

Abstract
This work describes the design and synthesis of a π‐conjugated telluro[3,2‐β][1]‐tellurophene‐based synthon that, embodying pyridyl and haloaryl chalcogen‐bonding acceptors, self‐assembles into nanoribbons through chalcogen bonds. The ribbons π‐stack in a multi‐layered architecture both in single crystals and thin films. Theoretical studies of the electronic states of chalcogen‐bonded material showed the presence of a local charge density between Te and N atoms. OTFT‐based charge transport measurements showed hole‐transport properties for this material. Its integration as a p‐type semiconductor in multi‐layered CuI‐based light‐emitting electrochemical cells (LECs) led to a 10‐fold increase in stability (38 h vs. 3 h) compared to single‐layered devices. Finally, using the reference tellurotellurophene congener bearing a C−H group instead of the pyridyl N atom, a herringbone solid‐state assembly is formed without charge transport features, resulting in LECs with poor stabilities (<1 h).
Keywords: Chalcogen Bond, Crystal Engineering, Organic Semiconductor, Supramolecular Architectures
Chalcogen‐bonded p‐type semiconducting ribbons have been engineered through the self‐assembly of tailored π‐conjugated modules. Theoretical studies of the electronic states showed the presence of a local charge density between Te and N atoms. Implementation of the ribbons as hole‐transport layers in light‐emitting electrochemical cells gave rise to devices with performances similar to those obtained with state‐of‐art semiconductors.
Introduction
Since the first observation in the 1960s [1] and conceptualization in the 2000s, [2] chalcogen‐bonding interactions (ChBIs, X−Ch⋅⋅⋅Y) [3] have raised great interest among chemists to gain control on the conformation of π‐conjugated polymers, [4] conceive new catalysts, [5] receptors for anion recognition, [6] and molecular tectons for crystal engineering. [7] In particular, in molecular crystals, ChBIs have been used to program discrete and polymeric architectures in the solid state. [8] Prior art in the field includes the use of benzo‐2,1,3‐chalcogenodiazoles to engineer infinite supramolecular ribbons. [9] In these architectures, the Ch and N atoms engage in multiple ChBIs, forming 2Ch‐2N squares recognition motifs. When mounted as walls on a resorcin[4]arene scaffold, the benzo‐2,1,3‐chalcogenodiazole units engage in 16 intramolecular ChBIs, templating the formation of a self‐assembled capsule. [10] Other solid‐state architectures include kinked supramolecular polymers, [11] in which 2‐substituted benzo‐1,3‐chalcogenazoles are wired through single ChBIs. Heteromolecular solid‐state polymers were also obtained by exploiting simultaneous chalcogen‐ and halogen‐bonding interactions [12] using the chalcogenazolo[5,4‐β]pyridine (CGP) unit developed by our group, [13] known to persistently form non‐covalent 2Ch‐2N hexagon motifs. [14] However, the exploitation of chalcogen‐bonded soft materials in optoelectronics has so far remained underdeveloped, [15] with the exclusion of few examples in non‐linear optics [16] and in organic thin‐film transistors (OTFTs). [17] Capitalizing on the strong orbital contribution (n2 (Y)→σ*(Ch‐X)), [18] one could envisage to exploit ChBIs to conceive supramolecular semiconductors in which the molecular units are electronically coupled. [19] Building on this idea, this work describes the first example of the design of a π‐conjugated molecular synthon that, by embodying two doubly chalcogen‐bonded units, assembles into supramolecular nanoribbons at the solid‐state (Scheme 1).
Scheme 1.

Schematic representation of supramolecular chalcogen‐based nanoribbons held by ChBIs in the solid‐state.
In thin films, the ribbon would π‐stack in a multi‐layered architecture, ultimately giving rise to a p‐type semiconducting material as depicted by charge‐transport measurement with TFTs. This was used to decouple hole injection/transport and exciton formation in multi‐layered red‐emitting CuI‐complex‐based light‐emitting electrochemical cells (LECs), realizing 10‐fold increased stabilities compared to reference devices with traditional hole‐transport layers. [20]
Results and Discussion
Design and Synthesis
Chalcogen‐bonded ribbons have been previously obtained with benzo‐1,2,5‐telluradiazole synthons exploiting the two Te‐centered σ‐holes to engage in bifurcated ChBIs. [21] In our approach, we considered the use of π‐conjugated scaffolds containing two chalcogen‐bonding recognition units at their extremities (Scheme 1). [22] Capitalizing on the persistent recognition behavior of the Te‐CGP unit, we conjectured that any polycyclic aromatic framework encoding chalcogen‐bonding donor Te and acceptor N atoms in a second nearest neighboring (SNN) topology, [13] i.e. a peripheral telluropheno[5,4‐β]pyridine‐like substructure, can undergo in‐plane self‐assembly. Anticipating scarce solubility and processability of any derivative exposing multiple SNN‐type recognition units, [23] we envisaged a π‐conjugated scaffold that would include heteromolecular chalcogen‐bonding recognition units. One moiety would develop strong 2Ch‐2N hexagons, while the second one would establish weak interactions, to self‐assemble into supramolecular ribbons at the solid state (Scheme 1). Building on the telluro[3,2‐β][1]‐tellurophene bicycle as the chalcogen‐bond donor framework, [24] we designed a self‐assembling polycyclic aromatic module that includes both a Te/N SNN‐type and a Te/halogen recognition pairs (Scheme 1b).
At the synthetic planning level, this design guided us to contemplate an organometallic‐mediated Te‐insertion reaction, followed by a one‐pot intramolecular cyclization as the key synthetic step to form the telluro[3,2‐β][1]‐tellurophene core from the relevant disubstituted alkyne derivative bearing both pyridyl and haloaryl moieties. A reference module bearing a C−H functionality at the place of the N atom, which is unable to undergo ribbon‐like assembly, has been also designed.
Taking advantage of the methodologies developed for preparing benzotellurophenes, [25] our synthetic efforts started with the investigations of the intramolecular cyclization reaction to obtain pyridinotellurophene derivatives from the corresponding 2‐bromo‐3‐alkynylpyridine (Scheme 2a). Selective deprotonation of commercially available 2‐bromopyridine 1 using LDA in THF at −95 °C, followed by the slow addition of I2, gave access to 2‐bromo‐3‐iodopyridine 2Pyr in high yield. [26] Sonogashira‐type cross‐coupling reaction of 2Pyr with the relevant ethynyl derivative in the presence of [Pd(PPh3)2Cl2] and CuI, gave molecules 3TMS and 3Ph in excellent and good yields, respectively. [27] Treatment of the bromo‐containing alkynes with a trialkyl magnesiate (prepared in situ by mixing i‐PrMgCl with n‐BuLi at 0 °C) [28] followed by the addition of freshly‐ground elemental Te powder led to pyridinotellurophenes 4TMS and 4Ph in 43 % and 41 % yields, respectively, through Te‐insertion and consecutive intramolecular cyclization (Scheme 2a).
Scheme 2.

a) Synthetic pathway to 4 and 7. Reagents and conditions: a) 1. LDA, THF, −95 °C, 1 h; 2. I2, THF, −95 °C to r.t., 4 h; b) R‐≡, 5 mol% [Pd(PPh3)2Cl2], 10 mol% CuI, NEt3, 1,4‐dioxane, r.t., 1.5 h; c) 1. i‐PrBu2MgLi⋅LiCl, THF, 0 °C, 1 h; 2. Te0, r.t., 3 h; 3. EtOH, r.t., overnight; d) 1. TMSA, 5 mol% [Pd(PPh3)2Cl2], 10 mol% CuI, NEt3, 1,4‐dioxane, r.t., 1.5 h; 2. K2CO3, MeOH, CH2Cl2, r.t., 1 h; e) the same conditions as those used for (d); f) for 6Pyr : 5 mol% [Pd(PPh3)2Cl2], 10 mol% CuI, C6F5I, i‐PrNH2, toluene, 80 °C, 3 h; for 6Benzo : 5 mol% [Pd(PPh3)2Cl2], 10 mol% CuI, C6F5I, NEt3, 40 °C, overnight; g) the same conditions as those used for (c); ORTEP representation of a single molecule of b) 4Ph , d) 7Pyr and f) 7Benzo drawn with 50 % displacement ellipsoid; [32] c) crystal structure of 4Ph showing the dimer formation in the solid state; 189.5 MHz 125Te NMR spectrum in C6D6 of (e) 7Pyr and g) 7Benzo .
X‐ray analysis of a single crystal of 4Ph obtained by slow evaporation of a CHCl3 solution confirmed the presence of the bicyclic tellurophenopyridine moiety. Each bicycle arranges in non‐covalent 2Ch‐2N hexagonal dimers through the formation of double ChBIs (d Te⋅⋅⋅N=3.044 Å<sum of vdW radii=3.67 Å, C7−Te1⋅⋅⋅N1=169.8°; Scheme 2c), validating our hypothesis, for which the tellurophenopyridine scaffold undergoes non‐covalent dimerization similarly to the CGP‐type motifs.[ 13 , 14 ] To prepare the target tellurotellurophene module, we conjectured that highly electron‐deficient perfluorinated aryl substituents could be used as favorable functionalities to induce an intramolecular SNAr cyclization reaction.[ 29 , 30 ] Thus, the synthesis of a disubstituted alkyne derivative bearing both a pyridyl and a pentafluoro aryl moiety was planned. TMS‐deprotection of 3TMS with K2CO3 in a 2 : 1 mixture of MeOH and CH2Cl2 followed by Sonogashira cross‐coupling reaction with iodopentafluorobenzene in the presence of [PdCl2(PPh3)2] and CuI led to the disubstituted alkynyl derivative 6Pyr in 40 % yield. Derivative 6Pyr was converted into bis tellurophenes 7Pyr by Te‐insertion followed by a double intramolecular cyclization reaction upon addition of i‐PrBu2MgLi⋅LiCl in THF and of elemental Te (Scheme 2a). Notably, an unexpected alkylation substitution reaction occurred at the 4‐position of the pentafluoroaryl moiety. [31] A similar synthetic pathway was also exploited to prepare congener 7Benzo . Bis‐substituted precursor 5Benzo was prepared in 97 % yield starting from 2‐bromoiodobenzene 2Benzo by Sonogashira cross‐coupling with TMS‐acetylene (TMSA) followed by TMS‐deprotection. Subsequent cross‐coupling reaction of iodopentafluorobenzene with 5Benzo gave 6Benzo in 65 % yield. Te‐insertion in the presence of i‐PrBu2MgLi⋅LiCl gave the tellurotellurophene derivative 7Benzo in 39 % yield. All compounds were fully characterized by 1H, 19F, 13C and 125Te (for 7Pyr and 7Benzo —Schemes 2e and 2g) NMR spectroscopy, IR, HR‐mass spectrometry, TGA (Figures S32, S33) and X‐ray diffraction analysis (Schemes 2b–f—see Supporting Information). [32] In particular, 125Te NMR spectra of 7Pyr and 7Benzo were measured in C6D6 to probe the presence of two chemically‐non‐equivalent chalcogen nuclei in the telluro[3,2‐b][1]‐tellurophene core, namely Te1 and Te2 (Schemes 2e and g). While a significant “through‐space” internuclear spin‐spin 19F‐125Te coupling constant (4TS J 19F‐125Te) value of 290 Hz was observed for the Te1 resonances, a weak through‐bond 3 J 19F‐125Te value of about 26 Hz was measured for the Te2 nuclei in both molecules, confirming the in‐plane molecular asymmetry. [33]
Photophysical and Redox Properties
The absorption spectra of derivatives 7Pyr and 7Benzo were recorded in CH2Cl2 at r.t. (Figure 1). Both compounds exhibit well‐structured absorption bands with two peaks centered at ≈330 and 380 nm associated to ϵ max≈10 000 M−1cm−1. Although both molecules are not emissive in CH2Cl2 at r.t. in both aerated and deaerated conditions, they revealed significant emissive signals in CHCl3/EtOH (1 : 1 v/v) glassy matrix at 77 K (Table S2, Supporting Information). [11a] The emission profiles of 7Pyr and 7Benzo depict identical well‐structured envelops with maxima at ≈552, 570 and 615 nm.
Figure 1.

Normalised absorbance (solid line), excitation (dotted line) and phosphorescence (dashed line) spectra of solutions of 7Pyr (a) and 7Benzo (b). Absorption was measured in CH2Cl2 at r.t., phosphorescence and excitation in CHCl3/EtOH (1 : 1 v/v) glassy matrix at 77 K.
Notably, they exhibit only phosphorescence emission with excited state lifetimes of ≈700 μs. This suggests a quantitative intersystem crossing (ISC) process induced by the presence of the Te atom (see below).[ 11a , 34 ]
Cyclic voltammetry (CV) experiments in CH2Cl2 and 1,1,2,2‐tetrachloroethane (TCE) were performed to unravel the redox properties of 7Pyr and 7Benzo (all potential values are given vs. Fc/Fc+). Both compounds exhibited quasi‐reversible redox behaviors (Table S3 and Figure S34, Supporting Information). The CV behavior was similar in different solvents, with the redox values measured in TCE being anodically shifted with respect to those in CH2Cl2. Moreover, a second irreversible oxidation peak (ca. 0.5 V) appeared for both molecules after multiple scans, suggesting the occurrence of side reactions and/or decomposition (Figures S32e and S32f in the Supporting Information). Finally, thin films of 7Pyr and 7Benzo showed quasi‐reversible oxidation events at +0.79 V and +0.87 V, respectively (scan rate of 100 mV s−1; Figure S35, Supporting Information). Upon repetitive CV scans, the current intensity increased due to a partial charging of the films. This led to a slight shift (≈10/20 mV) in the oxidation potential. No other redox event was detected after 20 scans, suggesting that these compounds are electrochemically stable in thin films.
Solid‐State Organization
Next, we studied the structural properties and the organization of solids made of 7Pyr and 7Benzo . The X‐ray analysis of single crystals of 7Pyr is marked by the presence of chalcogen‐bonded molecules, each developing into supramolecular ribbons (Figure 2a) held together through two types of ChBIs. While the proximal N and Te1 atoms (d Te1⋅⋅⋅N=2.961 Å<sum of vdW radii=3.67 Å, C7‐Te1⋅⋅⋅N1=172.6°) undergo double ChBIs through the typical doubly‐bonded 2Ch‐2N hexagon recognition motif of the CGP unit,[ 13 , 14 ] the Te2 atom establishes a weak ChBI with a F atom of a neighboring molecule (d Te2⋅⋅⋅F2=3.295 Å<sum of vdW radii=3.53 Å, C9−Te2⋅⋅⋅F2=172.6°). Each ribbon is organized into π–π stacks (d π‐π=3.390 Å; Figure S36b in the Supporting Information), ultimately leading to a two‐dimensional brickwork‐like organization (Figure 2b). Crystals of 7Benzo showed a different molecular packing organization, with only the Te2 atom engaging in bifurcated intermolecular ChBIs with two neighboring F atoms (d Te(α)⋅⋅⋅F3=3.259 Å, C10‐Te2⋅⋅⋅F3=164.7°; d Te(β)⋅⋅⋅F2=3.489 Å, C7‐Te2⋅⋅⋅F2=168.9°, both ChBIs<sum of vdW radii=3.53 Å; Figure 2c). These bifurcated interactions force the molecules to adopt a herringbone organization stabilized through intermolecular C−H⋅⋅⋅π contacts (d C−C=2.860 Å, Figure 2d).
Figure 2.

Left: X‐ray stick representation of a) the supramolecular nanoribbon developed by 7Pyr , and c) the herringbone organization of 7Benzo (n‐butyl chains omitted for clarity). ChBIs are highlighted with dashed lines. Right: space‐fill representation of the crystal packing of b) 7Pyr and d) 7Benzo . Crystallization solvents: CHCl3 and toluene for 7Pyr , petroleum ether for 7Benzo . Space groups: Pbca for 7Pyr , P21/c for 7Benzo . [32]
To further characterize the materials, we have investigated the local chemical and electronic environment nearby the chalcogen atoms by mean of solid‐state 125Te NMR spectroscopy (Figure 3). [35] The simulation (red line) provides isotropic chemical shift (δ iso, marked with * in Figure 3), the principal component (δ 11, δ 22 and δ 33), span (Ω) and skew (κ) values for both the chalcogen nuclei (Table S1 and comment in the Supporting Information). Building on the seminal 125Te ssNMR studies by Bryce and co‐workers, [35b] the different values shown by Te1 and Te2 nuclei for isotropic chemical shift δ iso (898.5 vs. 845.5 ppm), the largest principal component δ 11 (1854.7 vs. 2118.3 ppm), the shielded δ 33 (251.6 vs. 181.2 ppm) and the span values (1603.2 vs. 1937.1 ppm) easily suggest that the Te1 atom engages in stronger and more directional ChBIs than the Te2 nucleus due to the formation of the double 2Ch‐2N motif. To validate our interpretation, we performed 125Te ssNMR measurements with solids of reference 7Benzo . Here, the different trend of the Ω values for Te1 and Te2 (i.e., 2079.9 vs. 1825.4 ppm, respectively) confirms the X‐ray observations, for which only weak ChBIs are observed in solids of 7Benzo .
Figure 3.

Experimental (blue lines) and simulated (red lines) 125Te ssNMR spectra of a) 7Pyr and b) 7Benzo . Black spectra correspond to the simulated spectra of Te1 and Te2 atoms, with the isotropic peaks marked as black asterisks.
Computational Studies
To support our hypothesis regarding the bonding contribution of the strong N⋅⋅⋅Te ChBIs to the electronic properties of the supramolecular materials, the organization of 7Pyr and 7Benzo was investigated within the density functional theory (DFT) framework, starting from the experimental cell parameters of the crystal structure (Supporting Information for the calculation methods). Ab initio structural optimization has been performed considering a chalcogen‐bonded monolayer for 7Pyr structure, while a bilayer structure has been considered for 7Benzo to model every Te⋅⋅⋅F interaction. Te⋅⋅⋅N and Te⋅⋅⋅F intermolecular distances have been determined, as well as the interlayer π⋅⋅⋅π contact, being in good agreement with the experimental values (Figures S38a and S41a). The Partial Density of States (PDOS) has been evaluated (Figure 4). For both structures, we found that the highest states of the valence band are mainly based on Te p orbitals, whereas C p orbitals compose the lowest states of the conduction band.
Figure 4.

Partial Density of States (PDOS) of a) 7Pyr and b) 7Benzo structures at the DFT‐GGA level of theory. The Fermi energy has been normalized to zero. The purple line in (a) indicates the energy at which the Te⋅⋅⋅N local charge density has been extracted.
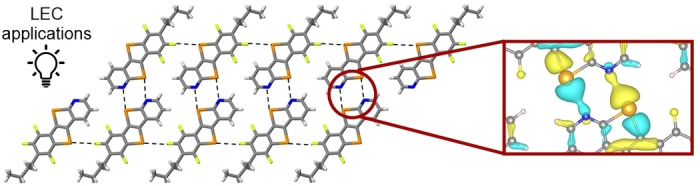
When investigating the local electronic states at Γ point for 7Pyr , a local charge density is found between Te and N atoms (Figure 5a), located at −3.56 eV below the Fermi energy. On the other hand, when the local electronic states at Γ point were calculated for 7Benzo (using the same isosurface value as for 7Pyr ), no localized charge density was observed between Te and F atoms (Figure 5b).
Figure 5.

Local charge densities for a) 7Pyr and b) 7Benzo structures, calculated at −3.56 and −3.74 eV, respectively. The isosurface value is chosen to be 0.001 eV Bohr−3. The positive (negative) values of the electronic wavefunctions are depicted in yellow (blue). C, Te, N, F and H atoms are respectively represented by grey, orange, blue, yellow and white spheres.
In order to complete the theoretical study, first‐principles simulations were also performed for π‐π stacked multilayers of 7Pyr and 7Benzo structures. In both cases, analogous results were obtained for the multi‐layered system of 7Pyr when the local electronic states at Γ point were investigated, showing the same type of Te⋅⋅⋅N interactions located at −3.71 eV. In accordance with the bilayer case, no localized charge density is found between Te and F atoms in 7Benzo multi‐layered structure.
Thin Film Organization
In view of the use of 7Pyr and 7Benzo in LECs (see below), we focused our gaze on the morphological characterization of thin 7Pyr ‐ and 7Benzo ‐containing films. They were prepared by either spin‐coating or drop‐casting techniques onto quartz, glass, silicon wafers and ITO/poly(3,4‐ethylenedioxythiophene)polystyrene sulfonate (PEDOT:PSS) coated glass (i.e., similar to those used in devices, see below). To get deeper insights into the molecular arrangements of the thin films, we complemented the characterization (for the macroscopic morphological characterization see Polarized Light Microscopy (PLM) and Scanning Electron Microscopy (SEM) measurements in Section 7 of the Supporting Information) with Grazing Incidence X‐ray Diffraction (GIXRD) using synchrotron radiation. In line with the observations provided by PLM and SEM, the morphology of thin 7Pyr ‐films is substrate‐independent (Figure 6). In Figure S45a, depicting the GIXRD pattern of a highly ordered 7Pyr ‐film spin‐coated on a silicon wafer, one can notice the presence of intense and high‐order reflections [0 0 2n] (n ε {0,1,2,…}) in the out‐of‐plane direction. This demonstrates a preferential orientation of molecules 7Pyr along the c‐axis in edge‐on configuration (Figure 6e and Figure S45c). This structural hypothesis is further supported by the full GIXRD indexing of the main reflections calculated from the crystal structure. Moreover, the lattice constant derived from the GIXRD reflection [0 0 4] c=44.12(2) Å corresponds to the crystal lattice constant within the experimental error bar.
Figure 6.

On the left, 2D‐GIXRD patterns of glass spin‐coated with thin films of a) 7Pyr and b) 7Benzo ; ITO/PEDOT:PSS covered glass with films of c) 7Pyr and d) 7Benzo ; schematic representation of the molecules on the surface for e) 7Pyr and f) 7Benzo . The indices of the main GIXRD reflections calculated from the crystal structure has been overlaid with their appropriate out‐of‐plane orientation and in‐plane random orientation.
The intense reflection [0 2 5] is the fingerprint for periodic π–π stacking (d π‐π=3.39 Å) as observed in the single crystal X‐ray structure, as well as visualized in the view perpendicular to the [0 2 5] reflection direction (Figure S45d). The same finding is observed in the spin‐coated film (Figures 6a, c), but with higher disorder. The corresponding drop‐casted film on ITO/PEDOT:PSS covered glass (Figure S45b, Supporting Information) shows additionally random orientation. This supports the idea for which the organization of 7Pyr ‐films is similar to that measured in the crystal structure, with the ChBIs ruling the solid‐state arrangement also in thin films. On the contrary, spin‐coated 7Benzo ‐films show a lower degree of order than those obtained from 7Pyr (Figure 6b, d). The 2D‐GIXRD patterns suggest the presence of both ordered lamellar stacks along the reciprocal out‐of‐plane direction [1 0 0] arranged in a head‐to‐tail fashion (as shown in Figure 6f and Figure S46) and partly randomly‐oriented molecular distribution on the substrate. Only the high intensity reflections [2 1 1] and [1 1 1] are visible in the in‐plane direction. The simulated GIXRD pattern from the crystal structure was overlaid with a modified lattice parameter a (determined from the out‐of‐plane diffraction peaks) of 26.56 Å compared to the 22.937 Å from the crystal structure.
Thin‐Film Transistor Fabrication
To shed light onto the charge transport properties of 7Pyr and 7Benzo , and to gain insight into the role of the order at the supramolecular level within the assemblies obtained by solution processing, we prepared and characterized thin‐film transistors in a bottom‐gate top‐contact configuration. In full accordance with the previously presented results, both optical microscopy (Figure S47a, b) and atomic force microscopy (Figure S47c) revealed that molecule 7Pyr self‐assembles into ordered structures whereas 7Benzo forms disordered clusters. More specifically, 7Pyr self‐assembles into crystals with a thickness of 40 nm and lateral size of several tens of μm, whereas 7Benzo forms disordered aggregates with a lateral size of a few μm and a thickness exceeding the hundreds of nm scale. The electrical characterization of the thin‐film transistors was carried out by recording transfer curves. While both materials revealed the absence of electron transport, a major difference has been observed in terms of hole mobility. Thin films of 7Benzo displayed a lack of hole transport whereas 7Pyr ‐based crystals exhibited a hole transport behavior. A representative transfer curve is displayed in Figure S47g. It exhibits an excellent I on/I off ratio of 105 and a low off current, indicating the low defect states in the vicinity of HOMO. The extracted hole mobility of 7Pyr was calculated as 8.8×10−5 cm2 V−1 s−1. While the device is obviously unoptimized for a specific application and there is room for enhancement of the device performance, e.g. via optimization of interfaces of 7Pyr with the dielectric gate and the metallic electrodes, this result represents the first demonstration of a semiconducting characteristic of supramolecular architecture held together via chalcogen bonds.
Implementation in LECs
Considering the ribbon‐type, chalcogen‐bond‐driven solid‐state organization, the p‐type semiconducting properties and the lack of electroluminescence (Supporting Information, Figure S48), we used thin films of 7Pyr as hole‐transport layers to build LECs.[ 20a , 20c , 36 ] 7Pyr ‐containing films were integrated as hole‐transport layers to decouple charge injection/transport and exciton formation in LECs with CuI‐complexes. These emitters are notoriously known to provide poorly stable LECs under device operation.[ 20b , 37 ] A 10‐fold enhancement in stability for blue‐, yellow‐ and red‐emitting LECs without affecting brightness and efficiency has recently been reported implementing a 10–20 nm thick hole‐transport layer (i.e., 4,4′‐bis(N‐carbazolyl)‐1,1′‐biphenyl or CBP), in multi‐layered LECs. [37] However, CBP layers tend to easily form micrometer‐size aggregates upon device fabrication and storage (e.g., either under ambient or inert conditions), compromising the device self‐stability.[ 37b , 38 ] Moreover, CBP‐LECs show a significant bluish green electroluminescence that limits their use in terms of device chromaticity. [36] 7Pyr ‐films are very robust with respect to morphology and fabrication, while they feature a similar electronic energy levels to those of CBP (Figure S49).
In view of the above lines, proof‐of‐concept LECs combining 7Pyr ‐, 7Benzo ‐ or CBP‐films as hole‐transporters and [Cu(dcbq)(Xantphos)]PF6 (Cu1; dcbq: 4,4′‐diethylester‐2,2′‐biquinoline and Xantphos: 4,5‐bis(diphenylphosphino)‐9,9‐dimethylxanthene) as the red emitter were fabricated with the architectures: i) ITO/PEDOT : PSS (70 nm)/7Pyr or 7Benzo (15 nm)/Cu1 (80 nm)/Al (90 nm) as targeted devices (Figure 7), and ii) ITO/PEDOT : PSS (70 nm)/CBP (15 nm)/Cu1 (80 nm)/Al (90 nm) and ITO/PEDOT : PSS (70 nm)/Cu1 (80 nm)/Al (90 nm) as reference devices. They were driven at the optimized pulsed current of 20 mA. [20b] As expected, 7Pyr ‐containing LECs outperformed the reference single‐layered LECs, [20b] showing similar efficiency values (ca. 0.18 lm/W), but one order of magnitude enhanced stability (3 h vs. 38 h for single‐ and multi‐layered LECs, respectively). Both devices showed the same deep‐red electroluminescence centered at 663 nm, associated to x/y CIE color coordinates of 0.65/0.32, stable over the entire device lifespan (Figure 7b). Thus, electron‐hole recombination efficiently occurs at the interface 7Pyr /Cu1, while hole injection/transport occurs firstly in the chalcogen‐bonded layer and it is further transferred to the Cu1 layer. As such, the formation of oxidized products (i.e., CuII complex species) is significantly reduced, leading to a significant enhancement of the device stability. At last, we compared the performances of 7Pyr ‐based with those integrating the benchmark CBP‐ and reference 7Benzo ‐film. In line with the I–V assays (see above), reference 7Benzo multi‐layered LECs showed a dramatic loss of stability (t 1/2 <1 h) at pulsed 20 mA (Figure 7c). In stark contrast, Figure 7 shows that similar irradiances and stabilities were noted for 7Pyr ‐ and CBP‐LECs, while the efficiency is slightly enhanced (0.18 vs. 0.15 lm W−1).
Figure 7.

a) Schematic representation of the multi‐layered LECs together with the chemical structure of Cu1. b) Electroluminescence spectra over time of ITO/PEDOT : PSS (60 nm)/7Pyr (15 nm)/Cu1 (80 nm)/Al at pulsed 20 mA. c) Average voltage and luminance vs. time of devices with 7Pyr /Cu1 (full squares), CBP/Cu1 (empty squares), 7Benzo /Cu1 (inset) active layers at pulsed 20 mA.
Conclusion
Herein we have reported on the design and synthesis of two telluro[3,2‐β][1]‐tellurophene π‐conjugated modules, 7Pyr and 7Benzo , which undergo programmed self‐assembly at the solid‐state through ChBIs. While 7Pyr was designed to undergo ribbon‐like organization through the formation of two double ChBIs, congener 7Benzo was edited in such a way that only herringbone‐type architectures could be formed at the solid‐state through weak ChBIs as displayed by single‐crystal X‐ray analysis. A computational investigation suggested that both 7Pyr and 7Benzo electronic structures were characterized by Te p orbitals in the highest states of the valence bands, and by C p orbitals in the lowest states of the conduction band but, only for 7Pyr , a local charge density was found between Te and N atoms. OTFT‐based charge transport measurements of single crystals of 7Pyr showed hole‐transport properties, whereas reference materials with 7Benzo showed no measurable mobility values. 2D‐GIXRD investigations showed that thin films containing 7Pyr display consistent molecular organization with that observed in single crystals. However, films containing 7Benzo depicted only short‐range order and no π‐stacking arrangements. Notably, the structural properties of films of 7Pyr revealed to be substrate‐independent and the same as those measured in single crystals.
Finally, it was demonstrated for the first time the use of chalcogen‐bonded compounds in multi‐layered LECs as a hole transport layer to decouple transport and emission using red‐emitting CuI complexes. LECs containing 7Pyr displayed a stability enhancement (ca. 40 h) when compared to devices integrating 7Benzo (<1 h), with performances similar to those obtained when traditional hole transport layers like CBP are used. Notably, the lack of electroluminescence signal is an interesting feature that paves the way towards its versatile implementation in LECs.
This work describes for the first time the design and preparation of a chalcogen‐bonded supramolecular semiconductor and its use in solid‐state lighting devices, establishing such intermolecular interactions as an effective supramolecular tool for designing functional materials, in general, and non‐electroluminescent hole transporters as effective strategy to enhance LECs, in particular.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
D.B. gratefully acknowledges the EU through the funding scheme projects MSCA‐RISE INFUSION (N° 734834), H2020‐NMBP‐2017 DECOCHROM (N° 760973) and the University of Vienna for the financial support. J.‐C.C., R.D.C. and D.B. acknowledge the MSCA‐ITN‐ETN STiBNite (N° 956923). R.D.C. acknowledges the EU Horizon 2020 research and innovation FET‐OPEN under grant agreement ARTIBLED (No 863170) and the ERC‐Co InOutBioLight (N° 816856). D.B. thanks CERIC‐ERIC for financial support (proposal n° 20207045) for accessing the Austrian SAXS beamline at Elettra. J.‐C.C. acknowledges the Fédération Wallonie‐Bruxelles (ARC grant N°21/26‐116), the European Union′s Horizon 2020 Research and Innovation program (Graphene Flagship Core3—N°881603), and the F.R.S. FNRS of Belgium. P.S. acknowledges the Labex project CSC (ANR‐10LABX‐0026 CSC) within the Investissement d'Avenir program ANR‐10‐IDEX‐0002‐02 the International Center for Frontier Research in Chemistry, the Institut Universitaire de France (IUF) and the Chinese Scholarship Council. Computational resources have been provided by the Consortium des Équipements de Calcul Intensif (CÉCI), funded by the Fonds de la Recherche Scientifique de Belgique (F.R.S.‐FNRS) under Grant No. 2.502 0.11
D. Romito, E. Fresta, L. M. Cavinato, H. Kählig, H. Amenitsch, L. Caputo, Y. Chen, P. Samorì, J.-C. Charlier, R. D. Costa, D. Bonifazi, Angew. Chem. Int. Ed. 2022, 61, e202202137; Angew. Chem. 2022, 134, e202202137.
Contributor Information
Prof. Dr. Rubén D. Costa, Email: ruben.costa@tum.de.
Prof. Dr. Davide Bonifazi, Email: davide.bonifazi@univie.ac.at.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Bent H. A., Chem. Rev. 1968, 68, 587–648; [Google Scholar]
- 1b. Alcock N. W., in Adv. Inorg. Chem. Radiochem. Elsevier, Amsterdam, 1972, pp. 1–58. [Google Scholar]
- 2.For some reviews on the topic see:
- 2a. Mahmudov K. T., Kopylovich M. N., da Silva M. F. C. G., Pombeiro A. J., Dalton Trans. 2017, 46, 10121–10138; [DOI] [PubMed] [Google Scholar]
- 2b. Biot N., Bonifazi D., Coord. Chem. Rev. 2020, 413, 213243; [Google Scholar]
- 2c. Gleiter R., Haberhauer G., Werz D. B., Rominger F., Bleiholder C., Chem. Rev. 2018, 118, 2010–2041; [DOI] [PubMed] [Google Scholar]
- 2d. Lim J. Y. C., Beer P. D., Chem 2018, 4, 731–783; [Google Scholar]
- 2e. Kolb S., Oliver G. A., Werz D., Angew. Chem. Int. Ed. 2020, 59, 22306–22310; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22490–22495. [Google Scholar]
- 3. Aakeroy C. B., Bryce D. L., Desiraju G. R., Frontera A., Legon A. C., Nicotra F., Rissanen K., Scheiner S., Terraneo G., Metrangolo P., Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar]
- 4.
- 4a. Hoover G. C., Seferos D. S., Chem. Sci. 2019, 10, 9182–9188; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Di Maria F., Zangoli M., Gazzano M., Fabiano E., Gentili D., Zanelli A., Fermi A., Bergamini G., Bonifazi D., Perinot A., Adv. Funct. Mater. 2018, 28, 1801946. [Google Scholar]
- 5.
- 5a. Vogel L., Wonner P., Huber S. M., Angew. Chem. Int. Ed. 2019, 58, 1880–1891; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1896–1907; for some selected examples in the field of catalysis see: [Google Scholar]
- 5b. Benz S., López-Andarias J., Mareda J., Sakai N., Matile S., Angew. Chem. Int. Ed. 2017, 56, 812–815; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 830–833; [Google Scholar]
- 5c. Wonner P., Dreger A., Vogel L., Engelage E., Huber S. M., Angew. Chem. Int. Ed. 2019, 58, 16923–16927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17079–17083; [Google Scholar]
- 5d. Young C. M., Elmi A., Pascoe D. J., Morris R. K., McLaughlin C., Woods A. M., Frost A. B., de la Houpliere A., Ling K. B., Smith T. K., Slawin A. M. Z., Willoughby P. H., Cockroft S. L., Smith A. D., Angew. Chem. Int. Ed. 2020, 59, 3705–3710; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 3734–3739; [Google Scholar]
- 5e. Wang W., Zhu H., Feng L., Yu Q., Hao J., Zhu R., Wang Y., J. Am. Chem. Soc. 2020, 142, 3117–3124. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Taylor M. S., Coord. Chem. Rev. 2020, 413, 213270; for selected examples with anion recognition see: [Google Scholar]
- 6b. Garrett G. E., Gibson G. L., Straus R. N., Seferos D. S., Taylor M. S., J. Am. Chem. Soc. 2015, 137, 4126–4133; [DOI] [PubMed] [Google Scholar]
- 6c. Lee L. M., Tsemperouli M., Poblador-Bahamonde A. I., Benz S., Sakai N., Sugihara K., Matile S., J. Am. Chem. Soc. 2019, 141, 810–814; [DOI] [PubMed] [Google Scholar]
- 6d. Garrett G. E., Carrera E. I., Seferos D. S., Taylor M. S., Chem. Commun. 2016, 52, 9881–9884; [DOI] [PubMed] [Google Scholar]
- 6e. Lim J. Y. C., Marques I., Thompson A. L., Christensen K. E., Félix V., Beer P. D., J. Am. Chem. Soc. 2017, 139, 3122–3133; [DOI] [PubMed] [Google Scholar]
- 6f. Bunchuay T., Docker A., Eiamprasert U., Surawatanawong P., Brown A., Beer P. D., Angew. Chem. Int. Ed. 2020, 59, 12007–12012; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 12105–12110; [Google Scholar]
- 6g. Docker A., Guthrie C. H., Kuhn H., Beer P. D., Angew. Chem. Int. Ed. 2021, 60, 21973–21978; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 22144–22149. [Google Scholar]
- 7.
- 7a. Cozzolino A. F., Elder P. J. W., Vargas-Baca I., Coord. Chem. Rev. 2011, 255, 1426–1438; [Google Scholar]
- 7b. Chivers T., Laitinen R. S., Chem. Soc. Rev. 2015, 44, 1725–1739; [DOI] [PubMed] [Google Scholar]
- 7c. Fanfrlík J., Přáda A., Padělková Z., Pecina A., Macháček J., Lepšík M., Holub J., Růžička A., Hnyk D., Hobza P., Angew. Chem. Int. Ed. 2014, 53, 10139–10142; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10303–10306; [Google Scholar]
- 7d. Huynh H.-T., Jeannin O., Fourmigué M., Chem. Commun. 2017, 53, 8467–8469. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Ho P. C., Szydlowski P., Sinclair J., Elder P. J. W., Kübel J., Gendy C., Lee L. M., Jenkins H., Britten J. F., Morim D. R., Vargas-Baca I., Nat. Commun. 2016, 7, 11299; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Ho P. C., Rafique J., Lee J., Lee L. M., Jenkins H. A., Britten J. F., Braga A. L., Vargas-Baca I., Dalton Trans. 2017, 46, 6570–6579; [DOI] [PubMed] [Google Scholar]
- 8c. Scilabra P., Terraneo G., Resnati G., Acc. Chem. Res. 2019, 52, 1313–1324; [DOI] [PubMed] [Google Scholar]
- 8d. Mullin W. J., Sharber S. A., S. W. Thomas III , J. Polym. Sci. 2021, 59, 1643–1663. [Google Scholar]
- 9.
- 9a. Bertini V., Dapporto P., Lucchesini F., Sega A., De Munno A., Acta Crystallogr. Sect. C 1984, 40, 653–655; [Google Scholar]
- 9b. Allan R. E., Gornitzka H., Kärcher J., Paver M. A., Rennie M., Russell C. A., Raithby P. R., Stalke D., Steiner A., Wright D. S., J. Chem. Soc. Dalton Trans. 1996, 1727–1730; [Google Scholar]
- 9c. Cozzolino A. F., Vargas-Baca I., Mansour S., Mahmoudkhani A. H., J. Am. Chem. Soc. 2005, 127, 3184–3190; [DOI] [PubMed] [Google Scholar]
- 9d. Cozzolino A. F., Britten J. F., Vargas-Baca I., Cryst. Growth Des. 2006, 6, 181–186. [Google Scholar]
- 10. Riwar L. J., Trapp N., Root K., Zenobi R., Diederich F., Angew. Chem. Int. Ed. 2018, 57, 17259–17264; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 17506–17512. [Google Scholar]
- 11.
- 11a. Kremer A., Aurisicchio C., De Leo F., Ventura B., Wouters J., Armaroli N., Barbieri A., Bonifazi D., Chem. Eur. J. 2015, 21, 15377–15387; [DOI] [PubMed] [Google Scholar]
- 11b. Kremer A., Fermi A., Biot N., Wouters J., Bonifazi D., Chem. Eur. J. 2016, 22, 5665–5675. [DOI] [PubMed] [Google Scholar]
- 12. Biot N., Bonifazi D., Chem. Eur. J. 2020, 26, 2904–2913. [DOI] [PubMed] [Google Scholar]
- 13. Biot N., Bonifazi D., Chem. Eur. J. 2018, 24, 5439–5443. [DOI] [PubMed] [Google Scholar]
- 14. Biot N., Romito D., Bonifazi D., Cryst. Growth Des. 2021, 21, 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ho P. C., Wang J. Z., Meloni F., Vargas-Baca I., Coord. Chem. Rev. 2020, 422, 213464. [Google Scholar]
- 16. Cozzolino A. F., Yang Q., Vargas-Baca I., Cryst. Growth Des. 2010, 10, 4959–4964. [Google Scholar]
- 17. Wang C., Abbas M., Wantz G., Kawabata K., Takimiya K., J. Mater. Chem. C 2020, 8, 15119–15127. [Google Scholar]
- 18.
- 18a. Politzer P., Murray J. S., Clark T., Phys. Chem. Chem. Phys. 2013, 15, 11178–11189; [DOI] [PubMed] [Google Scholar]
- 18b. Pascoe D. J., Ling K. B., Cockroft S. L., J. Am. Chem. Soc. 2017, 139, 15160–15167; [DOI] [PubMed] [Google Scholar]
- 18c. Bauzá A., Frontera A., ChemPhysChem 2020, 21, 26–31; [DOI] [PubMed] [Google Scholar]
- 18d. de Azevedo Santos L., van Der Lubbe S. C., Hamlin T. A., Ramalho T. C., Bickelhaupt F. M., ChemistryOpen 2021, 10, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Noh J., Jung S., Koo D. G., Kim G., Choi K. S., Park J., Shin T. J., Yang C., Park J., Sci. Rep. 2018, 8, 14448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. Fresta E., Costa R. D., J. Mater. Chem. C 2017, 5, 5643–5675; [Google Scholar]
- 20b. Fresta E., Weber M. D., Fernandez-Cestau J., Costa R. D., Adv. Opt. Mater. 2019, 7, 1900830; [Google Scholar]
- 20c. Matsuki K., Pu J., Takenobu T., Adv. Funct. Mater. 2020, 30, 1908641. [Google Scholar]
- 21.
- 21a. Esrafili M. D., Mohammadian-Sabet F., Chem. Phys. Lett. 2015, 634, 210–215; [Google Scholar]
- 21b. Benz S., Mareda J., Besnard C., Sakai N., Matile S., Chem. Sci. 2017, 8, 8164–8169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21c. Bao L., Kong X., Wang Y., Asian J. Org. Chem. 2020, 9, 757–760. [Google Scholar]
- 22. Romito D., Biot N., Babudri F., Bonifazi D., New J. Chem. 2020, 44, 6732–6738. [Google Scholar]
- 23.Te-containing compounds exposing multiple recognition units revealed to be poorly soluble in common organic solvents and displayed limited processability (Ref. [11a, 22]).
- 24. Sashida H., Yasuike S., J. Heterocycl. Chem. 1998, 35, 725–726. [Google Scholar]
- 25.
- 25a. Brandsma L., Hommes H., Verkruijsse H., De Jong R., Recl. Trav. Chim. Pays-Bas 1985, 104, 226–230; [Google Scholar]
- 25b. Sashida H., Sadamori K., Tsuchiya T., Synth. Commun. 1998, 28, 713–727; [Google Scholar]
- 25c. Engman L., Laws M. J., Malmström J., Schiesser C. H., Zugaro L. M., J. Org. Chem. 1999, 64, 6764–6770; [DOI] [PubMed] [Google Scholar]
- 25d. Muranaka A., Yasuike S., Liu C.-Y., Kurita J., Kakusawa N., Tsuchiya T., Okuda M., Kobayashi N., Matsumoto Y., Yoshida K., J. Phys. Chem. A 2009, 113, 464–473; [DOI] [PubMed] [Google Scholar]
- 25e. Rhoden C. R., Zeni G., Org. Biomol. Chem. 2011, 9, 1301–1313; [DOI] [PubMed] [Google Scholar]
- 25f. Jiang M., Guo J., Liu B., Tan Q., Xu B., Org. Lett. 2019, 21, 8328–8333. [DOI] [PubMed] [Google Scholar]
- 26. Duan X.-F., Li X.-H., Li F.-Y., Huang C.-H., Synthesis 2004, 2614–2616. [Google Scholar]
- 27. Chelucci G., Baldino S., Pinna G. A., Sechi B., Tetrahedron Lett. 2008, 49, 2839–2843. [Google Scholar]
- 28. Inoue A., Kitagawa K., Shinokubo H., Oshima K., J. Org. Chem. 2001, 66, 4333–4339. [DOI] [PubMed] [Google Scholar]
- 29. Coe P. L., Waring A. J., Yarwood T. D., J. Chem. Soc. Perkin Trans. 1 1995, 2729–2737. [Google Scholar]
- 30.When mono- and trifluorinated phenyl substituents are used as electrophilic aromatic sustrates, the telluro[3,2-b][1]-tellurophene is not formed and only alkylic addition of the butyl chain to the aryl ring is observed.
- 31. 1H NMR studies of the reaction mixture quenched after 50 minutes (Figure S30, Supporting Information), i.e. before the addition of freshly grounded Te, displayed the presence of the alkylated intermediate only. This suggests that the alkylation reaction have already occured within few minutes after the treatment of 6Benzo with the organomagnesium reagent. Furthermore, 19F NMR studies (Figure S31, Supporting Information) allowed to unambigously determined the position of the n-butyl chain on the perfluorinated ring.
- 32.Deposition numbers 2057472 (7 Pyr ), 2057473 (7 Benzo ) and 2063971 (4 Ph ) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 33. Hierso J.-C., Chem. Rev. 2014, 114, 4838–4867. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Montalti M., Credi A., Prodi L., Gandolfi M. T., Handbook of photochemistry, CRC press, Boca Raton, 2006; [Google Scholar]
- 34b. Baroncini M., Bergamini G., Ceroni P., Chem. Commun. 2017, 53, 2081–2093; [DOI] [PubMed] [Google Scholar]
- 34c. He G., Torres Delgado W., Schatz D. J., Merten C., Mohammadpour A., Mayr L., Ferguson M. J., McDonald R., Brown A., Shankar K., Rivard E., Angew. Chem. Int. Ed. 2014, 53, 4587–4591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 4675–4679; [Google Scholar]
- 34d. Parke S. M., Boone M. P., Rivard E., Chem. Commun. 2016, 52, 9485–9505; [DOI] [PubMed] [Google Scholar]
- 34e. Braun C. A., Ferguson M. J., Rivard E., Inorg. Chem. 2021, 60, 2672–2679. [DOI] [PubMed] [Google Scholar]
- 35.
- 35a. Kumar V., Xu Y., Leroy C., Bryce D. L., Phys. Chem. Chem. Phys. 2020, 22, 3817–3824; [DOI] [PubMed] [Google Scholar]
- 35b. Kumar V., Xu Y., Bryce D. L., Chem. Eur. J. 2020, 26, 3275–3286; [DOI] [PubMed] [Google Scholar]
- 35c. Southern S. A., Bryce D. L., Annu. Rev. NMR Spectrosc. 2021, 102, 1–80. [Google Scholar]
- 36.
- 36a. Elie M., Gaillard S., Renaud J., in Light-emitting electrochemical cells: concepts, advances and challenges, Springer International Publishing, New York, 2017; [Google Scholar]
- 36b. Fresta E., Monclús M. A., Bertz M., Ezquerro C., Molina-Aldareguia J. M., Berenguer J. R., Kunimoto M., Homma T., Costa R. D., Adv. Opt. Mater. 2020, 8, 2000295. [Google Scholar]
- 37.
- 37a. Weber M. D., Fresta E., Elie M., Miehlich M. E., Renaud J. L., Meyer K., Gaillard S., Costa R. D., Adv. Funct. Mater. 2018, 28, 1707423; [Google Scholar]
- 37b. Fresta E., Volpi G., Milanesio M., Garino C., Barolo C., Costa R. D., Inorg. Chem. 2018, 57, 10469–10479. [DOI] [PubMed] [Google Scholar]
- 38.
- 38a. Fresta E., Carbonell-Vilar J. M., Yu J., Armentano D., Cano J., Viciano-Chumillas M., Costa R. D., Adv. Funct. Mater. 2019, 29, 1901797; [DOI] [PubMed] [Google Scholar]
- 38b. Lundberg P., Lindh E. M., Tang S., Edman L., ACS Appl. Mater. Interfaces 2017, 9, 28810–28816; [DOI] [PubMed] [Google Scholar]
- 38c. Yu H., Zhang Y., Cho Y. J., Aziz H., ACS Appl. Mater. Interfaces 2017, 9, 14145–14152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.