

Abstract
Background
Tooth agenesis (TA) is a congenital abnormality that may present as syndromic or nonsyndromic. Considering its complex genetic aetiology, the aim of this study was to uncover the pathogenic mutants in patients with nonsyndromic TA and analyse the characteristics of these mutants.
Methods
Exome sequencing was performed to detect pathogenic variants in 72 patients from 43 unrelated families with nonsyndromic TA. All candidate variants were validated using Sanger sequencing. Bioinformatics and conformational analyses were performed to determine the pathogenic mechanisms of the mutants.
Results
The following eight mutations (six novel and two known) in six genes were identified in eight families: WNT10A [c.742C > T (p.R248*)], LRP6 [c.1518G > A (p.W506*), c.2791 + 1G > T], AXIN2 [c.133_134insGCCAGG (p.44_45insGQ)], PAX9 [c.439C > T (p.Q147*), c.453_454insCCAGC (p.L154QfsTer60)], MSX1 [c.603_604del (p.A203GfsTer10)] and PITX2 [c.522C > G (p.Y174*)]. Bioinformatics and conformational analyses showed that the protein structures were severely altered in these mutants, and indicated that these structural abnormalities may cause functional disabilities.
Conclusions
Our study extends the mutation spectrum in patients with nonsyndromic TA and provides valuable data for genetic counselling. The pathogenic mechanisms of TA in patients/families with unknown causative variants need to be explored further.
Keywords: exome sequencing, mutant, nonsyndromic tooth agenesis
Exome sequencing and Sanger sequencing revealed variations in known pathogenic genes in 19% (8/43) of 72 patients from 43 unrelated families with nonsyndromic tooth agenesis. This study extends the mutation spectrum in patients with nonsyndromic TA and provides valuable data for genetic counselling.

1. INTRODUCTION
Tooth agenesis (TA) is a common congenital abnormality that may occur in a nonsyndromic or syndromic form based on the presence of other accompanying symptoms. TA can be categorized as hypodontia (<6 teeth missing, excluding third molars), oligodontia (≥6 teeth missing, excluding third molars) and anodontia (missing all teeth). The prevalence of nonsyndromic TA varies greatly among different ethnic populations, ranging from 2.3 to 10% (Galluccio et al., 2012). TA, especially the absence of multiple teeth, may affect masticatory efficiency and the appearance of patients, causing physiological and psychological problems.
The aetiology of TA mainly involves genetic factors, but environmental factors may also contribute to TA. Mutations in PAX9, WNT10A, MSX1, EDA, AXIN2, WNT10B and LRP6, tend to be the main causative aetiological factors involved in nonsyndromic TA; other genes including BMP4, DKK1, EDAR, EDARADD, GREM2, KREMEN1, LTBP3, SMOC2 and PITX2, have also been associated with nonsyndromic TA (Yu, Wong, et al., 2019). Considering its complex aetiology, intensive efforts are needed to gain greater insights into TA.
In this study, 72 patients from 43 unrelated Chinese families with nonsyndromic TA were enrolled, and exome sequencing and Sanger sequencing were performed to analyse the pathogenic variants. Bioinformatics and conformational analyses were performed to investigate the characteristics of the obtained mutants.
2. SUBJECTS AND METHODS
2.1. Subjects and clinical diagnosis
This study included 72 patients from 43 Chinese families with nonsyndromic TA who presented to the School and Hospital of Stomatology, Wuhan University. Nonsyndromic TA was diagnosed based on clinical examinations and panoramic radiographs. The study was approved by the institutional ethics committee of the Hospital of Stomatology Wuhan University (2018‐A56), and informed consent was obtained from all participants.
2.2. Exome sequencing and data analysis
An improved salting‐out method was used to extract genomic DNA from peripheral blood. The DNA quality was analysed using a Qubit Assay Kit (Thermo Fisher Scientific) and agarose gel electrophoresis. High‐purity DNA samples with concentrations ≥20 ng/μl and a total amount ≥1.5 μg were used for exome sequencing on the HiSeq X Ten platform (Illumina, San Diego, CA, USA) at Genesky Biotechnologies Inc., Shanghai, China. The reads were mapped to the reference assembly (hg19) using the Burrows–Wheeler Aligner (Li & Durbin, 2010). Variant calling was carried out using the Genome Analysis Toolkit (GATK) and VarScan. All variants were further annotated using the 1000 Genomes Project (1000G), Exome Aggregation Consortium (ExAC), gnomAD, dbSNP, ClinVar, SIFT, PolyPhen‐2, MutationTaster, VarSome, OMIM, Gene Ontology and the KEGG Pathway analysis (Tang et al., 2017). All candidate pathogenic variants were validated by Sanger sequencing using a BigDye Terminator kit on an ABI 3730 XL sequencer (Foster City, CA, USA) at Tsingke Biotechnology (Wuhan, China).
2.3. Conservation analysis and variant pathogenicity prediction
Multi‐species amino acid sequence alignment was performed using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). Orthologues from humans to zebrafish were got from Ensembl. The variants were further assessed using the earlier mentioned bioinformatics databases, according to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpretation of sequence variants.
2.4. 3D structure modelling and bioinformatics analysis of mutants
Domain Graph (DOG, version 2.0) was used to generate schematic diagrams of protein domain structures with distribution of mutations. To predict the conformational changes in the mutants, the crystal structure of the Wnt signalling complex (SWISS‐MODEL template library, SMTL ID: 6ahy.2.B), E1 and E2 domains of LRP6 (SMTL ID: 4dg6.1.A) and Msx‐1 homoeodomain (HD)/DNA complex structure (SMTL ID: 1ig7.1.C) were used as templates to generate 3D models for the WNT10A, LRP6 and MSX1 mutants respectively. UCSF Chimera 1.16 was used for 3D structure visualization of wild‐type and mutant proteins.
The 3D structures of AXIN2 p.44_45insGQ, PAX9 p.Q147*, PAX9 p.L154QfsTer60 and PITX2 p.Y174* were not modelled because no suitable templates were available.
3. RESULTS
3.1. Clinical diagnosis and mutation detection
The tooth phenotypes and pedigree analysis of eight Chinese families are illustrated in Figures 1 and 2. In total, eight mutations in six genes were identified using exome sequencing, confirmed using Sanger sequencing (Table 1). Two LRP6 mutations, the AXIN2 mutation, two PAX9 mutations and the MSX1 mutation were novel whereas the mutations in WNT10A and PITX2 had been previously reported to be associated with TA (B. Zeng et al., 2017), odonto‐onycho‐dermal dysplasia (OODD) (Yu, Liu, et al., 2019) and Axenfeld–Rieger syndrome (ARS) (Vieira et al., 2006) respectively. Two LRP6 mutations, two PAX9 mutations and the MSX1 mutation were not found in dbSNP, ExAC 03, gnomAD_genome or HUABIAO (database of the HUABIAO project, https://www.biosino.org/wepd/).
FIGURE 1.

Clinical analyses of patients with nonsyndromic TA. (a–h) Panoramic radiographs and tooth phenotypes of patients in families 1–8. An asterisk (★) marks the congenital missing tooth.
FIGURE 2.
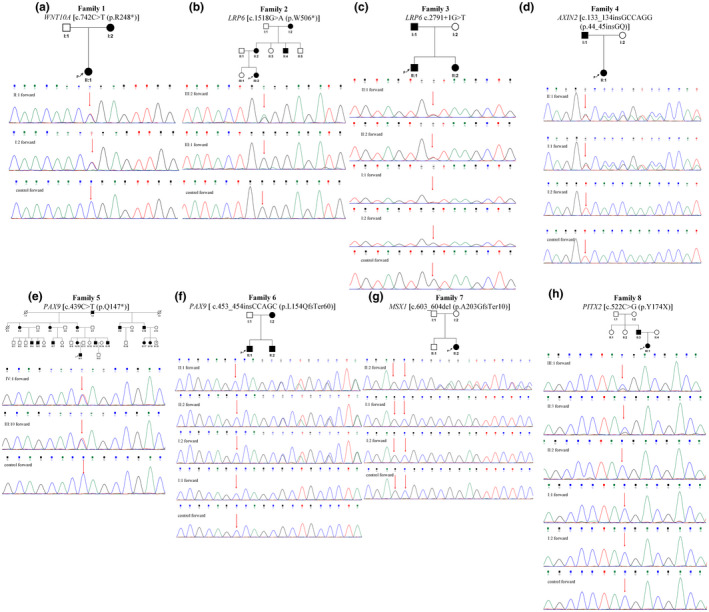
Pedigree and mutation analysis of families 1–8. (a) Sanger sequence chromatograms presenting a known heterozygous nonsense mutation WNT10A [c.742C > T (p.R248*)] in the proband (II:1) and her mother (I:2). (b) Sanger sequence chromatograms presenting a novel heterozygous nonsense mutation LRP6 [c.1518G > a (p.W506*)] in the proband (III:2) and her sister (III:1). (c) Sanger sequence chromatograms presenting a novel heterozygous splicing mutation LRP6 c.2791 + 1G > T in the proband (II:1), the sister (II:2) and the father (I:1). (d) Sanger sequence chromatograms presenting a novel heterozygous nonframeshift insertion AXIN2 [c.133_134insGCCAGG (p.44_45insGQ)] in the proband (II:1) and the father (I:1). (e) Sanger sequence chromatograms presenting a novel heterozygous nonsense mutation PAX9 [c.439C > T (p.Q147*)] in the proband (IV:1) and the mother (III:10). (f) Sanger sequence chromatograms presenting a novel heterozygous frameshift insertion PAX9 [c.453_454insCCAGC (p.L154QfsTer60)] in the proband (II:1) and the brother (II:2) and mother (I:2). (g) Sanger sequence chromatograms presenting a novel de novo heterozygous frameshift deletion MSX1 [c.603_604del (p.A203GfsTer10)] in the proband (II:2). (h) Sanger sequence chromatograms showing a known heterozygous nonsense mutation PITX2 [c.522C > G (p.Y174*)] in the proband (III:1) and her father (II:3). Filled symbols: Affected members; arrow: Proband.
TABLE 1.
Pathogenicity prediction of the eight mutations
| Families | Gene | Exon | Nucleotide change | Protein change | Mutation type | gnomAD_genome | MutationTaster Pred | VarSome_Predition | Pathogenicity classification (ACMG criteria) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | WNT10A | 3 | c.742C > T | p.R248* | nonsense | 8.425 × 10−6 | D | Pathogenic | Pathogenic (PVS1 + PS1 + PP1 + PP3 + PP4) |
| 2 | LRP6 | 7 | c.1518G > A | p.W506* | nonsense | – | A | Pathogenic | Pathogenic (PVS1 + PM2 + PP3 + PP4) |
| 3 | LRP6 | c.2791 + 1G > T | ? | splicing | – | D | Pathogenic | Pathogenic (PVS1 + PM2 + PP1 + PP3 + PP4) | |
| 4 | AXIN2 | 2 | c.133_134insGCCAGG | p.44_45insGQ | nonframeshift insertion | 2.409 × 10−5 | – | Uncertain significance | Uncertain significance (PM4 + PP1 + PP4) |
| 5 | PAX9 | 2 | c.439C > T | p.Q147* | nonsense | – | A | Pathogenic | Pathogenic (PVS1 + PM2 + PP1 + PP3 + PP4) |
| 6 | PAX9 | 2 | c.453_454insCCAGC | p.L154QfsTer60 | frameshift insertion | – | – | Pathogenic | Pathogenic (PVS1 + PM2 + PP1 + PP4) |
| 7 | MSX1 | 2 | c.603_604del | p.A203GfsTer10 | frameshift deletion | – | – | Pathogenic | Pathogenic (PVS1 + PS2 + PM2) |
| 8 | PITX2 | 3 | c.522C > G | p.Y174* | nonsense | – | D | Likely pathogenic | Pathogenic (PVS1 + PS1 + PS2 + PP1 + PP3 + PP4) |
Abbreviations: –, absent in gnomAD_genome; A, disease_causing_automatic; D, disease_causing; PM, moderate‐level evidence of pathogenic impact; PP, supporting‐level evidence of pathogenic impact; PS, strong evidence of pathogenic impact; PVS, very strong evidence of pathogenicity.
3.1.1. Family 1: WNT10A mutation
In family 1, the 17‐y‐old female proband and her mother were affected with TA. The proband (II:1) had 16 congenitally missing teeth (third molars excluded), with no other accompanying symptoms. Tooth 22 was cone shaped and some deciduous teeth were retained (Figure 1a). Her mother's clinical information is not described because she underwent dental restorations and a panoramic radiograph was unavailable. Exome sequencing and Sanger sequencing identified a previously reported heterozygous nonsense mutation (NM_025216: c.742C > T) in exon3 of WNT10A in the proband and her mother (Figure 2a).
3.1.2. Families 2 and 3: LRP6 mutation
The 26‐y‐old female proband (III:2) in family 2 showed oligodontia with 17 congenitally missing teeth (third molars excluded) and retention of deciduous teeth (Figure 1b). A pedigree investigation of the family is shown in Figure 2b. Exome sequencing and Sanger sequencing revealed a novel heterozygous nonsense mutation (NM_002336: c.1518G > A) in exon 7 of LRP6 in the proband, and verified the wild‐type genotype in her unaffected sister (Figure 2b). Unfortunately, the other family members were unavailable for testing.
In family 3, the 18‐y‐old male proband, his younger sister, and father, were affected with TA, whereas his mother was unaffected. The proband (II:1) had only four first molars, and all other teeth were congenitally missing. His 10‐y‐old sister (II:2) congenitally lacked 19 teeth, excluding the third molars, whereas his father (I:1) congenitally lacked four teeth, excluding the third molars (Figure 1c). Exome sequencing and Sanger sequencing revealed a novel heterozygous splicing mutation (NM_002336: c.2791 + 1G > T) in LRP6 in all three patients (Figure 2c).
3.1.3. Family 4: AXIN2 mutation
In family 4, a 19‐y‐old female proband and her father were affected with TA. The proband (II:1) had seven congenitally missing teeth (third molars excluded), with some retained deciduous teeth (Figure 1d). Her father's clinical information is not described because he underwent dental restorations and a panoramic radiograph was unavailable. Exome sequencing and Sanger sequencing revealed a novel heterozygous nonframeshift insertion (NM_004655: c.133_134insGCCAGG) in exon 2 of AXIN2 in both the proband and her father (Figure 2d). The mutation was found in dbSNP, ExAC 03 (2.52 × 10−5) and gnomAD_genome (2.409 × 10−5) (Table 1) but was not previously associated with any disease.
3.1.4. Families 5 and 6: PAX9 mutation
The 20‐y‐old male proband (IV:1) in family 5 had 17 congenitally missing teeth (third molars excluded), and his mother (III:10) congenitally lacked 14 teeth (third molars excluded) (Figure 1e). A pedigree investigation of family 5 is shown in Figure 2e. Exome sequencing and Sanger sequencing revealed a novel heterozygous nonsense mutation (NM_001372076: c.439C > T) in exon 2 of PAX9 in the proband and his mother (Figure 2e).
In family 6, the proband was a 9‐y‐old male and his younger brother and mother were also affected with TA. The proband (II:1) had nine congenitally missing teeth (third molars excluded) and his mother (I:2) congenitally lacked five teeth (third molars excluded) (Figure 1f). The clinical manifestations of his younger brother are not described because a panoramic radiograph was unavailable. Exome sequencing and Sanger sequencing revealed a novel heterozygous frameshift insertion (NM_001372076: c.453_454insCCAGC) in exon 2 of PAX9 in the proband, his younger brother, and mother (Figure 2f).
3.1.5. Family 7: MSX1 mutation
In family 7, the proband was a 13‐y‐old female; both her parents were unaffected. The proband (II:2) had 12 congenitally missing teeth (third molars excluded) (Figure 1g). Exome sequencing and Sanger sequencing identified a novel heterozygous frameshift deletion (NM_002448: c.603_604del) in exon 2 of MSX1 in the proband. The mutation was considered de novo based on Sanger sequencing of the proband's parents, after confirming their genetic relationship (Figure 2g).
3.1.6. Family 8: PITX2 mutation
The 5‐y‐old female proband (III:1) congenitally lacked at least 16 teeth (third molars excluded), and her father (II:3) congenitally lacked 12 teeth (third molars excluded) (Figure 1h). Both the proband and her father had no abnormalities in the eyes, abdomen or any other syndromic phenotypes. The pedigree investigation of family 8 is shown in Figure 2h. Exome sequencing and Sanger sequencing identified a heterozygous nonsense mutation (NM_000325: c.522C > G) in exon 3 of PITX2 in the proband and her father, whereas the grandparents and aunt of the proband showed a wild‐type genotype (Figure 2h). After confirming the genetic relationships among the family members, this mutation was considered de novo. Interestingly, this mutation has previously been described in patients with ARS (Vieira et al., 2006).
3.2. Conservation analysis and bioinformatics analyses
Conservation analysis showed that all eight mutations were evolutionarily conserved across many species (Supplementary Figure S1), implying their functional importance. The mutations were further assessed based on the ACMG variant pathogenicity guidelines (Table 1). WNT10A c.742C > T, LRP6 c.1518G > A and c.2791 + 1G > T, PAX9 c.439C > T and c.453_454insCCAGC, MSX1 c.603_604del and PITX2 c.522C > G were predicted to be pathogenic, whereas AXIN2 c.133_134insGCCAGG was of uncertain significance on the basis of present evidence that needs further exploration.
3.3. Mutation analysis
The protein encoded by WNT10A contains a WNT1 domain (Kirikoshi et al., 2001), as shown in Figure 3a and Supplementary Figure S2a. WNT10A c.742C > T produces a premature stop, resulting in truncated WNT10A (p.R248*). Compared with wild‐type WNT10A, p.R248* is distributed in the WNT1 domain (Supplementary Figure S2b).
FIGURE 3.

Diagram of protein structures indicating the distribution of mutations in WNT10A (a), LRP6 (b), AXIN2 (c), PAX9 (d), MSX1 (e) and PITX2 (f).
The extracellular fragment of wild‐type LRP6 contains four YWTD (Tyr, Trp, Thr and Asp)‐type β‐propeller domains comprising five YWTD repeats, each followed by an EGF‐like domain, designated as E1 to E4 from the N‐ to C‐terminus (Ren et al., 2021), as indicated in Figure 3b and Supplementary Figure S2c. LRP6 c.1518G > A produces a premature stop, resulting in truncated LRP6 (p.W506*). LRP6 p.W506* is located at the fourth YWTD repeat of the E2 domain and causes deletion of sequences from W506 through S1613 (Supplementary Figure S2d). As a splice donor site mutation in the canonical splice motif, LRP6 c.2791 + 1G > T, identified in family 3, may affect canonical GU‐AG dinucleotides, resulting in aberrant RNA splicing.
AXIN2 encodes an 843 amino acid polypeptide that includes a regulator of the G protein signalling (RGS) domain. AXIN2 [c.133_134insGCCAGG (p.44_45insGQ)] is located between the tankyrase‐binding motif and the RGS domain, resulting in the insertion of two novel amino acids, that is, glycine (Gly, G) and glutamine (Gln, Q), following Gln44 (Figure 3c).
PAX9 encodes a 341 amino acid polypeptide with a paired domain and octapeptide (Ogasawara et al., 1999). PAX9 c.439C > T produces a premature stop codon at Q147, whereas PAX9 c.453_454insCCAGC recodes the amino acids starting at L154, resulting in a premature stop codon after 60 codons. Bioinformatics analysis showed that the mutant‐PAX9 protein p.Q147* and PAX9 p.L154QfsTer60 were downstream of the paired domain, both lacking the octapeptide motif (Figure 3d).
MSX1 contains a highly conserved HD as indicated in Figure 3e and Supplementary Figure S2e. MSX1 [c.603_604del (p.A203GfsTer10)] located on the HD recodes the amino acids starting at E203, resulting in a premature stop codon after 10 codons. The HD domain is truncated in the mutant‐MSX1 protein p.A203GfsTer10 (Supplementary Figure S2f).
PITX2 has four isoforms, all of which include the OAR domain, a C‐terminal 14‐amino acid region (Cox et al., 2002). PITX2 [c.522C > G (p.Y174*)] produces a premature stop, resulting in truncated PITX2 (p. Y174*). PITX2 p. Y174* is located at the N‐terminus of the OAR domain and lacks the OAR domain (Figure 3f).
Therefore, WNT10A p.R248*, LRP6 p.W506*, LRP6 c.2791 + 1G > T, AXIN2 p.44_45insGQ, MSX1 p.A203GfsTer10, PAX9 p.Q147* and p.L154QfsTer60 and PITX2 p. Y174* cause structural and conformational changes, which may result in protein malfunction.
4. DISCUSSION
Tooth development is under strict genetic control, wherein signalling pathways, including Wnt/β‐catenin, TGF‐β/BMP and Eda/Edar/NF‐κB, play fundamental roles (Yu, Wong, et al., 2019). Notably, Wnt/β‐catenin signalling is a major pathway related to TA (Liu & Millar, 2010). Mutations in genes encoding members associated with the Wnt/β‐catenin pathway, including WNT10A, WNT10B, LRP6, KREMEN1, AXIN2, MSX1 and DKK1, have been detected in patients with TA (Yu, Wong, et al., 2019). WNT10A, LRP6, AXIN2 and MSX1 mutations were also identified in the present study.
WNT10A was first identified to be associated with OODD and was then delineated in other forms of ectodermal dysplasia, such as Schöpf–Schulz–Passarge syndrome (Bohring et al., 2009). Mutations in WNT10A were further identified as common causative factors contributing to the aetiology of TA (van den Boogaard et al., 2012). WNT10A mutants were found to inhibit Wnt signalling and dysregulate the expression levels of genes involved in protein folding and stability (Y. Zeng et al., 2021). The nonsense WNT10A mutation c.742C > T (p.R248*) detected in family 1 has been reported in two nonconsanguineous patients, one with nonsyndromic TA (B. Zeng et al., 2017) and the other with OODD (Yu, Liu, et al., 2019). This mutation produces a premature stop, resulting in a truncated WNT10A (p.R248*). This mutation is disease causing because of the C‐terminal truncation. However, the mechanism underlying the phenotypic variability in patients with WNT10A mutations remains unknown.
LRP6 mutations have been associated with early coronary disease (Mani et al., 2007), metabolic syndrome (Singh et al., 2013), neural tube defects (Shi et al., 2018), spina bifida (Lei et al., 2015) and nonsyndromic cleft lip and/or palate (Basha et al., 2018). Recently, LRP6 mutations have been reported to be related to nonsyndromic TA (Massink et al., 2015; Yu et al., 2021). Among these, a nonsense mutation (c.2292G > A; p.W764*) was reported to account for the hypohidrotic ectodermal dysplasia phenotype in the proband, while resulting in nonsyndromic TA in his father (Yu et al., 2021). LRP6 is a single‐pass transmembrane protein that acts as a co‐receptor of the Wnt/β‐catenin pathway. Wnt ligands, including WNT1, Wnt2, Wnt2b, Wnt6, Wnt8a, Wnt9a, Wnt9b and Wnt10b, bind to the E1E2 domain, whereas Wnt3 and Wnt3a prefer the E3E4 domain (Ren et al., 2021). W506 is located at the fourth YWTD repeat of the E2 domain in LRP6, and p.W506* causes deletion of sequences from W506 through S1613, disrupting the binding of Wnt ligands. LRP6 c.2791 + 1G > T is located in the canonical splice motif and probably results in aberrant transcript splicing. As LRP6 plays crucial roles in Wnt signal transduction, mutated LRP6 may disrupt the Wnt/β‐catenin pathway and cause TA. The molecular mechanisms underlying the LRP6 mutations that lead to different disease phenotypes remain to be elucidated.
AXIN2 encodes axis inhibition protein 2, which assembles a destruction complex and promotes β‐catenin degradation via the Wnt/β‐catenin pathway. Mutations in AXIN2 have been reported in both nonsyndromic TA and TA with colorectal cancer/polyposis. Several family studies indicated an association between TA and colorectal cancer/polyposis (Beard et al., 2019; Lammi et al., 2004; Marvin et al., 2011), while case‐controlled molecular studies and epidemiological data found no statistically significant evidence (Bonczek et al., 2021). In family 4, no colorectal cancer/polyposis was observed in the 19‐y‐old proband and her father. However, considering the young age of the patients, long‐term medical observation is needed. The nonframeshift insertion AXIN2 [c.133_134insGCCAGG (p.44_45GQ)] identified in family 4 causes the insertion of two novel amino acids, that is, glycine (Gly, G) and glutamine (Gln, Q), and may alter the structure and function of AXIN2, causing dysregulation of the Wnt/β‐catenin pathway and leading to TA.
Mutations in PAX9 and MSX1 were the first pathogenic factors discovered in TA (Stockton et al., 2000; Vastardis et al., 1996). Both these genes encode transcription factors that play crucial roles in tooth development. Deletion of Pax9 or Msx1 in mice causes an arrest at the bud stage (Jumlongras et al., 2001; Peters et al., 1998). Most PAX9 mutations cluster in and around the paired domain (PD) and mainly affect molars (Wong et al., 2018). The two patients in family 5 were similar as both lacked all molars, and the two patients in family 6 were similar as both lacked all maxillary molars. Interestingly, both mutations PAX9 c.439C > T (p.Q147*) and c.453_454insCCAGC (p.L154QfsTer60) identified in our study were located outside the paired domain and caused deletion of the octapeptide motif. We infer that the novel PAX9 p.Q147* and p.L154QfsTer60 impair the structure and function of PAX9, respectively, leading to tooth agenesis in families 5 and 6.
In addition to TA, MSX1 mutations have also been associated with nonsyndromic cleft lip with or without cleft palate (nsCL/P) (Suzuki et al., 2004) and the co‐occurrence of TA and nsCL/P (van den Boogaard et al., 2000), indicating that different MSX1 mutations may have distinct influences on orofacial development. However, the molecular mechanisms by which the MSX1 mutations cause different phenotypes remain unexplained. Exon 2 of MSX1 encodes a highly conserved HD, comprising an N‐terminal arm and three α‐helices (Isaac et al., 1995). HD plays a significant role in protein stability, DNA binding and interactions with the TATA‐binding protein (TBP) and Dlx families (Hu et al., 1998). MSX1 [c.603_604del (p.A203GfsTer10)] is located at helix II of the HD, incorporating a premature stop following unrelated 10 amino acid residues, which may cause MSX1 malfunction and lead to TA in family 7.
Pitx2 is specifically expressed in the epithelium during murine tooth development (Hjalt et al., 2000); tooth development is arrested at the placode or bud stage in Pitx2 −/− mice (Lin et al., 1999), indicating the fundamental role of Pitx2. In humans, PITX2 mutants are generally associated with ARS, which manly affects the development of the eyes, teeth and abdomen (Vieira et al., 2006). Notably, for the first time, a frameshift deletion, c.573_574delCA (p.L193QfsX5) in PITX2 was found to cause nonsyndromic dental anomalies in humans (Intarak et al., 2018). Interestingly, our study revealed a previously reported PITX2 nonsense mutation (Y121X), which was identified in patients with ARS (Vieira et al., 2006), that is NM_000325:c.522C > G (p.Y174*) in our study, was found to be related to nonsyndromic TA in family 8. This is the first study to uncover a PITX2 mutation in Chinese patients with nonsyndromic TA. This nonsense mutation affects the C‐terminal part of the PITX2 protein, including the OAR domain, which may cause protein malfunctions and affect tooth development. The mechanism underlying the phenotypic variability in patients with PITX2 mutations remains to be elucidated.
In conclusion, exome sequencing and Sanger sequencing revealed mutations in known pathogenic genes in 19% of nonsyndromic TA patients. Among these, seven mutations in WNT10A, LRP6, PAX9, MSX1 and PITX2 were classified as pathogenic, whereas the AXIN2 mutation was of uncertain significance on the basis of present evidence. Further analyses indicated that these mutants caused structural abnormalities and functional disabilities which resulted in TA in these families. Overall, our study extends the mutation spectrum in patients with nonsyndromic TA and provides valuable data for genetic counselling. However, further functional studies are required to elucidate the molecular pathogenesis of these mutations. The genetic cause of TA in patients with unknown causative variants, especially those from large pedigrees, implying the existence of undiscovered genes associated with TA, needs further analysis. Further research into the causes of TA will help to better understand the mechanisms of TA and enable early detection and clinical intervention to improve the oral health of patients with TA.
AUTHOR CONTRIBUTIONS
Haitang Yue: conceptualization, data curation, formal analysis, resources, investigation, methodology and writing—original draft. Jia Liang, Guangtai Song, Jing Cheng and Jiahui Li: resources, investigation and methodology. Yusheng Zhi: investigation and methodology. Zhuan Bian: conceptualization, data curation, funding acquisition, project administration, supervision, validation and writing—review and editing. Miao He: conceptualization, data curation, funding acquisition, investigation, project administration, supervision, validation and writing—review and editing.
FUNDING INFORMATION
This work was funded by the National Natural Science Foundation of China (Grant No. 81970904 to M.H., 81970923 and 82170944 to Z.B).
CONFLICT OF INTEREST
The authors declare no potential conflict of interest in the authorship and/or publication of this article.
ETHICS STATEMENT
The study was approved by the institutional ethics committee of the Hospital of Stomatology Wuhan University (2018‐A56), and informed consent was obtained from all participants.
Supporting information
Figure S1 Conservation analysis of the affected amino acids across orthologues. WNT10A R248 (a), LRP6 W506 (b), AXIN2 V45 (c), PAX9 Q147 and L154 (d), MSX1 A203 (e) and PITX2 Y174 (f) are well conserved across orthologues from chimpanzee to zebrafish. The numbers show the rightmost residue of each sequence
Figure S2 Conformational changes in the mutant WNT10A WNT1 domain (b), LRP6 E1E2 domain (d) and MSX1 HOX domain (f) compared with the respective wild‐type domains (a, c, e)
ACKNOWLEDGMENTS
We thank all the families for their enthusiastic participation in our study. This work was funded by the National Natural Science Foundation of China (Grant No. 81970904 to M.H., 81970923 and 82170944 to Z.B).
Yue, H. , Liang, J. , Song, G. , Cheng, J. , Li, J. , Zhi, Y. , Bian, Z. , & He, M. (2022). Mutation analysis in patients with nonsyndromic tooth agenesis using exome sequencing. Molecular Genetics & Genomic Medicine, 10, e2045. 10.1002/mgg3.2045
DATA AVAILABILITY STATEMENT
The data sets supporting the results of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Basha, M. , Demeer, B. , Revencu, N. , Helaers, R. , Theys, S. , Bou Saba, S. , Boute, O. , Devauchelle, B. , Francois, G. , Bayet, B. , & Vikkula, M. (2018). Whole exome sequencing identifies mutations in 10% of patients with familial non‐syndromic cleft lip and/or palate in genes mutated in well‐known syndromes. Journal of Medical Genetics, 55(7), 449–458. 10.1136/jmedgenet-2017-105110 [DOI] [PubMed] [Google Scholar]
- Beard, C. , Purvis, R. , Winship, I. M. , Macrae, F. A. , & Buchanan, D. D. (2019). Phenotypic confirmation of oligodontia, colorectal polyposis and cancer in a family carrying an exon 7 nonsense variant in the AXIN2 gene. Familial Cancer, 18(3), 311–315. 10.1007/s10689-019-00120-0 [DOI] [PubMed] [Google Scholar]
- Bohring, A. , Stamm, T. , Spaich, C. , Haase, C. , Spree, K. , Hehr, U. , Hoffmann, M. , Ledig, S. , Sel, S. , Wieacker, P. , & Röpke, A. (2009). WNT10A mutations are a frequent cause of a broad spectrum of ectodermal dysplasias with sex‐biased manifestation pattern in heterozygotes. American Journal of Human Genetics, 85(1), 97–105. 10.1016/j.ajhg.2009.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonczek, O. , Krejci, P. , Izakovicova‐Holla, L. , Cernochova, P. , Kiss, I. , & Vojtesek, B. (2021). Tooth agenesis: What do we know and is there a connection to cancer? Clinical Genetics, 99(4), 493–502. 10.1111/cge.13892 [DOI] [PubMed] [Google Scholar]
- Cox, C. J. , Espinoza, H. M. , McWilliams, B. , Chappell, K. , Morton, L. , Hjalt, T. A. , Semina, E. V. , & Amendt, B. A. (2002). Differential regulation of gene expression by PITX2 isoforms. The Journal of Biological Chemistry, 277(28), 25001–25010. 10.1074/jbc.M201737200 [DOI] [PubMed] [Google Scholar]
- Galluccio, G. , Castellano, M. , & La Monaca, C. (2012). Genetic basis of non‐syndromic anomalies of human tooth number. Archives of Oral Biology, 57(7), 918–930. 10.1016/j.archoralbio.2012.01.005 [DOI] [PubMed] [Google Scholar]
- Hjalt, T. A. , Semina, E. V. , Amendt, B. A. , & Murray, J. C. (2000). The Pitx2 protein in mouse development. Developmental Dynamics: An Official Publication of the American Association of Anatomists, 218(1), 195–200. [DOI] [PubMed] [Google Scholar]
- Hu, G. , Vastardis, H. , Bendall, A. J. , Wang, Z. , Logan, M. , Zhang, H. , Nelson, C. , Stein, S. , Greenfield, N. , Seidman, C. E. , Seidman, J. G. , & Abate‐Shen, C. (1998). Haploinsufficiency of MSX1: A mechanism for selective tooth agenesis. Molecular and Cellular Biology, 18(10), 6044–6051. 10.1128/mcb.18.10.6044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intarak, N. , Theerapanon, T. , Ittiwut, C. , Suphapeetiporn, K. , Porntaveetus, T. , & Shotelersuk, V. (2018). A novel PITX2 mutation in non‐syndromic orodental anomalies. Oral Diseases, 24(4), 611–618. 10.1111/odi.12804 [DOI] [PubMed] [Google Scholar]
- Isaac, V. E. , Sciavolino, P. , & Abate, C. (1995). Multiple amino acids determine the DNA binding specificity of the Msx‐1 homeodomain. Biochemistry, 34(21), 7127–7134. 10.1021/bi00021a026 [DOI] [PubMed] [Google Scholar]
- Jumlongras, D. , Bei, M. , Stimson, J. M. , Wang, W. F. , DePalma, S. R. , Seidman, C. E. , Felbor, U. , Maas, R. , Seidman, J. G. , & Olsen, B. R. (2001). A nonsense mutation in MSX1 causes witkop syndrome. American Journal of Human Genetics, 69(1), 67–74. 10.1086/321271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirikoshi, H. , Sekihara, H. , & Katoh, M. (2001). WNT10A and WNT6, clustered in human chromosome 2q35 region with head‐to‐tail manner, are strongly coexpressed in SW480 cells. Biochemical and Biophysical Research Communications, 283(4), 798–805. 10.1006/bbrc.2001.4855 [DOI] [PubMed] [Google Scholar]
- Lammi, L. , Arte, S. , Somer, M. , Jarvinen, H. , Lahermo, P. , Thesleff, I. , Pirinen, S. , & Nieminen, P. (2004). Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. American Journal of Human Genetics, 74(5), 1043–1050. 10.1086/386293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, Y. , Fathe, K. , McCartney, D. , Zhu, H. , Yang, W. , Ross, M. E. , Shaw, G. M. , & Finnell, R. H. (2015). Rare LRP6 variants identified in spina bifida patients. Human Mutation, 36(3), 342–349. 10.1002/humu.22750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2010). Fast and accurate long‐read alignment with burrows‐wheeler transform. Bioinformatics (Oxford, England), 26(5), 589–595. 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, C. R. , Kioussi, C. , O'Connell, S. , Briata, P. , Szeto, D. , Liu, F. , Izpisúa‐Belmonte, J. C. , & Rosenfeld, M. G. (1999). Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature, 401(6750), 279–282. 10.1038/45803 [DOI] [PubMed] [Google Scholar]
- Liu, F. , & Millar, S. E. (2010). Wnt/beta‐catenin signaling in oral tissue development and disease. Journal of Dental Research, 89(4), 318–330. 10.1177/0022034510363373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani, A. , Radhakrishnan, J. , Wang, H. , Mani, A. , Mani, M. A. , Nelson‐Williams, C. , Carew, K. S. , Mane, S. , Najmabadi, H. , Wu, D. , & Lifton, R. P. (2007). LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science (New York, N.Y.), 315(5816), 1278–1282. 10.1126/science.1136370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin, M. L. , Mazzoni, S. M. , Herron, C. M. , Edwards, S. , Gruber, S. B. , & Petty, E. M. (2011). AXIN2‐associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. American Journal of Medical Genetics. Part A, 155a(4), 898–902. 10.1002/ajmg.a.33927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massink, M. P. , Créton, M. A. , Spanevello, F. , Fennis, W. M. , Cune, M. S. , Savelberg, S. M. , Nijman, I. J. , Maurice, M. M. , van den Boogaard, M. , & van Haaften, G. (2015). Loss‐of‐function mutations in the WNT co‐receptor LRP6 cause autosomal‐dominant oligodontia. American Journal of Human Genetics, 97(4), 621–626. 10.1016/j.ajhg.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara, M. , Wada, H. , Peters, H. , & Satoh, N. (1999). Developmental expression of Pax1/9 genes in urochordate and hemichordate gills: Insight into function and evolution of the pharyngeal epithelium. Development (Cambridge, England), 126(11), 2539–2550. 10.1242/dev.126.11.2539 [DOI] [PubMed] [Google Scholar]
- Peters, H. , Neubüser, A. , Kratochwil, K. , & Balling, R. (1998). Pax9‐deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes & Development, 12(17), 2735–2747. 10.1101/gad.12.17.2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, Q. , Chen, J. , & Liu, Y. (2021). LRP5 and LRP6 in Wnt signaling: Similarity and divergence. Frontiers in Cell and Developmental Biology, 9, 670960. 10.3389/fcell.2021.670960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Z. , Yang, X. , Li, B. B. , Chen, S. , Yang, L. , Cheng, L. , Zhang, T. , Wang, H. , & Zheng, Y. (2018). Novel mutation of LRP6 identified in Chinese Han population links canonical WNT signaling to neural tube defects. Birth Defects Research, 110(1), 63–71. 10.1002/bdr2.1122 [DOI] [PubMed] [Google Scholar]
- Singh, R. , Smith, E. , Fathzadeh, M. , Liu, W. , Go, G. W. , Subrahmanyan, L. , Faramarzi, S. , McKenna, W. , & Mani, A. (2013). Rare nonconservative LRP6 mutations are associated with metabolic syndrome. Human Mutation, 34(9), 1221–1225. 10.1002/humu.22360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockton, D. W. , Das, P. , Goldenberg, M. , D'Souza, R. N. , & Patel, P. I. (2000). Mutation of PAX9 is associated with oligodontia. Nature Genetics, 24(1), 18–19. 10.1038/71634 [DOI] [PubMed] [Google Scholar]
- Suzuki, Y. , Jezewski, P. A. , Machida, J. , Watanabe, Y. , Shi, M. , Cooper, M. E. , Viet le, T. , Nguyen, T. D. , Hai, H. , Natsume, N. , Shimozato, K. , Marazita, M. L. , & Murray, J. C. (2004). In a Vietnamese population, MSX1 variants contribute to cleft lip and palate. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 6(3), 117–125. 10.1097/01.gim.0000127275.52925.05 [DOI] [PubMed] [Google Scholar]
- Tang, S. , Wang, X. , Li, W. , Yang, X. , Li, Z. , Liu, W. , Li, C. , Zhu, Z. , Wang, L. , Wang, J. , Zhang, L. , Sun, X. , Zhi, E. , Wang, H. , Li, H. , Jin, L. , Luo, Y. , Wang, J. , Yang, S. , & Zhang, F. (2017). Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. American Journal of Human Genetics, 100(6), 854–864. 10.1016/j.ajhg.2017.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boogaard, M. J. , Créton, M. , Bronkhorst, Y. , van der Hout, A. , Hennekam, E. , Lindhout, D. , Cune, M. , & Ploos van Amstel, H. K. (2012). Mutations in WNT10A are present in more than half of isolated hypodontia cases. Journal of Medical Genetics, 49(5), 327–331. 10.1136/jmedgenet-2012-100750 [DOI] [PubMed] [Google Scholar]
- van den Boogaard, M. J. , Dorland, M. , Beemer, F. A. , & van Amstel, H. K. (2000). MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nature Genetics, 24(4), 342–343. 10.1038/74155 [DOI] [PubMed] [Google Scholar]
- Vastardis, H. , Karimbux, N. , Guthua, S. W. , Seidman, J. G. , & Seidman, C. E. (1996). A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nature Genetics, 13(4), 417–421. 10.1038/ng0896-417 [DOI] [PubMed] [Google Scholar]
- Vieira, V. , David, G. , Roche, O. , de la Houssaye, G. , Boutboul, S. , Arbogast, L. , Kobetz, A. , Orssaud, C. , Camand, O. , Schorderet, D. F. , Munier, F. , Rossi, A. , Delezoide, A. L. , Marsac, C. , Ricquier, D. , Dufier, J.‐L. , Menasche, M. , & Abitbol, M. (2006). Identification of four new PITX2 gene mutations in patients with Axenfeld‐Rieger syndrome. Molecular Vision, 12, 1448–1460. [PubMed] [Google Scholar]
- Wong, S. W. , Han, D. , Zhang, H. , Liu, Y. , Zhang, X. , Miao, M. Z. , Wang, Y. , Zhao, N. , Zeng, L. , Bai, B. , Wang, Y. X. , Liu, H. , Frazier‐Bowers, S. A. , & Feng, H. (2018). Nine novel PAX9 mutations and a distinct tooth agenesis genotype‐phenotype. Journal of Dental Research, 97(2), 155–162. 10.1177/0022034517729322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, M. , Fan, Z. , Wong, S. W. , Sun, K. , Zhang, L. , Liu, H. , Feng, H. , Liu, Y. , & Han, D. (2021). Lrp6 dynamic expression in tooth development and mutations in oligodontia. Journal of Dental Research, 100(4), 415–422. 10.1177/0022034520970459 [DOI] [PubMed] [Google Scholar]
- Yu, M. , Liu, Y. , Liu, H. , Wong, S. W. , He, H. , Zhang, X. , Wang, Y. , Han, D. , & Feng, H. (2019). Distinct impacts of bi‐allelic WNT10A mutations on the permanent and primary dentitions in odonto‐onycho‐dermal dysplasia. American Journal of Medical Genetics. Part A, 179(1), 57–64. 10.1002/ajmg.a.60682 [DOI] [PubMed] [Google Scholar]
- Yu, M. , Wong, S. W. , Han, D. , & Cai, T. (2019). Genetic analysis: Wnt and other pathways in nonsyndromic tooth agenesis. Oral Diseases, 25(3), 646–651. 10.1111/odi.12931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, B. , Zhao, Q. , Li, S. , Lu, H. , Lu, J. , Ma, L. , Zhao, W. , & Yu, D. (2017). Novel EDA or EDAR mutations identified in patients with X‐linked Hypohidrotic ectodermal dysplasia or non‐syndromic tooth agenesis. Genes, 8(10), 259. 10.3390/genes8100259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Y. , Baugh, E. , Akyalcin, S. , & Letra, A. (2021). Functional effects of WNT10A rare variants associated with tooth agenesis. Journal of Dental Research, 100(3), 302–309. 10.1177/0022034520962728 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Conservation analysis of the affected amino acids across orthologues. WNT10A R248 (a), LRP6 W506 (b), AXIN2 V45 (c), PAX9 Q147 and L154 (d), MSX1 A203 (e) and PITX2 Y174 (f) are well conserved across orthologues from chimpanzee to zebrafish. The numbers show the rightmost residue of each sequence
Figure S2 Conformational changes in the mutant WNT10A WNT1 domain (b), LRP6 E1E2 domain (d) and MSX1 HOX domain (f) compared with the respective wild‐type domains (a, c, e)
Data Availability Statement
The data sets supporting the results of this study are available from the corresponding author upon reasonable request.