

Abstract
Background and purpose
Neuropathology plays a major role in deciphering disease mechanisms in multiple sclerosis (MS). This review article describes recent advances in neuropathological research related to inflammatory demyelinating diseases.
Methods
A retrospective review of neuropathological studies published during the last two decades was conducted.
Results
The importance of neuropathology is generally seen in its contribution to the diagnosis of diseases of the nervous system and, in particular, in neuro‐oncology. However, when it also includes analysis of the global three‐dimensional extension of brain damage and the temporal sequence of lesion evolution and relates this to molecular changes in the lesions, it offers the potential to decipher disease pathogenesis and to contribute to the development of effective and causative treatments. In MS research, neuropathology has been essential in discriminating the disease from other inflammatory autoimmune or demyelinating diseases, such as neuromyelitis optica spectrum disorders (NMOSD) or myelin oligodendrocyte glycoprotein antibody‐associated disease (MOGAD). It defined the hallmark of chronic progressive disease in MS patients as slowly expanding tissue damage, which occurs not only within and around lesions but also in the normal appearing white and gray matter. It showed that these changes occur in the course of a tissue‐resident immune response within the central nervous system, involving tissue‐resident effector memory cells and plasma cells. Molecular studies in neuropathologically defined micro‐dissected MS lesions identified a cascade of oxidative injury, mitochondrial damage and subsequent virtual hypoxia as a major pathway of tissue injury in MS.
Conclusions
The results of these studies were highly relevant for the identification of potential therapeutic targets in MS patients and the design of pivotal clinical trials.
Keywords: demyelination, inflammation, multiple sclerosis, neurodegeneration, neuropathology
This review article discusses the important role of neuropathology in research devoted to multiple sclerosis and other inflammatory demyelinating diseases during recent decades.

INTRODUCTION
Chronic disabling diseases of the central nervous system (CNS) in general develop on the basis of a highly complex interaction of different mechanisms, and they affect different regions of the brain and spinal cord, leading to diverse clinical manifestations. They frequently evolve in complex temporal patterns, giving rise to phases of high and low disease activity, which may be simultaneously different in different brain or lesion areas. Research on disease mechanisms must therefore take into account this complex situation, and neuropathology is currently the most suitable discipline to address this challenge. To accomplish this, however, neuropathology has to go beyond the two‐dimensional analysis of tissue sections generally performed in diagnostics, towards a multi‐dimensional analysis of structural changes in the spatial and temporal context of the lesions. This requires not only the reconstruction of the entire lesion spectrum in a single brain, but also in a virtual brain, providing information on the probability of lesion location in multiple patients. The temporal dimension can be seen by integrating information gained from known sequences of pathological events, such as the degradation of tissue components within macrophages, the dynamics of demyelination and remyelination or the time course of glial scar formation or fibrosis [1]. When this information is available, specific lesion areas can be selected for molecular studies by tissue microdissection or spatial transcriptomics, proteomics or metabolomics. These new techniques have revolutionized research strategies on brain diseases [2]. However, they can only be successful, when they are based on human brain material, which comes from a sufficiently large sample of patients with carefully defined clinical disease, and which is selected on the basis of stringent neuropathological criteria. In this review, examples are provided of how this strategy was applied to elucidate pathogenetic mechanisms of inflammation and brain damage in MS.
THE RELATIONSHIP BETWEEN MS AND OTHER INFLAMMATORY DEMYELINATING DISEASES
Although clinical features, which were later found to be typical of MS, were already described in the 18th century, MS was defined as a unique inflammatory demyelinating disease only in the second half of the 19th century [3]. Seminal contributions came from Eduard Rindfleisch, Joseph Babinski, Jean Marie Charcot, Otto Marburg and James Walker Dawson. These studies established MS as a chronic inflammatory disease, which leads to focal inflammatory demyelinating lesions in the brain and spinal cord, mainly formed around larger veins, and which are characterized by primary demyelination, sparing on axons and neurons, reactive astrocytic gliosis and spontaneous remyelination. Furthermore, it was already noted at this early time that reversible clinical deficits were associated with inflammation and demyelination, while permanent disability was related to axonal and neuronal degeneration. Detailed accounts on the historical perspective of MS pathology have been provided earlier [4, 5].
One aspect of MS pathology, which was for a long time difficult to understand, dealt with the mechanisms of active demyelination. Comparing initial changes of myelin in active lesions, as defined by stringent criteria, two fundamentally different mechanisms were noted. While a cellular attack on myelin sheaths, involving lymphocytes and macrophages, were described and illustrated by Babinski [6], the initial stages of active lesions described by Marburg [7] suggested the action of a soluble toxin. Since these observations were mainly made in patients with very severe and aggressive disease, this discrepancy could have different explanations. Mechanisms of demyelination may differ between specific patient subgroups, suggesting different disease entities hiding behind a common clinical and pathological phenotype. Alternatively, the mechanisms of demyelination may change in a given patient at different stages of lesion development and disease course. Finally, they may be attributable to differences in the cellular composition of immune cells occurring at random or being determined by the site and context of the lesions in the brain and spinal cord.
The only way to resolve this question is to analyze a large sample of such initial demyelinating lesions obtained at early disease stages from a broad spectrum of patients with inflammatory demyelinating diseases, focusing on brain biopsies taken in early disease stages or on autopsies of patients who died in the course of Marburg's type of acute MS. Such material is rare and only available in large neuropathological diagnostic reference centers. Sufficient numbers of cases and lesions can only be collected with international cooperation. When we performed such a study, the main result was the documentation of an interindividual heterogeneity of the pathology of initial MS‐like lesions, which suggested that demyelination in the brain and spinal cord followed patterns of tissue damage that were distinct among patient subgroups [8]. In all patients, active inflammatory demyelinating lesions developed on the background of an inflammatory reaction, dominated by (CD8+) T lymphocytes and B cells and demyelination was associated with activated macrophages or microglia. Despite the uniformity of the inflammatory reaction, four different patterns of demyelination were identified, suggesting differential involvement of antibodies, complement and macrophages or microglia in the initiation of demyelination. The two most prominent patterns of tissue injury were either reflecting antibody‐ and complement‐mediated tissue destruction (pattern II; [8]) or microglia activation associated with “virtual hypoxia” (pattern III; [9]).
The identification of a pattern of tissue damage, associated with immunoglobulin and complement deposition at sites of initial tissue damage, stimulated the search for potential pathogenic autoantibodies (Figure 1). Interestingly, patients with the most intense antibody and complement deposition in active lesions were those with a clinical phenotype of optico‐spinal MS or neuromyelitis optica [10]. The subsequent search for disease‐specific antibodies in such cases led to the discovery of neuromyelitis optica IgG [11], which is directed against the astrocytic water channel aquaporin 4 (AQP‐4) [12]. Transfer of such antibodies into rodents reproduced the neuropathological features of the disease in humans experimentally [13, 14]. With the advent of these antibodies, the disease was defined as a primary inflammatory astrocytopathy with secondary demyelination and axonal degeneration [15]. Furthermore, the high specificity and sensitivity of AQP‐4 antibodies as paraclinical disease markers allowed the broad spectrum of clinical phenotypes to be uncovered, which are now bunched together under the term neuromyelitis spectrum disorders (NMOSD) [16].
FIGURE 1.
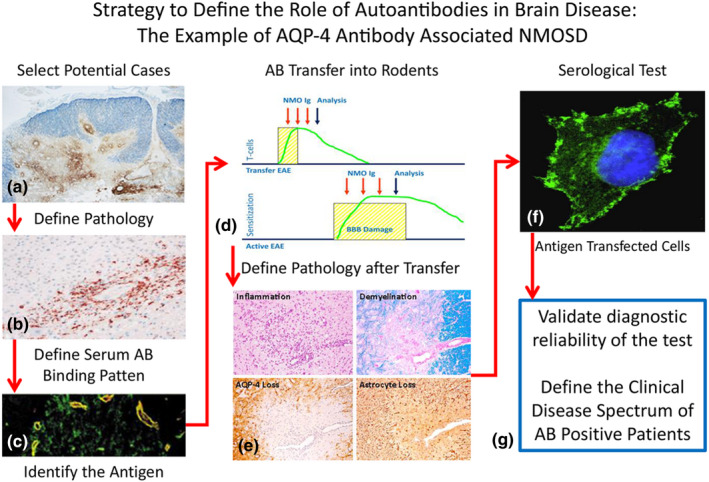
Strategy to define the role of autoantibodies in brain disease: the example of aquaporin 4 (AQP‐4) antibody‐associated neuromyelitis optica spectrum disorders (NMOSD). In the first step an inflammatory demyelinating disease with a clinical phenotype of optico‐spinal multiple sclerosis or neuromyelitis optica (NMO) has been identified to be associated with extensive antibody (a) and complement deposition in the lesions (b) [10]. This stimulated the search for autoantibodies in the patient sera, which revealed antibody binding to perivascular astrocyte processes (c) [11] and the identification of the target antigen as AQP‐4 [12]. These patient derived autoantibodies induced pathological alterations in the rodent nervous system after transfer into animals with T‐cell‐mediated brain inflammation (d), which were closely similar to those seen in NMO patients (e) [13, 14]. In parallel, a reliable paraclinical test was developed, which detects AQP‐4 antibodies in transfected cell lines (f). Using this paraclinical test for diagnosis it was possible to elucidate the clinical phenotype of NMOSD (g) [16]. EAE: experimental autoimmune encephalomyelitis [Colour figure can be viewed at wileyonlinelibrary.com]
A small subset of patients with optico‐spinal inflammatory demyelinating diseases, however, did not present with anti‐AQP‐4 autoantibodies. In some of these patients, other pathogenic autoantibodies were found, which are directed against myelin oligodendrocyte glycoprotein (MOG). As shown before in experimental models of autoimmune encephalomyelitis [17], these human anti‐MOG antibodies induced demyelination after transfer into rodents. Subsequently, the clinical spectrum of MOG antibody‐associated disease (MOGAD) was defined, including neuromyelitis optica, acute and relapsing disseminated encephalomyelitis, and some cases with atypical manifestations of MS [18]. The neuropathology of MOGAD more closely resembles acute or relapsing disseminated encephalomyelitis or transverse myelitis than classic MS [19, 20].
A major implication of these discoveries was that some diseases, which were classified until the end of the 20th century as part of the spectrum of MS, turned out to be separate disease entities with different clinical characteristics, pathology, pathogenesis and response to therapy. There is still a subset of patients with MS‐like disease with pathological evidence for antibody and complement involvement in whom no specific autoantibodies have been identified up to now. Whether these cases, too, are separate disease entities or are part of the disease spectrum of MS remains to be determined [21].
AUTOIMMUNITY IN MS AND OTHER INFLAMMATORY DEMYELINATING DISEASES
Multiple sclerosis and other inflammatory demyelinating diseases are widely believed to be autoimmune diseases [22]. This assumption is based on the detection of autoreactive T cells and antibodies in the immune repertoire of MS patients (but also of controls) and the beneficial effects of anti‐inflammatory and immunomodulatory treatments. However, in contrast to NMOSD and MOGAD, no MS‐specific autoimmune reaction has been convincingly identified until now. Currently the most compelling support for an autoimmune hypothesis is the observation that an inflammatory demyelinating disease, resembling some essential features of MS, can be induced in animals by sensitization with brain or spinal cord tissue [22]. Autoimmune‐mediated inflammatory diseases of the nervous system were originally observed in humans following rabies vaccination long before the first experimental studies documented their induction in animals. Neuropathological disease entities seen in humans after sensitization with brain tissue mainly consist of acute inflammatory demyelinating polyradiculoneuropathy (Guillain–Barre syndrome) and acute disseminated encephalomyelitis (ADEM) [23]. Only in exceptional cases was a chronic disease noted that reproduced the pathology of MS with large confluent inflammatory demyelinating lesions [24, 25]. Despite the rarity of this situation, this observation has been proposed as the most convincing evidence that MS is an autoimmune disease.
We recently had the chance to study one of these singular cases in more detail (Figure 2). Indeed, the pathology of this case closely resembled Marburg's type of acute MS [25]. However, one typical feature of chronic MS, the presence of chronic active slowly expanding lesions (see below) was missing. Since this patient died in the 1950s only some archival formaldehyde‐fixed and paraffin‐embedded material was available for further immunological studies. By combining RNA extraction from fixed and embedded formalin‐fixed material with deep sequencing and complex and highly demanding bioinformatic methods, we were able to reconstruct the antibody response within the brain lesions of this patient [26]. It turned out that the antibody response was clonally restricted and directed against a pathogenetically relevant epitope of the MOG molecule. The recombinant patient‐specific autoantibodies induced inflammatory demyelinating complement‐dependent lesions after transfer into rodents, thus proving a pathogenic autoimmune response similar to that seen in patients with MOGAD (Figure 2). Thus, our results provide further evidence of the autoimmune pathogenesis of MOGAD in humans, but not for MS.
FIGURE 2.
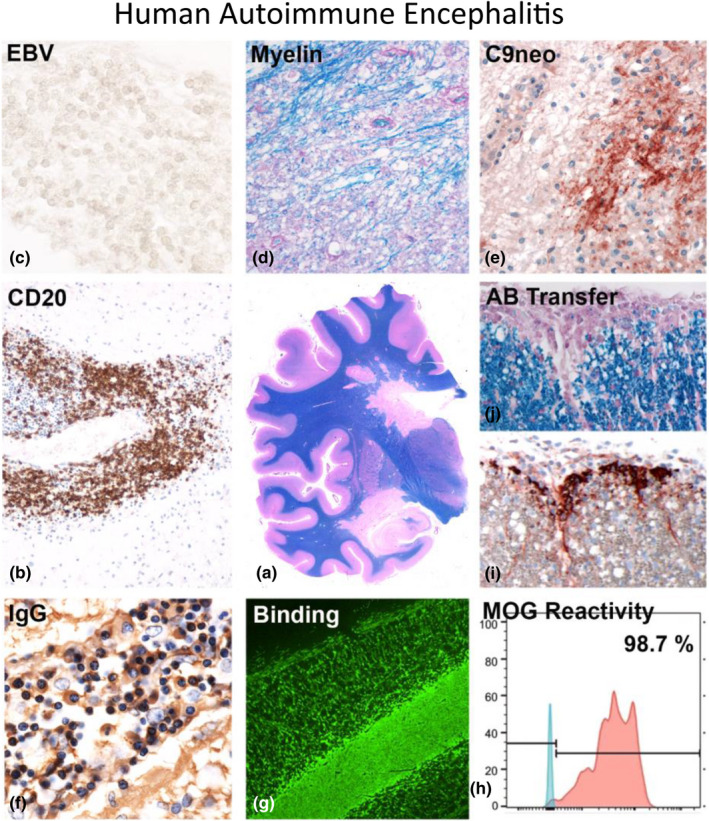
Human autoimmune enzephalomyelitis. Rabies vaccination, performed in the first half of the 20th century, was in some patients associated with a neuroparalytic complication, induced by autosensitization with brain tissue. Most of these patients developed acute inflammatory polyradiculoneuritis or acute disseminated encephalomyelitis [23], but exceptional cases showed a pathology very similar to Marburg's type of acute multiple sclerosis with periventricular plaques of demyelination [24], with perivenous extensions (so‐called Dawson fingers; central image (a) [25]). Demyelination in such cases was associated with profound inflammation with a very high content of B lymphocytes (b; CD20), which were negative for EBER‐Epstein–Barr virus reactivity (c; EBV). The lesions showed active demyelination (d; myelin) with profound deposition of activated complement (e; C9neo). The inflammatory infiltrates also contained numerous plasma cells, producing immunoglobulin G (f; IgG) [25]. Modern transcriptomic technologies allowed the identification of the pathogenic antibody response and the resurrection of a patient‐derived recombinant antibody, which bound to myelin (g; Binding), recognized a conformational epitope of myelin oligodendrocyte glycoprotein (h; MOG reactivity) and induced complement activation (i) and demyelination in vivo after passive transfer into rodents (j; Antibody Transfer) [26] [Colour figure can be viewed at wileyonlinelibrary.com]
SLOW PROPAGATION OF TISSUE DAMAGE AND “SILENT” PROGRESSION ARE HALLMARKS OF THE DISEASE PROCESS IN MS
As discussed above, recent data show that different disease entities can give rise to a clinical and neuropathological presentation that shares features of acute disseminated encephalomyelitis, neuromyelitis optica and acute or relapsing MS. What seems, however, to be specific for MS and is not shared with the other diseases is a chronic progressive disease phenotype [27, 28]. A characteristic pathological hallmark of MS is the presence of chronic active lesions [29] and, in particular, a subset that has been defined by “slowly expanding” or “smoldering” lesions [30]. These terms were based on the observation that the lesions were demarcated from the adjacent periplaque white matter by a rim of activated microglia, with only a very small percentage of the phagocytes containing initial stages of myelin degradation products. Such a pathological scenario is compatible with a very slow expansion of the lesions, but the term was criticized with the argument that pathology can only describe static but not dynamic changes. However, the accumulation of iron within activated microglia at the plaque edge recently allowed the dynamic development of these lesions to be observed with high accuracy in living patients using iron‐sensitive sequences in high‐field magnetic resonance imaging (MRI). In a prospective longitudinal study over 7 years, these iron rim lesions gradually expanded for more than 5 years and fused with other adjacent rim lesions, while plaques which lacked iron rims became smaller during the course of 2–3 years, before their size stabilized [31]. Such lesions are not only present in the progressive stage of MS, but are already prominent in relapsing MS, where they seem to be responsible for silent progression. Silent progression defines the progressive accumulation of brain damage before the threshold for functional compensation is passed, and before the patients convert to overt progressive disease. Neuropathology suggests that a similar pattern of slow progression of damage also occurs in cortical lesions and in the diffuse injury of the white and gray matter of MS patients (Figure 3). Neither slowly progressive lesions in neuropathology nor iron rim lesions in MRI have so far been identified in ADEM, NMOSD and MOGAD [19, 32].
FIGURE 3.
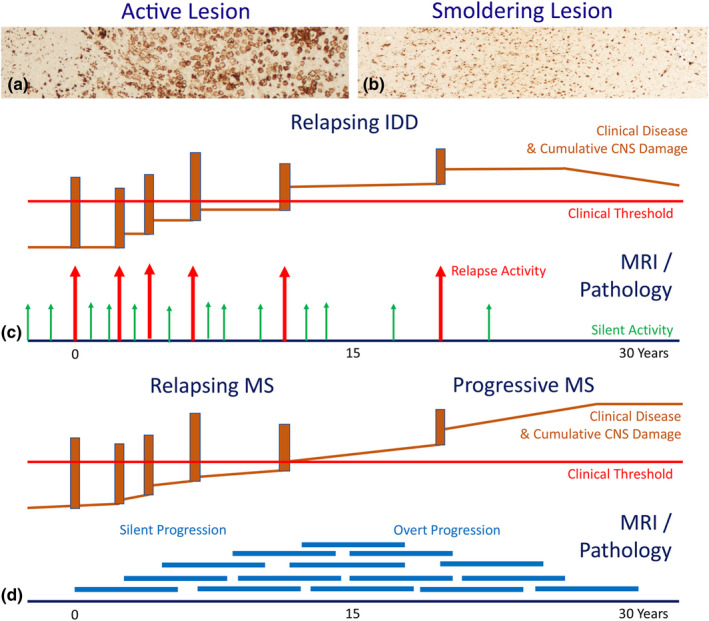
The difference between classic disease activity in acute or relapsing inflammatory demyelinating diseases and disease progression in chronic multiple sclerosis (MS). There are two different lesions types in inflammatory demyelinating diseases [30]: (a) The classic active lesions, characterized by profound inflammation, profound blood–brain barrier damage and synchronous active demyelination throughout the lesion with massive macrophage infiltration. (b) The chronic active (slowly expanding or smoldering) lesions, characterized by inflammation trapped behind a closed or repaired blood–brain barrier, profound microglia activation and moderate to minor ongoing active demyelination and axonal injury. These smoldering lesions gradually expand over years and fuse with adjacent smoldering lesions [31]. (c) Classic active lesions (arrows) are mainly seen in the relapsing stage of disease and are present in MS as well as in myelin oligodendrocyte glycoprotein antibody‐associated disease (MOGAD) and neuromyelitis optica spectrum disorders (NMOSD). When sufficiently severe and located in clinically eloquent regions, they are associated with a relapse of the disease, otherwise they contribute to silent disease activity. Neurodegeneration, associated with active lesions leads to relapse associated permanent non‐progressive neurological deficits. (d) Smoldering lesions (thick blue lines) show persistent low‐grade demyelination and neurodegeneration over several years. Such lesions accumulate in the late relapsing and early progressive stage of MS and give rise to disease progression, which may become clinically manifest as progressive MS, when the neurodegeneration exceeds the threshold of functional compensation [Colour figure can be viewed at wileyonlinelibrary.com]
PROGRESSIVE DEMYELINATION AND NEURODEGENERATION IN MS ARE ASSOCIATED WITH A TRAPPED INFLAMMATORY RESPONSE WITHIN THE CNS
Active lesions in the early stage of MS are depicted by contrast enhancement in MRI, reflecting blood–brain barrier damage in the course of active inflammation and the migration of leukocytes into the brain tissue. Such contrast‐enhancing lesions are rare in the progressive stage of the disease, despite the presence of profound perivascular inflammation and dispersion of the inflammatory infiltrates into the parenchyma [33]. These findings suggest a compartmentalized inflammatory response in the brain of patients with progressive disease [30].
Comprehensive characterization of the inflammatory response in the MS brain and its lesions, performed during the last years, now provides first insights into the nature of the compartmentalized immune response within the CNS. The inflammatory reaction within the MS brain is dominated by MHC Class I restricted CD8+ T lymphocytes and by B cells or plasma cells, irrespective of the stage of the disease or lesion evolution [34, 35, 36]. The CD8+ T cells display a phenotype of tissue resident memory T cells, but they show focal activation and proliferation. Regarding B lymphocytes CD20+ B cells are most numerous in active lesions, but they decrease in inactive disease stages and lesions [35, 37]. However, with lesion maturation CD38+ plasmablasts and CD138+ plasma cells increase in numbers. This also supports the tissue resident nature of the B‐cell response in the MS brain and provides an explanation for the long‐lasting and stable intrathecal antibody production.
The dominance of tissue‐resident effector memory cells in chronic MS lesions has several implications [38, 39]. First, these cells reside within the CNS and become activated, when they locally recognize their target. Thus, the entire inflammatory process is secluded within the CNS compartment. Secondly, most of these cells are in an inactive or resting state, while activation occurs after reappearance of their cognate antigen in a focally and temporally restricted manner. Thus, the vast majority of these cells cannot be eliminated by therapies, targeting activated immune cells. Thirdly, it is unlikely that these T cells recognize a classic autoantigen of the brain and spinal cord, which is continuously present and presented within this compartment. Such cells should be removed by activation‐induced cell death, as seen in acute models of autoimmune encephalomyelitis [40]. Potential candidate antigens for an intrathecally compartmentalized immune response by tissue‐resident effector memory cells are infections agents (such as for instance Epstein–Barr virus) or stress proteins, which are expressed in a temporally and focally restricted manner in the course of cell activation or tissue injury.
TISSUE DAMAGE IN CHRONIC MS IS ACCOMPLISHED BY A CASCADE OF MICROGLIA ACTIVATION, OXIDATIVE STRESS AND “VIRTUAL” HYPOXIA
Multiple different mechanisms of tissue injury have been proposed to occur in MS lesions, including the involvement of cytotoxic T cells, specific antibodies, microglia and macrophage activation. On a molecular basis, a variety of different toxic molecules liberated from activated immune cells or resident glia cells, such as for instance proteases, lipases, complement components, reactive oxygen and nitrogen species, and proinflammatory cytokines have been incriminated. It is likely that all of them contribute to some extent to tissue damage in different lesions and at different lesion stages, but their global importance in the disease process can only be judged on the basis of treatment responses or in an approach comparing different disease and lesion entities with a broad spectrum of novel molecular technologies.
An ideal experiment for this purpose must fulfill a number of requirements. First, a type of lesion should be selected which is unique to MS and which is carefully characterized neuropathologically for initial stages of its formation. Secondly, other lesions and diseases in humans must be identified that most closely share the phenotypes of inflammation or neurodegeneration with MS, but lack the disease‐specific features, such as primary demyelination. When this is achieved, technologies must be selected for molecular analysis that can be reliably performed on the material which is available [41].
To reach these goals, we selected active subpial cortical lesions because these are the most specific lesions in the MS brain that are not present in other human diseases of the nervous system [42]. Such subpial cortical lesions are associated with aggregates of inflammatory cells in the meninges with some features of tertiary lymph follicles [43]. It has been suggested that these inflammatory aggregates contain Epstein–Barr virus‐positive B cells and plasma cells [44], although this finding has not been confirmed by others so far. We then screened the pathological archives for brain diseases affecting the cortex and having follicle‐like meningeal inflammatory aggregates, which most closely resembled the composition of leukocytes seen in active cortical MS lesions with respect to T‐cell subsets, B cells and macrophages / microglia, but lacked the selective, plaque‐like primary demyelination. The best‐suited disease for this purpose was tuberculous meningitis. In addition, we attempted to control for plastic changes in the brain tissue, which occur as a consequence of neurodegeneration, and, thus, included samples from Alzheimer's disease in the analysis [41]. Finally, we were confronted with the problem that such tissue samples are only available in neuropathological archives. Thus, transcriptomic technology had to be adapted to be used on archival formalin‐fixed and paraffin‐embedded tissue samples [41].
When we used highly stringent cut‐off values for comparison of gene expression in these different conditions, only a limited number of genes appeared, which were differentially expressed in an MS‐specific manner, and these gene expression changes indicated microglia activation, oxidative injury and mitochondrial damage. Obviously, such an approach can only provide hypothesis‐generating results due to the very small number of cases and lesions which can be included in such a study and the technical limitations of transcriptomics performed in archival material. However, key molecules identified in this screen were also analyzed by conventional techniques in a large set of autopsy cases and the results were confirmed. In addition, similar mechanisms of tissue injury were identified in active lesions in early disease stages and chronic active slowly expanding lesions in progressive MS [45, 46, 47]. Thus, the data indicate that microglia, activated in the course of the intrathecal inflammatory response, produce reactive oxygen and nitric oxide intermediates, which provoke mitochondrial injury and result in disturbed cellular respiration and energy failure. “Virtual” hypoxia is, thus, one driving force for demyelination and neurodegeneration in MS, and this may provoke reversible functional deficit as well as irreversible tissue damage [47].
This pathogenetic concept can also explain a number of other features of cellular pathology in MS lesions, which occur in addition to demyelination and oligodendrocyte injury. A typical feature of tissue damage, induced by virtual hypoxia is that cell processes are more severely affected in comparison to the perinuclear cell body [48]. In oligodendrocytes this leads to a dying‐back oligodendrogliopathy. This is reflected by a primary loss of the most distal (peri‐axonal) cell processes visualized by a selective loss of myelin‐associated glycoprotein, which is followed by oligodendrocyte apoptosis [9]. In neurons, synapses are most severely impaired, followed by focal axonal injury or transection and dendritic fragmentation, which precedes neuronal apoptosis [46]. In astrocytes this process affects the most distal cell processes forming the perivascular glia limitans, which is a typical feature of protoplasmatic gliosis in active MS lesions [49]. In microglia, oxidative injury is associated with loss of cell processes and senescence, which is prominent in active MS lesions [50]. Finally, in oligodendrocyte progenitor cells, loss of processes results in remyelination failure, which is a typical feature of chronic MS lesions [51]. In summary, all the hallmarks of neuronal and glial pathology that are seen in active MS lesions can be explained by a mechanism of virtual hypoxia, which is induced by microglia activation, oxidative injury and mitochondrial damage.
CONCLUSIONS
Major progress has been achieved during the last decades in our understanding of pathogenetic mechanisms involved in inflammation, demyelination and neurodegeneration in MS. Obviously, this progress could only be reached by a close collaboration between clinical and basic research, the latter involving multiple different disciplines of neurobiology. However, neuropathology played a central role in this research by identifying the basic nature of the disease process in the CNS, by selecting the most suitable tissue samples for molecular research and by validating new molecular findings in relation to the type, nature and temporal sequence of events in the lesions. It is, thus, clear that without the direct analysis of diseased tissue, progress in our understanding of disease processes would have been very limited. It is currently argued that the application of new technologies of transcriptomics, proteomics and metabolomics will revolutionize our understanding of diseases, such as MS. Although this is in part true, the key bottle neck for such studies is the availability of suitable human tissue samples. For many diseases, including MS, such material will always be limited, in particular, material in a form which is perfectly suited for research. It will therefore be of critical importance to further modify and improve modern technologies so that they can be used reliably in archival fixed and embedded material as this is collected for decades in the archives of neuropathological departments.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTION
Hans Lassmann: Conceptualization (lead); Data curation (lead); Writing – original draft (lead); Writing – review and editing (lead).
Lassmann H. The contribution of neuropathology to multiple sclerosis research. Eur J Neurol. 2022;29:2869–2877. doi: 10.1111/ene.15360
Funding information
No funding was obtained for this article.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Brück W, Porada P, Poser S, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol. 1995;38(5):788‐796. doi: 10.1002/ana.410380514 [DOI] [PubMed] [Google Scholar]
- 2. Schwabenland M, Brück W, Priller J, Stadelmann C, Lassmann H, Prinz M. Analyzing microglial phenotypes across neuropathologies: a practical guide. Acta Neuropathol. 2021;142(6):923‐936. doi: 10.1007/s00401-021-02370-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murrey TJ. Multiple sclerosis: the history of disease. Demos Health. 2004:1‐576. ISBN: 9781888799804. [Google Scholar]
- 4. Kornek B, Lassmann H. Axonal pathology in multiple sclerosis. A historical note. Brain Pathol. 1999;9(4):651‐656. doi: 10.1111/j.1750-3639.1999.tb00547.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lassmann H. Multiple sclerosis pathology: evolution of pathogenetic concepts. Brain Pathol. 2005;15(3):217‐222. doi: 10.1111/j.1750-3639.2005.tb00523.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Babinski J. Recherches sur l´anatomie pathologique de la sclérose en plaques et étude comparative des diverses variétés de sclérose de la moelle. Arch Physiol (Paris). 1885;5‐6:186‐207. [Google Scholar]
- 7. Marburg O. Die sogenannte “akute Multiple Sklerose”. Jahrb Psychiatr. 1906;27:211‐312. [Google Scholar]
- 8. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol. 2000;47(6):707‐717. doi: 10.1002/1531-8249(200006)47:6<707:aid-ana3>3.0.co;2-q [DOI] [PubMed] [Google Scholar]
- 9. Aboul‐Enein F, Rauschka H, Kornek B, et al. Preferential loss of myelin‐associated glycoprotein reflects hypoxia‐like white matter damage in stroke and inflammatory brain diseases. J Neuropathol Exp Neurol. 2003;62(1):25‐33. doi: 10.1093/jnen/62.1.25 [DOI] [PubMed] [Google Scholar]
- 10. Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain. 2002;125(Pt 7):1450‐1461. doi: 10.1093/brain/awf151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364(9451):2106‐2112. doi: 10.1016/S0140-6736(04)17551-X [DOI] [PubMed] [Google Scholar]
- 12. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic‐spinal multiple sclerosis binds to the aquaporin‐4 water channel. J Exp Med. 2005;202(4):473‐477. doi: 10.1084/jem.20050304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol. 2009;66(5):630‐643. doi: 10.1002/ana.21837 [DOI] [PubMed] [Google Scholar]
- 14. Bennett JL, Lam C, Kalluri SR, et al. Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66(5):617‐629. doi: 10.1002/ana.21802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lucchinetti CF, Guo Y, Popescu BF, Fujihara K, Itoyama Y, Misu T. The pathology of an autoimmune astrocytopathy: lessons learned from neuromyelitis optica. Brain Pathol. 2014;24(1):83‐97. doi: 10.1111/bpa.12099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85(2):177‐189. doi: 10.1212/WNL.0000000000001729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. 1988;130(3):443‐454. [PMC free article] [PubMed] [Google Scholar]
- 18. Marignier R, Hacohen Y, Cobo‐Calvo A, et al. Myelin‐oligodendrocyte glycoprotein antibody‐associated disease. Lancet Neurol. 2021;20(9):762‐772. doi: 10.1016/S1474-4422(21)00218-0. Erratum. In: Lancet Neurol. 2021 Oct; 20(10):e6. Erratum in: Lancet Neurol. 2022 Jan; 21(1):e1. [DOI] [PubMed] [Google Scholar]
- 19. Höftberger R, Guo Y, Flanagan EP, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020;139(5):875‐892. doi: 10.1007/s00401-020-02132-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takai Y, Misu T, Kaneko K, et al. Myelin oligodendrocyte glycoprotein antibody‐associated disease: an immunopathological study. Brain. 2020;143(5):1431‐1446. doi: 10.1093/brain/awaa102 [DOI] [PubMed] [Google Scholar]
- 21. Tobin WO, Kalinowska‐Lyszczarz A, Weigand SD, et al. Clinical correlation of multiple sclerosis immunopathologic subtypes. Neurology. 2021;97(19):e1906‐e1913. doi: 10.1212/WNL.0000000000012782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rodríguez Murúa S, Farez MF, Quintana FJ. The immune response in multiple sclerosis. Annu Rev Pathol. 2022;17:121‐139. doi: 10.1146/annurev-pathol-052920-040318 [DOI] [PubMed] [Google Scholar]
- 23. Stuart G, Krikorian KS. The neuro‐paralytic accidents of anti‐rabies treatment. Ann Trop Med Parasitol. 1928;22:327‐377. [Google Scholar]
- 24. Uchimura I, Shiraki H. A contribution to the classification and the pathogenesis of demyelinating encephalomyelitis; with special reference to the central nervous system lesions caused by preventive inoculation against rabies. J Neuropathol Exp Neurol. 1957;16(2):139‐203; discussion, 203‐8. doi: 10.1097/00005072-195704000-00001 [DOI] [PubMed] [Google Scholar]
- 25. Höftberger R, Leisser M, Bauer J, Lassmann H. Autoimmune encephalitis in humans: how closely does it reflect multiple sclerosis? Acta Neuropath Commun. 2015;3:80. doi: 10.1186/s40478-015-0260-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Beltrán E, Paunovic M, Gebert D, et al. Archeological neuroimmunology: resurrection of a pathogenic immune response from a historical case sheds light on human autoimmune encephalomyelitis and multiple sclerosis. Acta Neuropathol. 2021;141(1):67‐83. doi: 10.1007/s00401-020-02239-2. Epub 2020 Oct 29. Erratum in: Acta Neuropathol. 2021 Apr;141(4):629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jarius S, Ruprecht K, Stellmann JP, et al. MOG‐IgG in primary and secondary chronic progressive multiple sclerosis: a multicenter study of 200 patients and review of the literature. J Neuroinflammation. 2018;15(1):88. doi: 10.1186/s12974-018-1108-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007;68(8):603‐605. doi: 10.1212/01.wnl.0000254502.87233.9a [DOI] [PubMed] [Google Scholar]
- 29. Luchetti S, Fransen NL, van Eden CG, Ramaglia V, Mason M, Huitinga I. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol. 2018;135(4):511‐528. doi: 10.1007/s00401-018-1818-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frischer JM, Weigand SD, Guo Y, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol. 2015;78(5):710‐721. doi: 10.1002/ana.24497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dal‐Bianco A, Grabner G, Kronnerwetter C, et al. Long‐term evolution of multiple sclerosis iron rim lesions in 7 T MRI. Brain. 2021;144(3):833‐847. doi: 10.1093/brain/awaa436 [DOI] [PubMed] [Google Scholar]
- 32. Misu T, Höftberger R, Fujihara K, et al. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013;125(6):815‐827. doi: 10.1007/s00401-013-1116-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hochmeister S, Grundtner R, Bauer J, et al. Dysferlin is a new marker for leaky brain blood vessels in multiple sclerosis. J Neuropathol Exp Neurol. 2006;65(9):855‐865. doi: 10.1097/01.jnen.0000235119.52311.16 [DOI] [PubMed] [Google Scholar]
- 34. van Nierop GP, van Luijn MM, Michels SS, et al. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. 2017;134(3):383‐401. doi: 10.1007/s00401-017-1744-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Machado‐Santos J, Saji E, Tröscher AR, et al. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue‐resident CD8+ T lymphocytes and B cells. Brain. 2018;141(7):2066‐2082. doi: 10.1093/brain/awy151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fransen NL, Hsiao CC, van der Poel M, et al. Tissue‐resident memory T cells invade the brain parenchyma in multiple sclerosis white matter lesions. Brain. 2020;143(6):1714‐1730. doi: 10.1093/brain/awaa117 [DOI] [PubMed] [Google Scholar]
- 37. Fransen NL, de Jong BA, Heß K, et al. Absence of B cells in brainstem and white matter lesions associates with less severe disease and absence of oligoclonal bands in MS. Neurol Neuroimmunol Neuroinflamm. 2021;8(2):e955. doi: 10.1212/NXI.0000000000000955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schenkel JM, Masopust D. Tissue‐resident memory T cells. Immunity. 2014;41(6):886‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Steinbach K, Vincenti I, Kreutzfeldt M, et al. Brain‐resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med. 2016;213(8):1571‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmied M, Breitschopf H, Gold R, et al. Apoptosis of T lymphocytes in experimental autoimmune encephalomyelitis. Evidence for programmed cell death as a mechanism to control inflammation in the brain. Am J Pathol. 1993;143(2):446‐452. [PMC free article] [PubMed] [Google Scholar]
- 41. Fischer MT, Wimmer I, Höftberger R, et al. Disease‐specific molecular events in cortical multiple sclerosis lesions. Brain. 2013;136(Pt 6):1799‐1815. doi: 10.1093/brain/awt110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Junker A, Wozniak J, Voigt D, et al. Extensive subpial cortical demyelination is specific to multiple sclerosis. Brain Pathol. 2020;30(3):641‐652. doi: 10.1111/bpa.12813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B‐cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14(2):164‐174. doi: 10.1111/j.1750-3639.2004.tb00049.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Serafini B, Rosicarelli B, Franciotta D, et al. Dysregulated Epstein‐Barr virus infection in the multiple sclerosis brain. J Exp Med. 2007;204(12):2899‐2912. doi: 10.1084/jem.20071030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mahad D, Ziabreva I, Lassmann H, Turnbull D. Mitochondrial defects in acute multiple sclerosis lesions. Brain. 2008;131(Pt 7):1722‐1735. doi: 10.1093/brain/awn105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haider L, Fischer MT, Frischer JM, et al. Oxidative damage in multiple sclerosis lesions. Brain. 2011;134(Pt 7):1914‐1924. doi: 10.1093/brain/awr128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183‐193. doi: 10.1016/S1474-4422(14)70256-X [DOI] [PubMed] [Google Scholar]
- 48. Lassmann H, van Horssen J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim Biophys Acta. 2016;1862(3):506‐510. doi: 10.1016/j.bbadis.2015.09.018 [DOI] [PubMed] [Google Scholar]
- 49. Sharma R, Fischer MT, Bauer J, et al. Inflammation induced by innate immunity in the central nervous system leads to primary astrocyte dysfunction followed by demyelination. Acta Neuropathol. 2010;120(2):223‐236. doi: 10.1007/s00401-010-0704-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hametner S, Wimmer I, Haider L, Pfeifenbring S, Brück W, Lassmann H. Iron and neurodegeneration in the multiple sclerosis brain. Ann Neurol. 2013;74(6):848‐861. doi: 10.1002/ana.23974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ziabreva I, Campbell G, Rist J, et al. Injury and differentiation following inhibition of mitochondrial respiratory chain complex IV in rat oligodendrocytes. Glia. 2010;58(15):1827‐1837. doi: 10.1002/glia.21052 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.