepsy can respond, either long term or transiently, to pyridoxine treatment, including hyperprolinemia II (P5CD deficiency, MIM#239510), familial hyperphosphatasia (Mabry syndrome), tissue nonspecific alkaline phosphatase (TNSALP) deficiency (OMIM#171760), antiquitin (ATQ) deficiency, West syndrome and other childhood generalized and focal epilepsy (Ohtahara et al., 2011).
We found four variants located on the ion transport domain and ankyrin‐G binding domain, and the variants from Emma S's case (Reid et al., 2015) were also located on the ion transport domain, similar to our Case 1. However, the variants from Klotz KA's case (Klunker, 2017) were not located in any domain. The median duration reported between pyridoxal therapy and cessation of spasms was 6 (5–13) days (Matsuura et al., 2019), which indicates that we can evaluate the efficacy of pyridoxal therapy within 1–2 weeks and stop the B6 supplement for nonresponders. At present, there is no relevant guideline or expert consensus to guide the dosage of pyridoxine for the treatment of KCNQ2 epileptic encephalopathy. Reviewing the relevant literature and combining our 4 cases, we recommend using a high dose of pyridoxine (50 mg/kg.d) first; once the seizures are controlled, low dose (40–60 mg/d) maintenance treatment can be used (Kwong et al., 2020; Mefford et al., 2012). It is necessary to maintain the treatment after the seizures are completely controlled; otherwise, the seizures will relapse after discontinuation. Long‐term use of high‐dose pyridoxine has been reported to cause some adverse drug reactions, including peripheral neuropathy and hepatotoxicity (Mills et al., 2014). At the same time, it is also necessary to prevent skin necrosis caused by leakage of pyridoxine during infusion, and oral treatment is recommended.
5. CONCLUSION
In summary, KCNQ2 epileptic encephalopathy has an early onset age (neonatal and infantile), and is often associated with psychomotor developmental disorders. At present, SCB is considered the first‐line drug for the treatment of KCNQ2 related epilepsy. Meanwhile, pyridoxine therapy might be a promising adjunct treatment option for intractable KCNQ2 epileptic encephalopathy patients. More research is needed to identify pyridoxine efficacy and its regimen.
AUTHOR CONTRIBUTIONS
J.C., Q.T.: Conceptualization, Methodology, Writing‐Original draft preparation; L.F., Y.S., J.L.: Writing‐Original draft preparation, Software, Data mining; H.L., Z.Y., M.L.: Data mining, Investigation; J.G.: Supervision, Writing‐Reviewing, and Editing.
FUNDING INFORMATION
This work was supported by the National Natural Science Foundation of China (No. 82071686), grant from the Science and Technology Bureau of Sichuan Province (No. 2021YFS0093), and a grant from the research funds of West China Second University Hospital (No. KL115, KL072).
CONFLICT OF INTEREST
The authors have no competing interests to declare. Jun Chen and Qiuji Tao contributed equally to this work.
ETHICAL COMPLIANCE
Written informed consent was obtained from the patient's legal guardians to participate in this study. This study was approved by the human ethics committees of West China Second University Hospital.
CONSENT FOR PUBLICATION
Written informed consent was obtained from the patients' parents for the publication of this article.
Supporting information
Figure S1
ACKNOWLEDGMENTS
We would like to thank the patients and their families for their cooperation.
Chen, J. , Tao, Q. , Fan, L. , Shen, Y. , Liu, J. , Luo, H. , Yang, Z. , Liang, M. , & Gan, J. (2022). Pyridoxine‐responsive KCNQ2 epileptic encephalopathy: Additional cases and literature review. Molecular Genetics & Genomic Medicine, 10, e2024. 10.1002/mgg3.2024
REFERENCES
- Allen, N. M. , Mannion, M. , Conroy, J. , Lynch, S. A. , Shahwan, A. , Lynch, B. , & King, M. (2015). The variable phenotypes of KCNQ‐related epilepsy. Epilepsia, 55(9), e99–e105. [DOI] [PubMed] [Google Scholar]
- Clayton, P. T. (2006). B6‐responsive disorders: A model of vitamin dependency. Journal of Inherited Metabolic Disease, 29(2–3), 317–326. [DOI] [PubMed] [Google Scholar]
- Footitt, E. J. , Heales, S. J. , Mills, P. B. , Allen, G. , Oppenheim, M. , & Clayton, P. T. (2011). Pyridoxal 5′‐phosphate in cerebrospinal fluid; factors affecting concentration. Journal of Inherited Metabolic Disease, 34(2), 529–538. [DOI] [PubMed] [Google Scholar]
- Garin Shkolnik, T. , Feuerman, H. , Didkovsky, E. , Kaplan, I. , Bergman, R. , Pavlovsky, L. , & Hodak, E. (2014). Blue‐gray mucocutaneous discoloration: A new adverse effect of ezogabine. JAMA Dermatology, 150(9), 984–989. [DOI] [PubMed] [Google Scholar]
- Goto, A. , Ishii, A. , Shibata, M. , Ihara, Y. , Cooper, E. C. , & Hirose, S. (2019). Characteristics of KCNQ2 variants causing either benign neonatal epilepsy or developmental and epileptic encephalopathy. Epilepsia, 60, 1870–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henshall, D. C. , Miguel, D. H. , Teresa, M. , & Tobias, E. (2013). P2X receptors as targets for the treatment of status epilepticus. Frontiers in Cellular Neuroscience, 7(s 7–8), 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez‐Pacheco, A. , Mesuret, G. , Sanz‐Rodriguez, A. , Tanaka, K. , Mooney, C. , Conroy, R. , Miras‐Portugal, M. T. , Diaz‐Hernandez, M. , Henshall, D. C. , & Engel, T. (2013). Increased neocortical expression of the P2X7 receptor after status epilepticus and anticonvulsant effect of P2X7 receptor antagonist A‐438079. Epilepsia Journal of the International League Against Epilepsy, 54(9), 1551–61. [DOI] [PubMed] [Google Scholar]
- Klunker, W. (2017). Vitamin B6‐responsive epilepsy due to a novel KCNQ2 mutation. Neuropediatrics, 48(3), 199–204. [DOI] [PubMed] [Google Scholar]
- Kuersten, M. , Tacke, M. , Gerstl, L. , Hoelz, H. , Stulpnagel, C. V. , & Borggraefe, I. (2020). Antiepileptic therapy approaches in KCNQ2 related epilepsy: A systematic review. European Journal of Medical Genetics, 63(1), 103628. [DOI] [PubMed] [Google Scholar]
- Kwong, C. , Ming, H. , & Na, S. (2020). KCNQ2 encephalopathy and responsiveness to Pyridoxal‐5′‐phosphate. Journal of Pediatric Genetics. 10.1055/s-0040-1721384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura, R. , Hamano, S. I. , Kubota, J. , Daida, A. , Ikemoto, S. , Hirata, Y. , & Koichihara, R. (2019). Efficacy and safety of pyridoxal in west syndrome: A retrospective study. Brain and Development, 41(5), 413–419. [DOI] [PubMed] [Google Scholar]
- Mefford, H. C. , Cook, J. , & Gospe, S. M. (2012). Epilepsy due to 20q13.33 subtelomere deletion masquerading as pyridoxine‐dependent epilepsy. American Journal of Medical Genetics Part A, 158A(12), 3190–3195. [DOI] [PubMed] [Google Scholar]
- Millichap, J. J. , Park, K. L. , Tsuchida, T. , Ben‐Zeev, B. , Carmant, L. , Flamini, R. , Joshi, N. , Levisohn, P. M. , Marsh, E. , Nangia, S. , Narayanan, V. , Ortiz‐Gonzalez, X. R. , Patterson, M. C. , Pearl, P. L. , Porter, B. , Ramsey, K. , McGinnis, E. L. , Taglialatela, M. , Tracy, M. , … Cooper, E. C. (2016). KCNQ2 encephalopathy: Features, mutational hot spots, and ezogabine treatment of 11 patients. Neurology Genetics, 2(5), e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills, P. B. , Camuzeaux, S. S. M. , Footitt, E. J. , Mills, K. A. , Paul, G. , Laura, F. , Das, K. B. , Varadkar, S. M. , Sameer, Z. , & Robert, M. W. (2014). Epilepsy due to PNPO mutations: Genotype, environment and treatment affect presentation and outcome. Brain, 137(5), 1350–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtahara, S. , Yamatogi, Y. , & Ohtsuka, Y. (2011). Vitamin B(6) treatment of intractable seizures. Brain & Development, 33(9), 783–789. [DOI] [PubMed] [Google Scholar]
- Orhan, G. , Bock, M. , Schepers, D. , Ilina, E. I. , Reichel, S. N. , Loffler, H. , Jezutkovic, N. , Weckhuysen, S. , Mandelstam, S. , Suls, A. , Danker, T. , Guenther, E. , Scheffer, I. E. , De Jonghe, P. , Lerche, H. , & Maljevic, S. (2014). Dominant‐negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Annals of Neurology, 75(3), 382–394. [DOI] [PubMed] [Google Scholar]
- Parra, M. , Stahl, S. , & Hellmann, H. (2018). Vitamin B6 and its role in cell metabolism and physiology. Cell, 7(7), 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisano, T. , Numis, A. L. , Heavin, S. B. , Weckhuysen, S. , Angriman, M. , Suls, A. , Podesta, B. , Thibert, R. L. , Shapiro, K. A. , Guerrini, R. , Scheffer, I. E. , Marini, C. , & Cilio, M. R. (2015). Early and effective treatment of KCNQ2 encephalopathy. Epilepsia, 56(5), 685–691. [DOI] [PubMed] [Google Scholar]
- Reid, E. S. , Williams, H. , Stabej, P. , James, C. , & Clayton, P. T. (2015). Seizures due to a KCNQ2 mutation: Treatment with vitamin B6. Springer Berlin Heidelberg. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvo, M. , Safo, M. K. , & Contestabile, R. (2012). Biomedical aspects of pyridoxal 5′‐phosphate availability. Frontiers in Bioscience (Elite Edition), 4(3), 897–913. [DOI] [PubMed] [Google Scholar]
- Theriault, O. , Poulin, H. , Thomas, G. R. , Friesen, A. D. , Al‐Shaqha, W. A. , & Chahine, M. (2014). Pyridoxal‐5′‐phosphate (MC‐1), a vitamin B6 derivative, inhibits expressed P2X receptors. Canadian Journal of Physiology and Pharmacology, 92(3), 189–196. [DOI] [PubMed] [Google Scholar]
- Weckhuysen, S. , Mandelstam, S. , Suls, A. , Audenaert, D. , & Jonghe, P. D. (2012). KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Annals of Neurology, 71(1), 15–25. [DOI] [PubMed] [Google Scholar]
- Wilson, M. (2019). The biochemical investigation of genetic disorders responsive to vitamin B6 supplementation. University College London. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1