

SUMMARY
Momilactone A, an important plant labdane‐related diterpenoid, functions as a phytoalexin against pathogens and an allelochemical against neighboring plants. The genes involved in the biosynthesis of momilactone A are found in clusters, i.e., momilactone A biosynthetic gene clusters (MABGCs), in the rice and barnyardgrass genomes. In addition, we know little about the origin and evolution of MABGCs. Here, we integrated results from comprehensive phylogeny and comparative genomic analyses of the core genes of MABGC‐like clusters and MABGCs in 40 monocot plant genomes, providing convincing evidence for the birth and evolution of MABGCs in grass species. The MABGCs found in the PACMAD clade of the core grass lineage (including Panicoideae and Chloridoideae) originated from a MABGC‐like cluster in Triticeae (BOP clade) via lateral gene transfer (LGT) and followed by recruitment of MAS1/2 and CYP76L1 genes. The MABGCs in Oryzoideae originated from PACMAD through another LGT event and lost CYP76L1 afterwards. The Oryza MABGC and another Oryza diterpenoid cluster c2BGC are two distinct clusters, with the latter originating from gene duplication and relocation within Oryzoideae. Further comparison of the expression patterns of the MABGC genes between rice and barnyardgrass in response to pathogen infection and allelopathy provides novel insights into the functional innovation of MABGCs in plants. Our results demonstrate LGT‐mediated origination of MABGCs in grass and shed lights into the evolutionary innovation and optimization of plant biosynthetic pathways.
Keywords: biosynthetic gene cluster, diterpenoid momilactone, lateral gene transfer, grass, phylogeny
Significance Statement
How biosynthetic gene clusters (BGCs) originate and evolve is less known in eukaryotes. Momilactone A, an important defensive and allelopathic secondary chemical in plants, is synthesized by momilactone A BGCs (MABGCs). By exploiting the phylogeny and comparative genomics of the core genes of MABGC‐like clusters and MABGCs across 40 plant species, we reconstruct the evolutionary trajectory of MABGCs in grass, i.e., the PACMAD clade (including Panicoideae and Chloridoideae) acquired MABGCs from Triticeae via lateral gene transfer (LGT) of a MABGC‐like cluster and another LGT event passed the PACMAD MABGC on to Oryza. The composition of MABGCs is dynamic with gain and/or loss of genes in the recipient species after LGT. The study demonstrates that, like prokaryotes, plants are capable of moving a cluster of genes involved in the same biosynthesis pathway by LGT. The results shed light into the evolutionary innovation of BGCs and optimization of a biosynthetic pathway via synthetic biology.
INTRODUCTION
Secondary metabolites, particularly phytoalexins, play essential roles in defense against pathogens, pests, herbivores, and neighboring plants. Many of them are synthesized by enzymes encoded by genes arranged as a cluster, called biosynthetic gene cluster (BGC), e.g., benzoxazinoid DIMBOA in maize (Zea mays), monoterpene β‐phellandrene in tomato (Solanum lycopersicum), diterpene momilactones in rice (Oryza sativa), and triterpene thalianol in Arabidopsis thaliana (Field & Osbourn, 2008; Frey et al., 1997; Guo et al., 2018; Matsuba et al., 2013; Shimura et al., 2007; Zhan et al., 2022). A BGC is composed by three or more non‐homologous genes that are located close to each other and encode enzymes participating in the same biosynthesis pathway. Co‐regulation and co‐inheritance indicate selective advantages of BGCs (Nützmann & Osbourn, 2014; Rokas et al., 2018). At least two BGCs, involved in the biosynthesis of diterpenoid phytoalexins, have been identified in the genome of the cultivated Asian rice (O. sativa) (Guo et al., 2018). They are c4BGC associated with momilactone A (MA) production (hereafter MABGC) on chromosome 4 and c2BGC for phytocassane production on chromosome 2 (Miyamoto et al., 2016; Shimura et al., 2007; Toyomasu et al., 2020). Biosynthesis of momilactone requires a series of catalytic reactions, involving enzymes from not only MABGC (CPS4, KSL4, CYP99A2/3, and MAS1/2) but also CYP76M8 from c2BGC, indicating interdependent evolution of the two BGCs (De La Peña & Sattely, 2021; Kitaoka et al., 2021; Shimura et al., 2007) (Figure 1a). A cytochrome P450 enzyme encoded by CYP701A8 on chromosome 6 is also involved in rice momilactone biosynthesis (Figure 1a) (Kitaoka et al., 2021). The rice MABGC was reported to evolve within Oryza through duplication and assembly of ancestral biosynthetic genes before the divergence of the BB genome (Miyamoto et al., 2016). MABGCs have also been found in the genomes of paddy weed barnyardgrass Echinochloa crus‐galli and bryophyte Calohypnum plumiforme (Guo et al., 2017; Mao et al., 2020). The functional similarity of MABGCs in grass and bryophyte is likely a result of convergent evolution (Mao et al., 2020; Zhang & Peters, 2020). Compared with the O. sativa MABGC, the E. crus‐galli MABGC has an extra copy of CYP76L1 (originally wrongly assigned as a member of the CYP76M subfamily) (Guo et al., 2017). Although the divergence time of the two core grass clades, BOP clade (Bambusoideae, Oryzoideae, and Pooideae) and PACMAD clade (Panicoideae, Arundinoideae, Chloridoideae, Micrairoideae, Aristidoideae, and Danthonioideae) to which rice and barnyardgrass respectively belong, is more than 50 million years ago (Ma, Liu, et al., 2021) (Figure 1b), sexual introgression or vertical inheritance from Oryza to Echinochloa was proposed to result in the patchy occurrence of MABGCs in grass (Peters, 2020; Zhang & Peters, 2020). Despite these findings, the origin and evolutionary relationship of MABGCs in grass remains mysterious.
Figure 1.
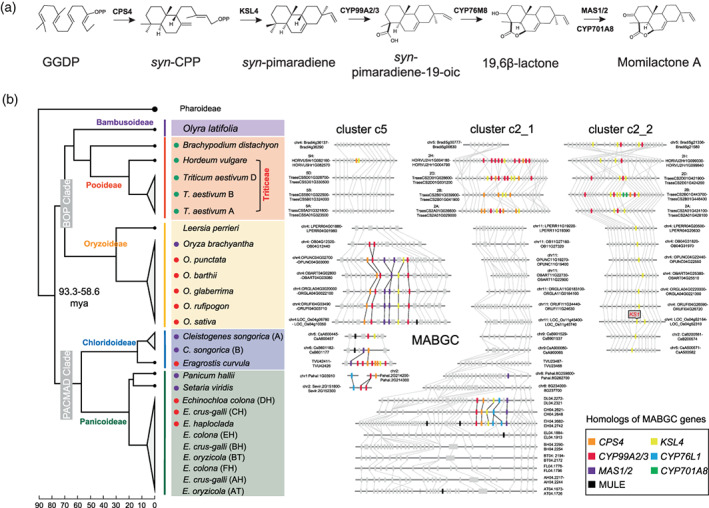
Momilactone biosynthesis pathway and clusters of momilactone A biosynthetic genes (MABGCs) in grass genomes.
(a) Momilactone A biosynthesis pathway in rice. Key enzymes for each catalysis reaction are indicated.
(b) Genomic distribution of momilactone biosynthetic genes and their homologs in grass genomes. Left panel shows the phylogenetic topology of grass species. Red, purple, and green dots in front of the species scientific names represent the presence of intact MABGCs, partial MABGCs, and other MABGC‐like clusters, respectively. Right panel displays the micro‐genomic synteny of MABGCs and MABGC‐like genes among multi‐genomes. Gray rectangle elements represent other genes near the genomic regions of MABGC and MABGC‐like clusters. [Colour figure can be viewed at wileyonlinelibrary.com]
The gene clustering structure observed in MABGCs is a common phenomenon in bacteria and fungi, in which the origination and evolution of operons have been extensively studied (Nützmann et al., 2018). Gene duplication, neofunctionalization, and relocation are the main routes for BGC assembly in most fungi and plants (Nützmann et al., 2018; Rokas et al., 2018). Lateral gene transfer (LGT) is widespread in prokaryotes and eukaryotes, including grass species (Hibdige et al., 2021). LGT is an important shortcut to the acquisition of gene clusters, particularly in microorganisms (Kominek et al., 2019; Slot & Rokas, 2011). Although high‐quality genomes generated in recent years have offered good opportunities to trace the evolutionary trajectory of gene clusters in plants (Liu et al., 2020; Yang et al., 2021), we still know little about the mechanisms underlying the birth and evolution of BGCs in plants and whether LGT plays a role in the acquisition of BGCs in grass.
The sporadic distribution of MABGCs in the two divergent grass genera Oryza and Echinochloa provides an opportunity for investigating the detailed evolutionary trajectory of BGCs in plants, with the utilization of high‐quality genomes in the grass family. Here, we analyzed the individual core genes of MABGCs in 40 monocot genomes using phylogenetic and comparative genomics approaches. We found that it is likely that the grass MABGCs originated from a BGC in Triticeae, which was passed on to the PACMAD and Oryza clades subsequently via two LGT events, leading to the formation of the MABGCs observed in rice and barnyardgrass through further gene loss and gain. Our work sheds new insights into the evolutionary innovation of momilactone biosynthetic pathway in grass.
RESULTS
Identification of momilactone biosynthetic genes in grass
Using amino acid sequences of the core MABGC genes (CPS4, KSL4, CYP99A2/3, and MAS1/2) from rice (O. sativa) and CYP76L1 from barnyardgrass (E. crus‐galli) as queries, we screened out homologs of all the core MABGC genes from 40 monocot genomes, including 21 species from the PACMAD clade (15 in subfamily Panicoideae and six in subfamily Chloridoideae), 17 species from the BOP clade (two from Bambusoideae, eight from Oryzoideae, and seven from Pooideae), and two outgroup species Pharus latifolius and Ananas comosus (Table S1). The intact MABGCs (defined as harboring at least one copy of KSL4, MAS1/2, CPS4, and CYP99A2/3 or CYP76L1 homologs within a 200‐kb genomic window) were identified in Oryza from Oryzoideae, Echinochloa from Panicoideae, and Eragrostis from Chloridoideae (Figure 1b, Table S2). In Oryza, while the intact MABGCs were found on chromosome 4 in the AA and BB genomes, only two tandemly duplicated CYP99A2/3 homologs were found in the FF genome (Oryza brachyantha), indicating that MABGCs in Oryza had been clustered or generated at least before the divergence of the AA and BB genomes (approximately 6.76 million years ago, mya) (Stein et al., 2018). In Echinochloa, MABGCs were found on chromosome 4 in three (sub)genomes (subgenome CH of hexaploid E. crus‐galli, subgenome DH of hexaploid Echinochloa colona, and diploid Echinochloa haploclada), which formed a monoclade in Echinochloa genome phylogeny (Wu, Shen, et al., 2022). A candidate MABGC in the genome of weeping lovegrass Eragrostis curvula was identified but only partial MABGCs were found in other Chloridoideae species (e.g., Cleistogenes songorica, Eragrostis nindensis, and Eragrostis tef) (Figure 1b, Table S2). Although MABGCs were found in three genera (Oryza, Eragrostis, and Echinochloa), they are not syntenic in physical genomic positions, implying the dynamic evolution of MABGCs in grass (Figure 1b). The positional non‐synteny among the MABGC loci may be related to the activities of the mutator‐like transposable elements found near the locations of MABGCs (Figure 1b).
Several MABGC‐like clusters were found on chromosome 2 (clusters c2_1 and c2_2) and chromosome 5 (cluster c5) in Pooideae based on the homology search of MABGC genes (Figure 1b, Table S2). These clusters are composed of CYP99A2/3, KSL4, and CPS4 homologs and without MAS1/2 homologs. The c2_2 clusters are mainly composed of CYP99A2/3 and KSL4 homologs, and were assembled before the divergence of Triticeae and Brachypodium (Figure 1b). Based on the analysis of the syntenic regions in the genomes of Oryzoideae and Chloridoideae, KSL4 homologs in the c2_2 clusters are likely to be derived from tandem duplication of KS1, a gene responsible for gibberellin biosynthesis in rice (Toyomasu et al., 2020), and CYP99A2/3 homologs are embedded among KSL4 homologs in Pooideae (Figure 1b). Intriguingly, the c2_2 cluster in subgenome B of hexaploid Triticum aestivum or tetraploid Triticum dicoccoides contains an additional CPS4 (TraesCS2B01G445500 in T. aestivum subgenome B) and two CYP701A8 homologs (TraesCS2B01G445300 and TraesCS2B01G445400 in T. aestivum subgenome B, TRIDC2BG065020 and TRIDC2BG065030 in T. dicoccoides subgenome B), which are absent in other c2_2 clusters (Figure 1b, Table S2). Phylogeny and genomic synteny revealed that the two CYP701A8 copies were specifically retained in subgenome B of Triticeae and evolved from the duplication and relocation of the grass‐range native CYP701A8 homologs (native genes are a set of conserved syntenic orthologs within highly syntenic blocks from multiple genomes) (e.g., Pl06g08600 from P. latifolius [N0], three tandem duplicates Bradi1g37560, Bradi1g37576, and Bradi1g37547 from B. distachyon [N1], TraesCS7B01G265800 from T. aestivum subgenome B [N2], and CYP701A8 or LOC_Os06g37300 from O. sativa [N3] in Figure S1). It should be noted that CYP701A8 is required for the production of momilactone in rice (O. sativa) (Kitaoka et al., 2021); however, located away from the MABGC, while in the cluster c2_2 of T. aestivum subgenome B, two CYP701A8 homologs were found to be embedded in the MABGC‐like cluster. Cluster c5 composed of only CPS4 and KSL4 homologs was found in the genome of barley Hordeum vulgare (Figure 1b). Cluster c2_1 is absent in the Brachypodium genome, implying a recent origination of c2_1 within Triticeae. In T. aestivum, while the cluster c2_1 of subgenome B has no CYP99A2/3 homolog, both c2_1 clusters from subgenomes A and D contain at least one copy of KSL4, CPS4, and CYP99A2/3 (Figure 1b).
To investigate whether these MABGC‐like clusters in Triticeae function in plant pathogen resistance as rice MABGCs, the expression levels of the genes from those clusters in T. aestivum were profiled to explore their responses to pathogen infection (Figure S2). The expression levels of two KSL4, one CYP99A2/3, two CYP701A8, and one CPS4 from the cluster c2_2 of subgenome B increased dramatically in response to infection of Fusarium head blight (Fusarium graminearum), crown rot (Fusarium pseudograminearum), powdery mildew (Blumeria graminis), and tan spot (Pyrenophora tritici‐repentis), while almost no expression change was observed for the genes of the c2_2 clusters from subgenomes A and D (Figure S2). Compared with the c2_2 clusters, the three c2_1 clusters from three subgenomes displayed opposite responses upon pathogen infection, where the clusters from subgenomes A and D exhibited significantly increased gene expression, while almost no response was observed for cluster c2_1 from subgenome B (Figure S2). Comparing the gene compositions of the clusters from three subgenomes, c2_1 from subgenome B harbors additional CPS4 and CYP701A8 homologs and c2_2 from subgenome B includes no CYP99A2/3 (Figure 1b). Opposite responses of the clusters c2_2 and c2_1 from subgenome B and of those from subgenomes A and D upon infection of the same pathogens suggest functional redundancy of the two clusters, leading to differential loss of core genes in the clusters of different subgenomes and to retaining only a single copy of intact functional cluster (with CPS4, KSL4, and CYP99A2/3). In addition, within each functional cluster (c2_2 from subgenome B and c2_1 from subgenomes A and D), only a subcluster of genes functioned in response to pathogen infection (Figure S2). For example, of the 10 genes in the cluster c2_2 from subgenome B, only six continuously distributed genes (homologs: two KSL4, two CYP701A8, one CPS4, and one CYP99A2/3) responded to pathogen infection. Taken together, the MABGC‐like clusters in wheat showed the subgenome‐biased response to a range of pathogen stresses.
Lateral transfer of MABGC genes among grass
The divergence time between the BOP and PACMAD clades is more than 50 mya (Ma, Liu, et al., 2021). The distribution of MABGCs and MABGC‐like clusters in different grass species reveals complex evolutionary relationship among these clusters (Figure 1b). Whether the MABGC‐like clusters in Triticeae are related to the origin of MABGCs in grass has never been investigated. Thus, we performed phylogenetic analyses across the whole grass family to infer the evolution of core biosynthetic genes in MABCGs and MABGC‐like clusters and to trace the potential trajectory of MABGC evolution in grass.
Evolution of CPS4
CPS4, syn‐copalyl diphosphate synthase 4, catalyzes geranylgeranyl diphosphate (GGDP) into syn‐copalyl diphosphate (syn‐CPP), the first reaction of the momilactone biosynthesis pathway (Figure 1a). Based on the rooted phylogeny tree, the CPS4 homologs in MABGCs from Oryza, Echinochloa, and Eragrostis are clustered in a monoclade (MABGC clade), nested within CPS4 homologs from Pooideae (Figure 2a). The CPS4 homologs in Pooideae are mainly assigned separately in three lineages, corresponding to clusters c2_1, c2_2, and c5, of which the CPS4 homologs from cluster c2_2 is located at the basal position in the rooted phylogeny. CPS2 (LOC_Os02g36210) from c2BGC and CPS3 (LOC_Os09g15050) were identified as homologs of CPS4 (LOC_Os04g09900) in rice. Except the homologs from the MABGC clade and CPS3 clade, the CPS4 homologs in grass were found to have a congruent relationship as revealed by the species phylogeny, thus homologs at the subfamily level are grouped together and have conserved physical positions among Poaceae genomes (Figure 2b). CPS2 from O. sativa (N0 in Figure 2b, LOC_Os02g36210) is syntenic to Pl02g21350 (N1) from P. latifolius (Pharoideae, basal group in grass family), Zlat_10013826 (N3) from Zizania latifolia (Oryzoideae), LPERR02G17320 (N4) from Leersia perrieri (Oryzoideae), Ola021376 (N2) from Olyra latifolia (Bambusoideae), Et_1A_005482 (N5) from Eragrostis tef subgenome A (Chloridoideae), and eh_chr7.2132 (N6) from Echinochloa haploclada (Panicoideae). Therefore, both phylogeny and genomic synteny indicate that the CPS4 homologs of the MABGC monoclade are neither native copies nor duplicated paralogs of native copies (e.g., CPS2 in O. sativa) but instead possibly originated by LGT. It is noticed that CPS3 genes in Oryza are nested within PACMAD lineage, implying the possibility of another LGT event. To test the LGT hypothesis, we employed four topology test approaches on constrained trees, including resampling of estimated log‐likelihoods bootstrapping (bp‐RELL) method, Kishino–Hasegawa (KH) test, Shimodaira–Hasegawa (SH) test, and expected likelihood weight (ELW) test, to determine whether the LGT tree (tree 1 in Figure 2c) could statistically explain the data better than non‐LGT (native origin, tree 2 and tree 3) phylogenies. The result revealed that the LGT phylogeny of CPS4 was strongly supported and non‐LGT phylogenies were rejected (Test 1 in Figure 2c). Within the MABGC clade, CPS4 genes from three subfamilies (Oryzoideae, Panicoideae, and Chloridoideae) form three distinct groups (Figure 2a). We constructed constrained trees to test the topology robustness using Pooideae CPS4 homologs as outgroup genes and found that none of the three topologies could be rejected, suggesting that the variations in CPS4 homologs are not sufficient to decipher the phylogenetic relationship among MABGCs from Oryza, Echinochloa, and Eragrostis (Test 2 in Figure 2c).
Figure 2.
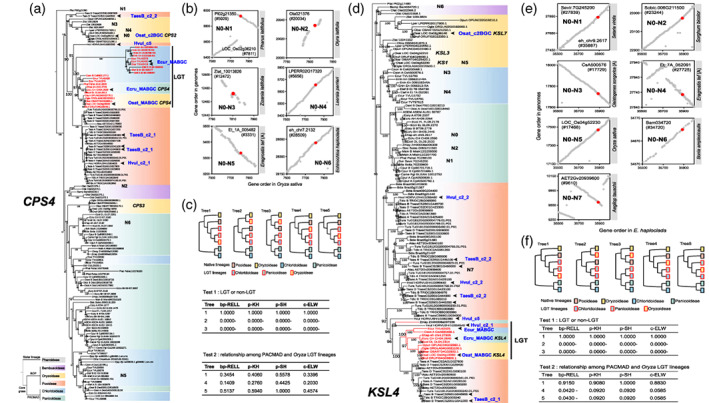
Phylogeny and genomic synteny analyses of CPS4 and KSL4 genes.
(a) Maximum‐likelihood phylogenetic tree of CPS4 and its homologs across the grass family. Homolog in Pharus latifolius is set as an outgroup. Different background colors represent different subfamilies. Homologs from momilactone A biosynthetic gene cluster (MABGC) clade are in red. Some homologs from MABGC or MABGC‐like clusters are highlighted by horizontal arrowheads.
(b) Genomic synteny among the conserved native CPS4 homologs. Red dots suggest that the two CPS4 homologs from two genomes are in good genomic synteny.
(c) Topology tests on five constrained trees. Top panel shows the topologies of constrained trees. Middle (Test 1) and bottom (Test 2) panels display the results of the two tests on the lateral gene transfer (LGT) event of CPS4 from Pooideae to PACMAD and the LGT from PACMAD to Oryza, respectively. bp‐RELL, bootstrap proportion using RELL (resampling of estimated log‐likelihoods) method; c‐ELW, confidence using expected likelihood weight method; p‐KH, p‐value of Kishino–Hasegawa test; p‐SH, p‐value of Shimodaira–Hasegawa test.
(d) Phylogenetic tree of KSL4 and its homologs.
(e) Genomic synteny among the conserved native KSL4 homologs.
(f) Topology tests on five constrained trees to test the robustness of the hypothesis of LGT for KSL4. Minus signs “−” in the topology tests represent that the corresponding topology could be rejected significantly (P < 0.05). [Colour figure can be viewed at wileyonlinelibrary.com]
Evolution of KSL4
KSL4, ent‐kaurene synthase‐like 4, cyclizes syn‐CPP into syn‐pimaradiene (Figure 1a). KSL4 genes from MABGCs form a highly supported monoclade nested within the Triticeae lineage (Figure 2d). The clade composed of KSL4 homologs from the c2_2 clusters of Pooideae is located at the base within the Pooideae lineage, and the KSL4 clade composed by KSL4 homologs in the c2_1 clusters is a sister to the MABGC clade. The genomic positions of two tandem duplicates KS1 and KSL3 from rice c2BGC are highly conserved in grass family with perfect genomic synteny among grass genomes (Figure 2e). KS1 (N5, LOC_Os04g52230) or KSL3 (LOC_Os04g52210) from O. sativa (Oryzoideae), Bam034720 (N6) from Bonia amplexicaulis (Bambusoideae), AET2Gv20939600 (N7) from Aegilop tauschii (Pooideae), CsA500576 (N3) from Cleistogenes songorica subgenome A (Chloridoideae), Et_7A_052091 (N4) from E. tef subgenome A (Chloridoideae), Sobic.006G211500 (N2) from Sorghum bicolor (Panicoideae), and Sevir.7G245200 (N1) from Setaria viridis (Panicoideae) are all syntenic to the native KSL4 homolog eh_chr9.2617 (N0) from E. haploclada (Panicoideae) (Figure 2e). KSL7 was integrated into rice c2BGC as a duplicate of KS1/KSL3. These results indicate that KS1 or KSL3 is a natively conserved KSL gene in the grass family, with the fundamental function of KS1 in synthesizing gibberellins (Miyamoto et al., 2016), and KSL4 is probably inherited from Triticeae by LGT, rather than gene duplication, convergent evolution, or in‐complete lineage sorting. Topology tests strongly support the LGT origination of KSL4 (Test 1 in Figure 2f). Within the LGT lineage, three clades from three subfamilies are formed. Topology tests based on bp‐RELL rejected the topology that KSL4 lineage in Oryza is sister to the common ancestor of Panicoideae and Chloridoideae lineages (Test 2 in Figure 2f), indicating that KSL4 in Oryza was likely derived from Panicoideae or Chloridoideae.
Evolution of CYP99A2/3
CYP99A2/3 functions as a C19 oxidase (Figure 1a). The CYP99A2/3 homologs are separated into two main lineages in the phylogeny tree, consistent with species phylogeny (PACMAD and BOP clades) (Figure S3a). In the BOP lineage, two monoclades (LGT1 and LGT2) are nested within CYP99A2/3 homologs from Pooideae. Monoclade LGT2 composed of genes from Panicoideae is a sister to one clade from Pooideae. Monoclade LGT1, including CYP99A2/3 genes in MABGCs, composed of three subclades from Oryza, Panicoideae and Chloridoideae, is a sister to the Triticeae clade containing CYP99A2/3 homologs in clusters c2_1 (Figure S3a). CYP99A2 and CYP99A3 in Oryza arose from tandem duplication after the divergence of the AA/BB and FF Oryza genomes. Besides Echinochloa, the CYP99A2/3 genes were found in other species in Panicoideae and particularly expanded in Setaria. In Chloridoideae, the copies of CYP99A2/3 were expanded in Eragrostis. Topology tests strongly support that clade LGT1 arose from Pooideae lineage (Test 1 in Figure S3b), but the phylogenetic relationship among clades from the three subfamilies within the LGT1 clade could not be clearly deciphered (Test 2 in Figure S3b).
Evolution of MAS1/2
MAS1/2 catalyzes the oxidation of 3β‐hydroxy‐syn‐pimaradien‐19,6β‐olide to form the characteristic C3 keto group in rice (Figure 1a). MAS1 and MAS2 genes are tandem duplicates in Oryza, nested within PACMAD linage, and are sisters to the clade from Chloridoideae (Figure S4a). MAS3 genes in grass form a congruent topology just like that in the subfamily‐level species phylogeny. Their genomic positions are conserved across grass genomes (Figure S4b). The conserved homologs of MAS1/2 are absent in Pooideae and Bambusoideae, indicating that MAS1/2 homologs are specific to the PACMAD clade. Topology tests revealed that MAS1/2 in Oryza were derived from PACMAD via LGT (Figure S4c).
Based on the above phylogenetic and comparative genomics analyses of the core genes in MABGCs across grass genomes, we propose that the genomic position of KSL4 homolog in the cluster c2_2 of Pooideae, which is syntenic to KS1 in rice, is the eventual origin of MABGC‐like clusters in Pooideae, such as cluster c5 and cluster c2_1 formed by assembly of KSL4 homologs with CPS4 and CYP99A2/3 (Figure 1b). MABGC‐like cluster was then passed on to the common ancestor of Panicoideae and Chloridoideae by LGT, where the MABGC, with the indispensable core genes required for momilactone biosynthesis, was formed by further integration of MAS1/2 (Figure 3). The Oryza MABGCs were acquired from Panicoideae or Chloridoideae via another LGT event because the genes from the Oryza MABGC are sisters to or nested within PACMAD homologs and topology tests (e.g., bp‐RELL) on KSL4 and MAS1/2 homologs rejected the topology that Oryza genes are sisters to common ancestors of Panicoideae and Chloridoideae (Figure 2f, Figure S4c).
Figure 3.
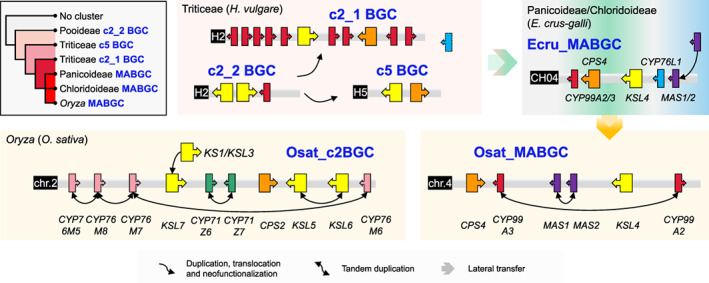
Proposed evolutionary trajectory of clusters of momilactone A biosynthetic genes (MABGCs) in grass.
BGC structures are illustrated for representative species (Hordeum vulgare from Pooideae or Triticeae, Echinochloa crus‐galli from PACMAD clade, Oryza sativa from Oryza or Oryzoideae). Cluster c2_2 in Pooideae or Triticeae is the eventual origin of MABGCs. Clusters c2_1 and c5 are originated through gene duplication and translocation of cluster c2_2. Subsequently, the ancient c2_1 or c5 (MABGC‐like) cluster was transferred into the PACMAD clade via lateral gene transfer, where the MABGC‐like recruited CYP76L1 and MAS1/2, leading to the birth of MABGC. Oryza species acquire MABGC from PACMAD clade via lateral gene transfer, followed by loss of CYP76L1. Oryza MABGC is not the duplicate of c2BGC, another diterpenoid BGC for the biosynthesis of phytocassane. Recurrent tandem duplication took place during the evolution of MABGC and c2BGC in rice. [Colour figure can be viewed at wileyonlinelibrary.com]
Different to the origination of MABGC, c2BGC in Oryzoideae was formed via gene duplication, translocation, and neofunctionalization, rather than LGT (Figure 3). CPS2 is conserved across the whole grass family (Figure 2) and is the ancestor of CPS4 in MABGC. KSL7 was translocated along with CPS2 as a duplicate of KS1/KSL3 (Figure 3). CYP76M7 and CYP76M8 were recently duplicated in Oryza (Figure S5), with CYP76M7 playing essential role in phytocassane biosynthesis and CYP76M8 being mainly responsible for momilactone production (Kitaoka et al., 2021). CYP71Z6 and CYP71Z7 are tandem duplicates, phylogenetically neighboring to four CYP71Z genes from c7BGC (a cluster on chromosome 7 associated with the production of the casbane‐type diterpenoid phytoalexin ent‐10‐oxodepressin) (Liang et al., 2021; Zhan et al., 2020) (Figure S6). KSL5 and KSL6 are Oryza‐specific tandem duplicates, phylogenetically neighboring to KSL8 (responsible for the biosynthesis of Oryzalexin S) and KSL10 (responsible for the biosynthesis of Oryzalexins A–F) (Miyamoto et al., 2016; Nemoto et al., 2004) (Figure S7).
Comparison of MABGCs between Oryza and Echinochloa species
In comparison with Oryza MABGCs, Echinochloa MABGCs have an extra copy of cytochrome P450 gene CYP76L1 (Figure 1b). The phylogenetic tree of CYP76L1 homologs is composed of two major lineages, in line with species phylogeny (BOP and PACMAD) (Figure S8a). CYP76L1 genes in MABGCs are nested within the Pooideae lineage and found in the genomes of Setaria and Panicum. Genome synteny was used to rule out the possibility of convergent evolution and incomplete lineage sorting, which could result in the discordance between gene topology and species phylogeny. Taking O. sativa genome as a reference, we scanned the genomic synteny around the native CYP76L1 regions among grass genomes (Figure S8b). Lper_LPERR09G08730 (N1) in Leersia perrieri (Oryzoideae), eh_chr3.2603 (N2) in E. haploclada (Panicoideae), Et_2A_016179 (N3) in the E. tef subgenome A (Chloridoideae), Ola025389 (N4) from O. latifolia (Bambusoideae), and AET5Gv20559300 (N5) from A. tauschii (Pooideae) are all highly syntenic to LOC_Os09g27500 (N0) from O. sativa, indicating that Paniceae CYP76L1 genes nested within Pooideae genes in the gene tree were acquired via LGT as extra copies of the native CYP76L1. Topology tests strongly support the LGT origination of CYP76L1 in Echinochloa MABGCs from Pooideae (Figure S8c). To distinguish the two types of CYP76L1, we named the native CYP76L1 homologs and CYP76L1 acquired by LGT as CYP76L1‐native and CYP76L1‐lgt, respectively. CYP76L1‐native (LOC_Os9g27500), but not CYP76L1‐lgt, is present in the O. sativa genome, while both CYP76L1‐native and CYP76L1‐lgt are present in Echinochloa MABGCs (Figure 1b, Figure S8a). Given the presence of CYP76L1 in barnyardgrass MABGC and its strong LGT signal based on the phylogenetic tree, we speculate that CYP76L1 was transferred from Pooideae to PACMAD along with the MABGC core genes as a cluster, and subsequently lost in Oryza (Figure 3).
In barnyardgrass (E. crus‐galli), CYP76L1‐lgt is co‐expressed with other MABGC genes (Sultana et al., 2019). A recent study in rice demonstrated that CYP76L1‐lgt from barnyardgrass is functionally equivalent to CYP76M8 in catalyzing the 6β‐hydroxylation of syn‐pimaradiene (Kitaoka et al., 2021), implying a function of CYP76L1‐lgt in momilactone biosynthesis. CYP76L1‐native may not participate in momilactone production. First, gene co‐expression network analysis revealed that CYP76L1‐native is not a component of the momilactone production pathway in rice (Figure 4a). Second, upon treatment of jasmonic acid (JA), the content of momilactone in rice roots was highly induced because of increased expression of the genes of the momilactone‐biosynthesis‐related network, but no upregulation of CYP76L1‐native was found (Figure 4a, Figure S9a).
Figure 4.
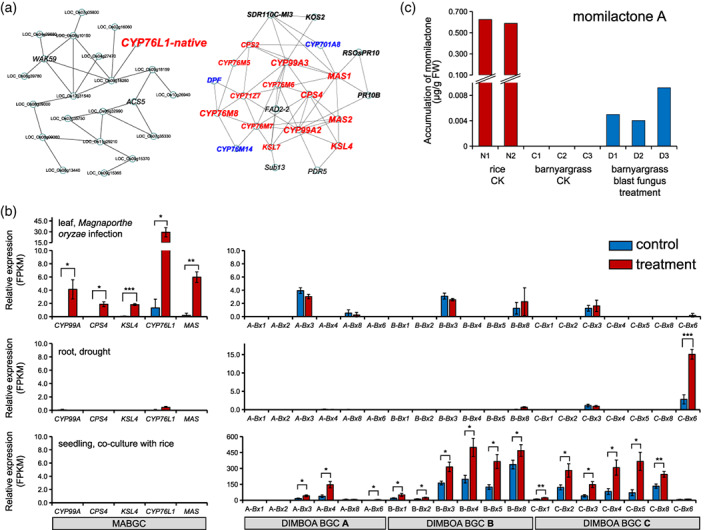
Expression profiling of clusters of momilactone A biosynthetic genes (MABGCs) from rice (Oryza sativa) and barnyardgrass (Echinochloa crus‐galli).
(a) Gene co‐expression network of native CYP76L1 (CYP76L1‐native) reveals the independent relationship between CYP76L1‐native and the momilactone biosynthetic genes in rice.
(b) Transcriptomic profiling of barnyardgrass (E. crus‐galli) MABGC and the gene clusters for DIMBOA biosynthesis (Bx clusters and Bx6 genes from three subgenomes) in response to blast pathogen Magnaporthe oryzae inoculation, drought, and co‐culture with rice. *P < 0.05; **P < 0.01; ***P < 0.001, Student's t‐test.
(c) Comparison of momilactone A accumulation in leaves from rice (O. sativa) and barnyardgrass (E. crus‐galli) under control (CK) and blast fungus M. oryzae infection conditions. N1–N2, C1–C3, and D1–D3 are biological replicates of the three experiments. [Colour figure can be viewed at wileyonlinelibrary.com]
Infection by blast pathogen Magnaporthe oryzae induces the production of momilactone in rice (Hasegawa et al., 2010), likely due to upregulation of MABGC genes (except MAS2) (Figure S9b). In leaves of barnyardgrass (E. crus‐galli), consistent with significant upregulation (P < 0.05) of MABGC genes, including CYP76L1‐lgt (Figure 4b), the content of momilactone A was also significantly induced by M. oryzae infection, but its level was approximately 100 times lower than that observed in rice leaves without pathogen infection (Figure 4c, Figure S10).
Remarkably, barnyardgrass is one of the most detrimental weeds in paddy fields (Ye et al., 2020). Momilactone secreted by rice roots plays a critical role in rice allelopathy, by which growth of barnyardgrass in the neighboring environments of rice but not the rice plants themselves is inhibited by momilactone because barnyardgrass is much more sensitive to momilactone than rice (Kato‐Noguchi & Peters, 2013). Barnyardgrass induces rice to secrete momilactone but the barnyardgrass‐induced rice allelopathy is not associated with the MABGC in barnyardgrass, as no genes from the barnyardgrass MABGC were differentially regulated under the rice and barnyardgrass co‐planting conditions (Figure 4b). Instead, genes in the barnyardgrass DIMBOA cluster, involved in the production of benzoxazinoids secondary metabolites, were significantly upregulated under the co‐planting conditions (Figure 4b) (Guo et al., 2017). While the DIMBOA cluster is only approximately 359 kb away from the MABGC in the E. crus‐galli genome (Figure S11), the two barnyardgrass BGCs are not co‐expressed (Sultana et al., 2019) and have distinct functions based on their expression profiles responding to pathogen infection and co‐cultivation with rice (Figure 4b), with the MABGC being involved in the response to pathogen infection and the DIMBOA cluster involved in the allelopathic interaction.
DISCUSSION
Evolution of MABGCs in grass
Gene duplication, neo‐functionalization and translocation have been considered as the primary routes to BGC assembling (Nützmann et al., 2018). The origination of MABGC has been extensively studied and discussed, particularly after the discovery of MABGCs in E. crus‐galli and bryophyte C. plumiforme (Guo et al., 2017; Kitaoka et al., 2021; Mao et al., 2020; Miyamoto et al., 2016; Peters, 2020; Smit & Lichman, 2022; Zhang & Peters, 2020). MABGC in bryophyte C. plumiforme was evolved independently and convergently (Mao et al., 2020) and it was speculated that the rice MABGC emerged within Oryza, first by the addition of CYP99A2/3 to the syntenic locus, followed by recruitment of CPS4, KSL4, and MAS1/2 (Miyamoto et al., 2016). MABGC in Echinochloa was considered to be transferred from Oryza presumably through hybridization and introgression (Peters, 2020; Smit & Lichman, 2022; Zhang & Peters, 2020). Our results appear not to support the introgression‐origin hypothesis for the Echinochloa MABGC. First, the divergence time between Echinochloa and Oryza was more than 50 mya (Ma, Liu, et al., 2021). The genomic incompatibility would prevent pairing of their chromosomes and inter‐hybridization between Pooideae and Panicoideae is impossible without embryo rescues (Mahelka et al., 2021). Secondly, besides Echinochloa species, we also identified MABGCs in weeping lovegrass Eragrostis curvula from Chloridoideae, and non‐clustered MABGC genes in other PACMAD species (e.g., CPS4 in Cleistogenes songorica, Eragrostis tef, Eragrostis nindensis, and Panicum hallii; KSL4 in Cleistogenes songorica) (Figure 2, Table S2). The homologous genes of each of the MABGC core genes form a monoclade. The distribution pattern implies that MABGC genes have arisen in the common ancestor of PACMAD and subsequently lost some individual gene(s) differentially. Thirdly, the MABGC genes in Oryza are not clustered with native Oryza genes but nested in Triticeae or PACMAD lineage, which rejects the hypothesis that rice MABGC was assembled within Oryza by duplication and relocation (Figure 2). Further phylogeny, genome synteny and topology tests support the transfer of MABGC from PACMAD to Oryza.
We propose a novel evolutionary model for grass MABGCs, in which lateral transfer acts as the main force driving the dispersal of MABGCs in the grass family (Figure 3), because by integrating phylogeny, genome synteny, topology tests, and other evidence, the possibilities of sexual hybridization, incomplete lineage sorting or convergent evolution could all be ruled out. The model includes two LGT events, the transfer of MABGC‐like clusters (including CPS4, KSL4, and CYP99A2/3) from Triticeae to PACMAD and another one from PACMAD to Oryza with the addition of MAS1/2. Transcriptomic analysis showed the responsiveness of the MABGC‐like clusters (c2_1 and c2_2) in Triticeae upon pathogen stress (Figure S2). Notably, a recent study by Polturak et al. (2022) has found that cluster c2_2 from subgenome B (i.e., cluster 2[2B] by Polturak et al. 2022) and clusters c2_1 from subgenomes A and D (i.e., cluster 1[2A]/1[2D] by Polturak et al. 2022) produce isopimara‐7,15‐diene‐derived diterpenoids and pimara‐8(14),15‐diene‐derived diterpenoids from GGDP (the substrate of CPS4 in the momilactone pathway), respectively. The functional diversification between MABGC‐like clusters and MABGCs is related to their differences in gene composition (Figure 1b), suggesting innovations of BGCs by addition and/or deletion of different enzymes. The second LGT is presumed to lead to the direct acquisition of MABGCs in Oryza. MAS1/2 genes were likely assembled into MABGCs in ancestral PACMAD. Interesting, the biosynthesis of momilactone in rice is dramatically more active than that in E. crus‐galli (Figure 4c, Figure S10). Copy number variations in MAS1/2 and CYP99A2/3 between the two species may contribute to their differences in momilactone biosynthesis and accumulation, and the catalytic efficiency difference between CYP76M8 and CYP76L1‐lgt may be another factor, which requires future experimental validations. Whether the loss of CYP76L1‐lgt was due to genetic drift or the competing advantage from CYP76M8 in catalyzing synpimaradiene‐19‐oic to 19,6β‐lactone could not be determined so far.
LGT events have been observed in grass species (Dunning et al., 2019; Hibdige et al., 2021; Mahelka et al., 2021; Park et al., 2021; Wu, Jiang, et al., 2022). Recently, Hibdige et al. (2021) performed an Poaceae‐scale LGT detection using coding sequences from 17 grass genomes and transcriptomic data. While, in total, 135 LGT candidates were identified, the MABGC genes identified in this study and some other known LGT fragments (Dunning et al., 2019; Wu, Jiang, et al., 2022) were not included, likely due to the limited number of genomes sampled and use of stringent filtering criteria. It was hypothesized that LGT is prevalent in perennial and rhizomatous species benefiting from their vegetative propagation (Hibdige et al., 2021; Mahelka et al., 2021). However, most MABGC‐recipient grass species used in the present study are annual and non‐rhizomatous. To have a solid conclusion on the prevalence of LGT, further investigation is required by including more species with different characteristics (only three rhizomatous grass genomes used by Hibdige et al., 2021). Ours and other studies indicate that the sizes of LGT fragments are sometimes large, e.g., a DNA fragment containing multiple genes and even a BGC (Dunning et al., 2019; Mahelka et al., 2021; Wu, Jiang, et al., 2022). In addition, a large Panicum‐derived fragment (>200 kb), harboring several stress‐related protein‐coding genes, was found in the genomes of wild Hordeum species (Mahelka et al., 2021); and the fragments including the Bx clusters in Triticeae were reported to be acquired from ancestral Panicoideae via LGT (Wu, Jiang, et al., 2022).
Potential mechanisms of LGT
How DNA fragments transfer between two phylogenetically distant plant species is still debatable. Here we assigned the LGT mechanisms that have been proposed so far into two categories: direct‐contact and vector‐mediated. Direct‐contact mechanisms include parasitism, illegitimate pollination, and grafting. The most commonly described direct contact pathway is parasitism, for instance, a sorghum gene has been reported to be moved into Striga hermonthica, a eudicot parasite weed infecting many grass species (Yoshida et al., 2010). No parasitic grass has been reported so far, so it is unlikely that the LGT observed in this study is achieved by this pathway. Illegitimate pollination has been discussed widely, such as the acquisition of C4 genes of Alloteropsis in the grass family (Christin et al., 2012). Essentially, illegitimate pollination is an extremely fortuitous sexual hybridization between two divergent species. In the present case, hybridization between PACMAD and BOP species, which diverged more than 50 mya, seems unlikely, although the possibility could not be ruled out because partial hybrids between PACMAD and BOP species have indeed been generated under controlled conditions (Riera‐Lizarazu et al., 1996). The absence of vascular cambium and a scattered arrangement of vascular bundles in grass are thought to preclude grafting among grass species (Melnyk & Meyerowitz, 2015). A recent work has overturned the consensus that vascular cambium is a prerequisite for graft formation in plants and proved that embryonic hypocotyl allows grafting in most monocotyledonous orders, including grasses (Reeves et al., 2022). Although the study by Reeves et al. was conducted under controlled conditions and there are limitations to the emergence of graft union, the root‐to‐root interactions could provide opportunities for natural grafting in grasses, just like in trees (Gaut et al., 2019; Melnyk & Meyerowitz, 2015).
Vector‐mediated LGT could be facilitated by parasitic plants, insects, or pathogens. Even when both the donor and recipient species are not parasitic, their shared parasitic plants could act as intermediate LGT vectors. The recurrent DNA exchanges between hosts and their parasitic plants in the long‐term evolution period offer a possible route to the transfer of genetic materials between divergent species. Transfers of DNA between insect or fungi and plants have been reported recently. Whitefly has acquired the BtPMaT1 gene from a host plant, enabling it to neutralize plant toxin phenolic glucosides (Xia et al., 2021). Similarly, the Fhb7 gene of the fungus Epichloë was transferred to Thinopyrum wheatgrass (Triticeae), providing resistance to Fusarium head blight and crown rot in wheat (Wang et al., 2020). The mutual transfers of DNA between vectors and plants imply that the vectors have built a DNA‐transfer bridge to overcome the barrier preventing exchange of genetic materials between divergent species. By integrating more genomes across kingdoms, it is expected that the footprints of vector‐mediated LGT would be discovered.
Relationship of multiple BGCs in the same genome
BGCs are not only widely distributed in the plant kingdom, but also with multiple copies encoding diverse metabolites in a single genome. How multiple BGCs within the same genome interact has been little investigated and our study provides certain new insights on it.
In polyploid genomes, homeologous genes often show biases in expression, selection and/or epigenetic modification (Cheng et al., 2018; Ye et al., 2020). The biased expression and selection observed in homeologous BGC genes in polyploids may be due to their functional redundancy. Genes of the Bx cluster from subgenome B of hexaploid bread wheat (T. aestivum) show dominant expression (Nomura et al., 2005). In E. crus‐galli, genes of the Bx cluster from subgenome AH are suppressed and display relaxed purifying selection, leading to loss of more genes compared with its two homeologous Bx clusters (Wu, Jiang, et al., 2022). In this study, we found that each subgenome of the hexaploid bread wheat (T. aestivum) contains a cluster c2_1 and a cluster c2_2. Gene expression profiling reveals predominance of cluster c2_2 from subgenome B and clusters c2_1 from subgenomes A and D in response to pathogen infection (Figure S2). In subgenome B, the cluster c2_2 is relatively intact (with CPS4 and CYP701A8 homologs) compared with the other two copies of cluster c2_2, while cluster c2_1 is fragmentary (without CYP99A2/3 genes) compared with the other two homeologous copies (Figure 1b). The presence–absence variation pattern of the core genes of the wheat MABGC‐like clusters is in line with the gene expression profiles in response to pathogen infection. Generally, BGCs with multiple copies in a polyploid genome display biased expression in response to pathogen stress and biased integrity in the composition of core genes. The suppressed copy tends to lose more core genes under less constrained selection. In short, genome polyploidization provides more opportunities for BGC diversification and innovation.
In rice, both c2BGC and MABGC encode enzymes catalyzing the reactions of biosynthesizing labdane‐related diterpenoids (phytocassane by c2BGC and momilactone by MABGC) (Miyamoto et al., 2016). Although the two BGCs are located separately on different chromosomes, CYP76M8 from c2BGC is recruited to the momilactone biosynthetic pathway, indicating interdependent evolution and co‐operation between c2BGC and MABGC (Kitaoka et al., 2021). Most genes in the two BGCs also display a similar expression pattern in response to pathogen infection or JA treatment (Figure S9), despite their different origination revealed by family‐scale phylogenetic analysis. Unlike MABGC that originates from two LGT events, c2BGC was assembled within the Oryza genus at the native genomic position of CPS2, which is conserved across the grass family (Figure 2a), with subsequent recruitment of other genes (e.g., KSL7) via gene duplication, neofunctionalization, and relocation. Owing to the putative loss of CYP76L1 inherited from PACMAD in rice MABGC, MABGC has cooperated with c2BGC by employing CYP76M8 and displaying gene co‐expression. However, the co‐expression pattern is not observed for clusters c2_1 and c2_2 in wheat, although both clusters are terpene‐related and located on the same chromosome (Polturak et al., 2022). In each of the three wheat subgenomes, one cluster is dramatically activated by pathogen stress while another remains silent almost all the time (Figure S2), suggesting independence of the two clusters. The polyploidization of wheat has complicated the relationship among the clusters from different subgenomes. Generally, most genes of cluster c2_2 from subgenome B and cluster c2_1 from subgenomes A and D display co‐regulation (Figure S2). Why clusters from one subgenome selectively keep silenced or sensitive to stress awaits further investigations.
Most characterized BGCs so far are located independently (Guo et al., 2018; Zhan et al., 2022). However, in opium poppy (Papaver somniferum) genome, 15 genes have been assembled into a compact super‐BGC called BIA cluster (a total length of 584 kb), which encodes enzymes of two distinct pathways related to biosynthesis of noscapine and morphinan (Yang et al., 2021). The two BGCs display coordinated gene expression and regulation. Similarly, in a lineage of Echinochloa, while MABGC is neighboring to the Bx cluster (Figure S11), the two clusters produce distinct metabolites (momilactone and benzoxazinoids, respectively). No co‐expression was observed for the genes of MABGC and Bx (Sultana et al., 2019), in line with their different stress responses (Figure 4b). It is noticed that the distance between the MABGC and Bx cluster in polyploid genomes (359 kb in E. crus‐galli and 515 kb in E. colona) has become much shorter than that in diploid E. haploclada (975 kb) (Figure S11). Whether the clustering of the two BGCs is a result of selective advantage or just genetic drift remains unclear. The burst of sequenced plant genomes provides opportunities to investigate more super‐BGCs and the potential mechanisms underlying clustering of super‐BGCs.
CONCLUSIONS
By integrating gene phylogeny and comparative genomics analyses using 40 monocot genomes, we showed that intact MABGCs are present in Oryza from Oryzoideae (BOP clade), Echinochloa from Panicoideae (PACMAD clade), and Eragrostis from Chloridoideae (PACMAD clade), and propose the evolution trajectory of MABGCs in grass, i.e., MABGCs in PACMAD had arisen from LGT from MABGC‐like clusters in Triticeae followed by further integration of MAS genes, which was then acquired by Oryzoideae via another LGT event. Our study demonstrates essential roles of LGT in the origination, dispersal, and innovation of plant BGCs.
EXPERIMENTAL PROCEDURES
Homolog identification of biosynthetic genes
The protein sequences of momilactone and phytocassane biosynthetic genes of O. sativa (CPS4, LOC_Os04g09900; KSL4, LOC_Os04g10060; CYP99A2/3, LOC_Os04g09920, and LOC_Os04g10160; MAS1/2, LOC_Os04g10000 and LOC_Os04g10010; KSL5/6, LOC_Os02g36220 and LOC_Os02g36264; CYP76M5/6/7/8, LOC_Os02g36030, LOC_Os02g36280, LOC_Os02g36110, and LOC_Os02g36070; CYP71Z6/7, LOC_Os02g36150 and LOC_Os02g36190; CYP701A8, LOC_Os06g37300) and of E. crus‐galli (CYP76L1, CH04.2641) were set as reference baits. For the genes with multiple transcripts, the longest ones were selected. BLASTP was performed using the reference baits against more than 1.95 million protein sequences from the genomes of 39 grass species and A. comosus (Table S1). Raw homologs were selected with the filtering criteria of BLAST e‐value less than 1e‐30 and identity greater than 50%. Raw homologs were aligned using MAFFT (v7.310) (Katoh & Standley, 2013) and the phylogenetic trees were built using IQ‐TREE (v1.6.6) under the best substitution model with 1000 replicates for bootstrap (Nguyen et al., 2015). Using A. comosus and P. latifolius as outgroup species, the final repository of MABGC homologs were determined according to the phylogeny and used in the following analysis.
Phylogeny analysis
Homologous sequences of each biosynthetic gene were re‐aligned using mafft (v7.310) and trimmed using gblocks (v0.91b) with the parameter “‐b4 = 5 ‐b5 = h” (Castresana, 2000). The best substitution model for each trimmed alignment was determined by ModelFinder and the phylogenetic tree was constructed by iq‐tree (v1.6.6) for 1000 bootstrap replicates (Nguyen et al., 2015). Constrained trees were searched and built in iq‐tree (v1.6.6) and topology tests on them were performed with 10 000 times of bootstrapping using four approaches, including resampling of estimated bp‐RELL method, KH test, SH test, and ELW test (Kishino et al., 1990; Kishino & Hasegawa, 1989; Shimodaira & Hasegawa, 1999; Strimmer & Rambaut, 2002). The KH and SH tests return P‐values, a tree is rejected if P < 0.05 (marked with a “−” sign). Tests bp‐RELL and confidence‐ELW return posterior weights, which are not P‐value and the weights sum up to 1 across all the trees tested.
Genomic synteny
Protein sequences from the grass genomes were compared pairwise using BLASTP and the best hits with thresholds of e‐value less than 1e‐30 and identity greater than 50% were kept. According to their physical positions in each genome, the genes were ordered. Each blast best‐hit had a pair of coordinates and the gene‐to‐gene synteny was plotted pairwise for grass genomes.
Gene expression analysis
For hexaploid wheat (T. aestivum) gene expression profiling, the quantified relative expression values (TPM, Transcripts Per kilobase of exon model per Million mapped reads) of genes in different tissues and under different pathogen infections (Fusarium head blight pathogen Fusarium graminearum, crown rot pathogen Fusarium pseudograminearum, powdery mildew pathogen Blumeria graminis, and tan spot pathogen Pyrenophora tritici‐repentis) were obtained from WheatOmics 1.0 database (http://wheatomics.sdau.edu.cn/) (Ma, Wang, et al., 2021) (Table S2). For barnyardgrass (E. crus‐galli), the RNA‐seq datasets under the treatment of co‐culture with rice seedlings (Guo et al., 2017), infection by blast fungus M. oryzae and drought induced by polyethylene glycol (Ye et al., 2020) were mapped against the latest version of E. crus‐galli reference genome STB08 (Wu, Shen, et al., 2022) and quantified using tophat (v2.1.1) and cufflinks (v2.2.1) (Trapnell et al., 2012). For rice (O. sativa), the co‐expression network was built in the RiceFREND database (https://ricefrend.dna.affrc.go.jp/) (Sato, Namiki, et al., 2013) and the expression data from samples subjected to JA treatment, drought stress, and blast fungus M. oryzae infection were obtained from RiceXPro (https://ricexpro.dna.affrc.go.jp/) (Sato, Takehisa, et al., 2013) and Plant Public RNA‐seq Database (http://ipf.sustech.edu.cn/pub/ricerna/) (Yu et al., 2022).
Momilactone A quantification
Echinochloa leaves infected by blast fungus or mock treatment were used in the quantification of momilactone A. Each treated sample (roughly 100 mg) was submerged in 4 ml of 80% methanol at 4°C for 24 h, and 5 μl of the extract was subjected to liquid chromatography–tandem mass spectrometry analysis using API‐3000 with an electrospray ion source (Applied Biosystems Instruments, Foster City, CA, USA) and an Agilent 1100 high‐performance liquid chromatography instrument (Agilent Technologies, Palo Alto, CA, USA) equipped with a PEGASIL C18 column (150 mm long, 2.1 mm in diameter; Senshu Scientific, Tokyo, Japan) with the selected reaction monitoring transitions (for momilactone A, m/z 315/271), as described previously (Miyamoto et al., 2016). Extracts from fresh rice leaves (Nipponbare) and momilactone A authentic sample were quantified and analyzed using the same procedure. In the chromatograms, retention times for extracts from treated barnyardgrass leaves and rice leaves matched with that of momilactone A standard (Figure S10).
AUTHOR CONTRIBUTIONS
LF and DW conceived the research. DW and YH performed the data analysis. SA, HN, and KO performed the experimental quantification of momilactone A in rice and barnyardgrass. C‐YY, KO, Q‐HZ, LG, and LF discussed the findings. Q‐HZ and LF edited the manuscript. DW wrote the manuscript. All authors read and contributed to the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
Supporting information
Table S1. A list of the plant genomes used in this study.
Table S2. Homologs of the key genes (CPS4, KSL4, CYP99A2/3, CYP76L1, MAS1/2, and CYP701A8) involved in momilactone biosynthesis in grass species.
Figure S1. Phylogeny and genomic synteny of CYP701A8 homologs in grass. Genes in different subfamilies are marked in different color backgrounds. Genes used in synteny analysis are marked in the phylogenetic tree from N0 to N3. Homologs in Triticeae subgenomes B and four CYP701A8 homologs in O. sativa are highlighted. The red syntenic dots represent pairs of native homologs between genomes.
Figure S2. Transcriptomic profiling of wheat genes in MABGC‐like clusters under pathogen infections. Genes in different colors are from different subgenomes (blue, subgenome A; red, subgenome B; green, subgenome D). Homolog information is shown by red (CYP99A2/3), yellow (KSL4), green (CYP701A8), and orange (CPS4). Dataset1, the spikelet (SP) and rachis (RACH) from two wheat accessions 2618 and 2890 were infected by Fusarium head blight (Fusarium graminearum) (FG) and water (control). Dataset2, coleoptile sheaths of wheat accession Chara were infected by crown rot (Fusarium pseudograminearum) (Fp). Dataset3, leaves from accession N9134 were inoculated by powdery mildew (Blumeria graminis). Dataset4, leaves from Glenlea and Salamouni were infected by tan splot (Pyrenophora tritici‐repentis). The quantified gene expression (TPM) levels were obtained from WheatOmics 1.0 (Ma, Wang, et al., 2021).
Figure S3. Phylogeny of CYP99A2/3 homologs in grass and topology tests. (a) Homolog phylogeny of CYP99A2/3 homologs. Genes in different subfamilies are marked in different color backgrounds. Cluster information of some homologs in Triticeae and MABGCs are suggested. (b) Topology tests. Three (Tree 1, 2, 3) and three (Tree 1, 4, 5) constrained trees were set for Test 1 and Test 2, respectively. Minus signs “−” represent that the corresponding topology could be rejected significantly (P < 0.05).
Figure S4. Phylogeny and genomic synteny of MAS1/2 homologs in grass and topology tests. (a) A maximum‐likelihood tree of MAS1/2 and homologs across the grass family. The homolog in A. comosus is set as an outgroup. Different background colors represent different subfamilies. (b) Genomic synteny among the native MAS3 homologs. Red dots represent that the two MAS3 homologs from two genomes are in good synteny. (c) Topology tests on three constrained trees. The top panel shows the topologies of constrained trees used in tests. The bottom panels show the results of the test on the LGT event of MAS1/2 from PACMAD to Oryza. Minus signs “−” represent that the corresponding topology could be rejected significantly (P < 0.05).
Figure S5. A maximum‐likelihood phylogenetic tree of CYP76M5/6/7/8 homologs in grass. Different background colors represent different subfamilies. The branch containing CYP76M5/6/7/8 from O. sativa is zoomed in.
Figure S6. A maximum‐likelihood phylogeny of CYP71Z6/7 homologs in grass. Different background colors represent different subfamilies. The branch containing CYP71Z genes from O. sativa is zoomed in and their cluster information (c2BGC and c7BGC) is shown.
Figure S7. A maximum‐likelihood phylogeny of KSL5/6 homologs in grass. Different background colors represent different subfamilies.
Figure S8. Phylogeny and genomic synteny of CYP76L1 homologs in grass and topology tests. (a) A maximum‐likelihood tree of CYP76L1 and its homologs across the grass family. Different background colors represent different subfamilies. Genes used in synteny analysis are marked in the phylogenetic tree from N0 to N5. (b) Genomic synteny among the native CYP76L1 homologs. Red dots represent that the two homologs from two genomes are in good synteny. (c) Topology tests on two constrained trees. The top panel shows the topologies of constrained trees under tests. The bottom panels show the test results on the LGT of CYP76L1 from Pooideae to Panicoideae. Minus signs “−” represent that the corresponding topology could be rejected significantly (P < 0.05).
Figure S9. Expression of MABGC, c2BGC, and related genes in rice under JA treatment (a), drought and rice blast fungus M. oryzae infection (b).
Figure S10. Liquid chromatography–tandem mass spectrometry (LC‐MS/MS) analyses of momilactone A in rice and barnyardgrass leaves. Extracts from fresh leaves of rice (O. sativa) Nipponbare and barnyardgrass (E. crus‐galli) STB08 under mock and blast fungus infection treatment were analyzed. Momilactone A was detected with the selected reaction monitoring (m/z 315/271).
Figure S11. The structures of MABGCs and DIMBOA Bx clusters on chromosomes 4 in nine Echinochloa subgenomes.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation (31971865 and 32170621), the Department of Science and Technology of Zhejiang Province (2022C02032), Zhejiang Natural Science Foundation (LZ17C130001), and Jiangsu Collaborative Innovation Center for Modern Crop Production.
DATA AVAILABILITY STATEMENT
Genome accession numbers or versions used in this study are given in supplementary Table S1.
REFERENCES
- Castresana, J. (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 17, 540–552. [DOI] [PubMed] [Google Scholar]
- Cheng, F. , Wu, J. , Cai, X. , Liang, J. , Freeling, M. & Wang, X. (2018) Gene retention, fractionation and subgenome differences in polyploid plants. Nature Plants, 4, 258–268. [DOI] [PubMed] [Google Scholar]
- Christin, P.‐A. , Edwards, E.J. , Besnard, G. , Boxall, S.F. , Gregory, R. , Kellogg, E.A. et al. (2012) Adaptive evolution of C4 photosynthesis through recurrent lateral gene transfer. Current Biology, 22, 445–449. [DOI] [PubMed] [Google Scholar]
- De La Peña, R. & Sattely, E.S. (2021) Rerouting plant terpene biosynthesis enables momilactone pathway elucidation. Nature Chemical Biology, 17, 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunning, L.T. , Olofsson, J.K. , Parisod, C. , Choudhury, R.R. , Moreno‐Villena, J.J. , Yang, Y. et al. (2019) Lateral transfers of large DNA fragments spread functional genes among grasses. Proceedings of the National Academy of Sciences of the United States of America, 116, 4416–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field, B. & Osbourn, A.E. (2008) Metabolic diversification—independent assembly of operon‐like gene clusters in different plants. Science, 320, 543–547. [DOI] [PubMed] [Google Scholar]
- Frey, M. , Chomet, P. , Glawischnig, E. , Stettner, C. , Grün, S. , Winklmair, A. et al. (1997) Analysis of a chemical plant defense mechanism in grasses. Science, 277, 696–699. [DOI] [PubMed] [Google Scholar]
- Gaut, B.S. , Miller, A.J. & Seymour, D.K. (2019) Living with two genomes: grafting and its implications for plant genome‐to‐genome interactions, phenotypic variation, and evolution. Annual Review of Genetics, 53, 195–215. [DOI] [PubMed] [Google Scholar]
- Guo, L. , Qiu, J. , Li, L.‐F. , Lu, B. , Olsen, K. & Fan, L. (2018) Genomic clues for crop‐weed interactions and evolution. Trends in Plant Science, 23, 1102–1115. [DOI] [PubMed] [Google Scholar]
- Guo, L. , Qiu, J. , Ye, C. , Jin, G. , Mao, L. , Zhang, H. et al. (2017) Echinochloa crus‐galli genome analysis provides insight into its adaptation and invasiveness as a weed. Nature Communications, 8, 1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa, M. , Mitsuhara, I. , Seo, S. , Imai, T. , Koga, J. , Okada, K. et al. (2010) Phytoalexin accumulation in the interaction between rice and the blast fungus. Molecular Plant‐Microbe Interactions, 23, 1000–1011. [DOI] [PubMed] [Google Scholar]
- Hibdige, S.G.S. , Raimondeau, P. , Christin, P. & Dunning, L.T. (2021) Widespread lateral gene transfer among grasses. The New Phytologist, 230, 2474–2486. [DOI] [PubMed] [Google Scholar]
- Katoh, K. & Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato‐Noguchi, H. & Peters, R.J. (2013) The role of momilactones in rice allelopathy. Journal of Chemical Ecology, 39, 175–185. [DOI] [PubMed] [Google Scholar]
- Kishino, H. & Hasegawa, M. (1989) Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. Journal of Molecular Evolution, 29, 170–179. [DOI] [PubMed] [Google Scholar]
- Kishino, H. , Miyata, T. & Hasegawa, M. (1990) Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. Journal of Molecular Evolution, 31, 151–160. [Google Scholar]
- Kitaoka, N. , Zhang, J. , Oyagbenro, R.K. , Brown, B. , Wu, Y. , Yang, B. et al. (2021) Interdependent evolution of biosynthetic gene clusters for momilactone production in rice. Plant Cell, 33, 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kominek, J. , Doering, D.T. , Opulente, D.A. , Shen, X.X. , Zhou, X. , DeVirgilio, J. et al. (2019) Eukaryotic acquisition of a bacterial operon. Cell, 176, 1356–1366.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, J. , Shen, Q. , Wang, L. , Liu, J. , Fu, J. , Zhao, L. et al. (2021) Rice contains a biosynthetic gene cluster associated with production of the casbane‐type diterpenoid phytoalexin ent‐10‐oxodepressin. The New Phytologist, 231, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Cheema, J. , Vigouroux, M. , Hill, L. , Reed, J. , Paajanen, P. et al. (2020) Formation and diversification of a paradigm biosynthetic gene cluster in plants. Nature Communications, 11, 5354. 10.1038/s41467-020-19153-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, P.‐F. , Liu, Y.‐L. , Jin, G.‐H. , Liu, J.‐X. , Wu, H. , He, J. et al. (2021) The Pharus latifolius genome bridges the gap of early grass evolution. Plant Cell, 33, 846–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, S. , Wang, M. , Wu, J. , Guo, W. , Chen, Y. , Li, G. et al. (2021) WheatOmics: a platform combining multiple omics data to accelerate functional genomics studies in wheat. Molecular Plant, 14, 1965–1968. [DOI] [PubMed] [Google Scholar]
- Mahelka, V. , Krak, K. , Fehrer, J. , Caklová, P. , Nagy Nejedlá, M. , Čegan, R. et al. (2021) A panicum‐derived chromosomal segment captured by Hordeum a few million years ago preserves a set of stress‐related genes. The Plant Journal, 105, 1141–1164. [DOI] [PubMed] [Google Scholar]
- Mao, L. , Kawaide, H. , Higuchi, T. , Chen, M. , Miyamoto, K. , Hirata, Y. et al. (2020) Genomic evidence for convergent evolution of gene clusters for momilactone biosynthesis in land plants. Proceedings of the National Academy of Sciences of the United States of America, 117, 12472–12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuba, Y. , Nguyen, T.T.H. , Wiegert, K. , Falara, V. , Gonzales‐Vigil, E. , Leong, B. et al. (2013) Evolution of a complex locus for terpene biosynthesis in solanum . Plant Cell, 25, 2022–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnyk, C.W. & Meyerowitz, E.M. (2015) Plant grafting. Current Biology, 25, R183–R188. [DOI] [PubMed] [Google Scholar]
- Miyamoto, K. , Fujita, M. , Shenton, M.R. , Akashi, S. , Sugawara, C. , Sakai, A. et al. (2016) Evolutionary trajectory of phytoalexin biosynthetic gene clusters in rice. The Plant Journal, 87, 293–304. [DOI] [PubMed] [Google Scholar]
- Nemoto, T. , Cho, E.‐M. , Okada, A. , Okada, K. , Otomo, K. , Kanno, Y. et al. (2004) Stemar‐13‐ene synthase, a diterpene cyclase involved in the biosynthesis of the phytoalexin oryzalexin S in rice. FEBS Letters, 571, 182–186. [DOI] [PubMed] [Google Scholar]
- Nguyen, L. , Schmidt, H.A. , Haeseler, A. von and Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Molecular Biology and Evolution, 32, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura, T. , Ishihara, A. , Yanagita, R.C. , Endo, T.R. & Iwamura, H. (2005) Three genomes differentially contribute to the biosynthesis of benzoxazinones in hexaploid wheat. Proceedings of the National Academy of Sciences of the United States of America, 102, 16490–16495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nützmann, H.‐W. & Osbourn, A. (2014) Gene clustering in plant specialized metabolism. Current Opinion in Biotechnology, 26, 91–99. [DOI] [PubMed] [Google Scholar]
- Nützmann, H.‐W. , Scazzocchio, C. & Osbourn, A. (2018) Metabolic gene clusters in eukaryotes. Annual Review of Genetics, 52, 159–183. [DOI] [PubMed] [Google Scholar]
- Park, M. , Christin, P. & Bennetzen, J.L. (2021) Sample sequence analysis uncovers recurrent horizontal transfers of transposable elements among grasses. Molecular Biology and Evolution, 38, 3664–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, R.J. (2020) Doing the gene shuffle to close synteny: dynamic assembly of biosynthetic gene clusters. The New Phytologist, 227, 992–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polturak, G. , Dippe, M. , Stephenson, M.J. , Chandra Misra, R. , Owen, C. , Ramirez‐Gonzalez, R.H. et al. (2022) Pathogen‐induced biosynthetic pathways encode defense‐related molecules in bread wheat. Proceedings of the National Academy of Sciences of the United States of America, 119, e2123299119. 10.1073/pnas.2123299119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, G. , Tripathi, A. , Singh, P. , Jones, M.R.W. , Nanda, A.K. , Musseau, C. et al. (2022) Monocotyledonous plants graft at the embryonic root–shoot interface. Nature, 602, 280–286. [DOI] [PubMed] [Google Scholar]
- Riera‐Lizarazu, O. , Rines, H.W. & Phillips, R.L. (1996) Cytological and molecular characterization of oat × maize partial hybrids. Theoretical and Applied Genetics, 93–93, 123–135. [DOI] [PubMed] [Google Scholar]
- Rokas, A. , Wisecaver, J.H. & Lind, A.L. (2018) The birth, evolution and death of metabolic gene clusters in fungi. Nature Reviews. Microbiology, 16, 731–744. [DOI] [PubMed] [Google Scholar]
- Sato, Y. , Namiki, N. , Takehisa, H. , Kamatsuki, K. , Minami, H. , Ikawa, H. et al. (2013) RiceFREND: a platform for retrieving coexpressed gene networks in rice. Nucleic Acids Research, 41, D1214–D1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, Y. , Takehisa, H. , Kamatsuki, K. , Minami, H. , Namiki, N. , Ikawa, H. et al. (2013) RiceXPro version 3.0: expanding the informatics resource for rice transcriptome. Nucleic Acids Research, 41, D1206–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira, H. & Hasegawa, M. (1999) Multiple comparisons of log‐likelihoods with applications to phylogenetic inference. Molecular Biology and Evolution, 16, 1114–1116. [Google Scholar]
- Shimura, K. , Okada, A. , Okada, K. , Jikumaru, Y. , Ko, K.W. , Toyomasu, T. et al. (2007) Identification of a biosynthetic gene cluster in rice for momilactones. The Journal of Biological Chemistry, 282, 34013–34018. [DOI] [PubMed] [Google Scholar]
- Slot, J.C. & Rokas, A. (2011) Horizontal transfer of a large and highly toxic secondary metabolic gene cluster between fungi. Current Biology, 21, 134–139. [DOI] [PubMed] [Google Scholar]
- Smit, S.J. & Lichman, B.R. (2022) Plant biosynthetic gene clusters in the context of metabolic evolution. Natural Product Reports. 10.1039/D2NP00005A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein, J.C. , Yu, Y. , Copetti, D. , Zwickl, D.J. , Zhang, L. , Zhang, C. et al. (2018) Genomes of 13 domesticated and wild rice relatives highlight genetic conservation, turnover and innovation across the genus Oryza . Nature Genetics, 50, 285–296. [DOI] [PubMed] [Google Scholar]
- Strimmer, K. & Rambaut, A. (2002) Inferring confidence sets of possibly misspecified gene trees. Proceedings of the Royal Society of London. Series B, Biological Sciences, 269, 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana, M.H. , Liu, F. , Alamin, M. , Mao, L. , Jia, L. , Chen, H. et al. (2019) Gene modules co‐regulated with biosynthetic gene clusters for allelopathy between rice and barnyardgrass. International Journal of Molecular Sciences, 20, 3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyomasu, T. , Shenton, M.R. & Okada, K. (2020) Evolution of labdane‐related diterpene synthases in cereals. Plant & Cell Physiology, 61, 1850–1859. [DOI] [PubMed] [Google Scholar]
- Trapnell, C. , Roberts, A. , Goff, L. , Pertea, G. , Kim, D. , Kelley, D.R. et al. (2012) Differential gene and transcript expression analysis of RNA‐seq experiments with TopHat and cufflinks. Nature Protocols, 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. , Sun, S. , Ge, W. , Zhao, L. , Hou, B. , Wang, K. et al. (2020) Horizontal gene transfer of Fhb7 from fungus underlies fusarium head blight resistance in wheat. Science, 368, eaba5435. [DOI] [PubMed] [Google Scholar]
- Wu, D. , Jiang, B. , Ye, C.‐Y. , Timko, M.P. & Fan, L. (2022) Horizontal transfer and evolution of the biosynthetic gene cluster for benzoxazinoids in plants. Plant Communications, 3, 100320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, D. , Shen, E. , Jiang, B. , Feng, Y. , Tang, W. , Lao, S. et al. (2022) Genomic insights into the evolution of Echinochloa species as weed and orphan crop. Nature Communications, 13, 689. 10.1038/s41467-022-28359-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia, J. , Guo, Z. , Yang, Z. , Han, H. , Wang, S. , Xu, H. et al. (2021) Whitefly hijacks a plant detoxification gene that neutralizes plant toxins. Cell, 184, 1693–1705.e17. [DOI] [PubMed] [Google Scholar]
- Yang, X. , Gao, S. , Guo, L. , Wang, B. , Jia, Y. , Zhou, J. et al. (2021) Three chromosome‐scale Papaver genomes reveal punctuated patchwork evolution of the morphinan and noscapine biosynthesis pathway. Nature Communications, 12, 6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, C.‐Y. , Wu, D. , Mao, L. , Jia, L. , Qiu, J. , Lao, S. et al. (2020) The genomes of the allohexaploid Echinochloa crus‐galli and its progenitors provide insights into polyploidization‐driven adaptation. Molecular Plant, 13, 1298–1310. [DOI] [PubMed] [Google Scholar]
- Yoshida, S. , Maruyama, S. , Nozaki, H. & Shirasu, K. (2010) Horizontal gene transfer by the parasitic plant Striga hermonthica . Science, 328, 1128. [DOI] [PubMed] [Google Scholar]
- Yu, Y. , Zhang, H. , Long, Y. , Shu, Y. & Zhai, J. (2022) Plant public RNA‐seq database: a comprehensive online database for expression analysis of ~45 000 plant public RNA‐seq libraries. Plant Biotechnology Journal, 20, 806–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, C. , Lei, L. , Liu, Z. , Zhou, S. , Yang, C. , Zhu, X. et al. (2020) Selection of a subspecies‐specific diterpene gene cluster implicated in rice disease resistance. Nature Plants, 6, 1447–1454. [DOI] [PubMed] [Google Scholar]
- Zhan, C. , Shen, S. , Yang, C. , Liu, Z. , Fernie, A.R. , Graham, I.A. et al. (2022) Plant metabolic gene clusters in the multi‐omics era. Trends in Plant Science. 10.1016/j.tplants.2022.03.002 [DOI] [PubMed] [Google Scholar]
- Zhang, J. & Peters, R.J. (2020) Why are momilactones always associated with biosynthetic gene clusters in plants? Proceedings of the National Academy of Sciences of the United States of America, 117, 13867–13869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. A list of the plant genomes used in this study.
Table S2. Homologs of the key genes (CPS4, KSL4, CYP99A2/3, CYP76L1, MAS1/2, and CYP701A8) involved in momilactone biosynthesis in grass species.
Figure S1. Phylogeny and genomic synteny of CYP701A8 homologs in grass. Genes in different subfamilies are marked in different color backgrounds. Genes used in synteny analysis are marked in the phylogenetic tree from N0 to N3. Homologs in Triticeae subgenomes B and four CYP701A8 homologs in O. sativa are highlighted. The red syntenic dots represent pairs of native homologs between genomes.
Figure S2. Transcriptomic profiling of wheat genes in MABGC‐like clusters under pathogen infections. Genes in different colors are from different subgenomes (blue, subgenome A; red, subgenome B; green, subgenome D). Homolog information is shown by red (CYP99A2/3), yellow (KSL4), green (CYP701A8), and orange (CPS4). Dataset1, the spikelet (SP) and rachis (RACH) from two wheat accessions 2618 and 2890 were infected by Fusarium head blight (Fusarium graminearum) (FG) and water (control). Dataset2, coleoptile sheaths of wheat accession Chara were infected by crown rot (Fusarium pseudograminearum) (Fp). Dataset3, leaves from accession N9134 were inoculated by powdery mildew (Blumeria graminis). Dataset4, leaves from Glenlea and Salamouni were infected by tan splot (Pyrenophora tritici‐repentis). The quantified gene expression (TPM) levels were obtained from WheatOmics 1.0 (Ma, Wang, et al., 2021).
Figure S3. Phylogeny of CYP99A2/3 homologs in grass and topology tests. (a) Homolog phylogeny of CYP99A2/3 homologs. Genes in different subfamilies are marked in different color backgrounds. Cluster information of some homologs in Triticeae and MABGCs are suggested. (b) Topology tests. Three (Tree 1, 2, 3) and three (Tree 1, 4, 5) constrained trees were set for Test 1 and Test 2, respectively. Minus signs “−” represent that the corresponding topology could be rejected significantly (P < 0.05).
Figure S4. Phylogeny and genomic synteny of MAS1/2 homologs in grass and topology tests. (a) A maximum‐likelihood tree of MAS1/2 and homologs across the grass family. The homolog in A. comosus is set as an outgroup. Different background colors represent different subfamilies. (b) Genomic synteny among the native MAS3 homologs. Red dots represent that the two MAS3 homologs from two genomes are in good synteny. (c) Topology tests on three constrained trees. The top panel shows the topologies of constrained trees used in tests. The bottom panels show the results of the test on the LGT event of MAS1/2 from PACMAD to Oryza. Minus signs “−” represent that the corresponding topology could be rejected significantly (P < 0.05).
Figure S5. A maximum‐likelihood phylogenetic tree of CYP76M5/6/7/8 homologs in grass. Different background colors represent different subfamilies. The branch containing CYP76M5/6/7/8 from O. sativa is zoomed in.
Figure S6. A maximum‐likelihood phylogeny of CYP71Z6/7 homologs in grass. Different background colors represent different subfamilies. The branch containing CYP71Z genes from O. sativa is zoomed in and their cluster information (c2BGC and c7BGC) is shown.
Figure S7. A maximum‐likelihood phylogeny of KSL5/6 homologs in grass. Different background colors represent different subfamilies.
Figure S8. Phylogeny and genomic synteny of CYP76L1 homologs in grass and topology tests. (a) A maximum‐likelihood tree of CYP76L1 and its homologs across the grass family. Different background colors represent different subfamilies. Genes used in synteny analysis are marked in the phylogenetic tree from N0 to N5. (b) Genomic synteny among the native CYP76L1 homologs. Red dots represent that the two homologs from two genomes are in good synteny. (c) Topology tests on two constrained trees. The top panel shows the topologies of constrained trees under tests. The bottom panels show the test results on the LGT of CYP76L1 from Pooideae to Panicoideae. Minus signs “−” represent that the corresponding topology could be rejected significantly (P < 0.05).
Figure S9. Expression of MABGC, c2BGC, and related genes in rice under JA treatment (a), drought and rice blast fungus M. oryzae infection (b).
Figure S10. Liquid chromatography–tandem mass spectrometry (LC‐MS/MS) analyses of momilactone A in rice and barnyardgrass leaves. Extracts from fresh leaves of rice (O. sativa) Nipponbare and barnyardgrass (E. crus‐galli) STB08 under mock and blast fungus infection treatment were analyzed. Momilactone A was detected with the selected reaction monitoring (m/z 315/271).
Figure S11. The structures of MABGCs and DIMBOA Bx clusters on chromosomes 4 in nine Echinochloa subgenomes.
Data Availability Statement
Genome accession numbers or versions used in this study are given in supplementary Table S1.