

Abstract
New biomarkers indicating the abuse of drugs and alcohol are still of major interest for clinical and forensic sciences. The endogenous neurotransmitter and approved drug, gamma‐hydroxybutyric acid (GHB), is often illegally used for drug‐facilitated crimes by spiking GHB into alcoholic beverages. Analytical detection windows of only 6 h in blood and 12 h in urine are often too short to provide reliable proof of GHB ingestion. Therefore, new biomarkers are needed to prove exogenous GHB administration. Previously, amino acid GHB conjugates were discovered in an untargeted metabolomics screening and fatty acid esters with GHB were recently discussed as promising biomarkers to enlarge the analytical detection time windows. However, the development of analytical methods is still slowed down since reference compounds for targeted screenings are still missing. In this paper, we describe simple procedures for the rapid synthesis and purification of amino acid GHB conjugates as well as fatty acid esters, which can be adopted in analytical and clinical/forensic laboratories. Structural characterization data, together with IR, 1H‐nuclear magnetic resonance (NMR), 13C‐NMR, high‐resolution mass spectra (MS), and MS/MS spectra in positive and negative ionization mode are reported for all obtained GHB conjugates and GHB conjugate precursors.
Keywords: amino acid, biomarker synthesis, fatty acid, GHB, targeted screening
The availability of reference compounds is a fundamental prerequisite for targeted methods in forensics. In here, we describe a simple synthetic procedure for the rapid synthesis of amino acid and fatty acid gamma‐hydroxybutyric acid (GHB) conjugates suitable as possible biomarkers for GHB intake. The procedure can ubiquitously be adopted in forensic laboratories.

1. INTRODUCTION
Gamma‐hydroxybutyric acid (GHB) is a short chain fatty acid (SCFA) which occurs as an endogenous compound and serves as precursor of the neurotransmitter gamma‐aminobutyric acid (GABA) in the human body. GHB binds as an agonist to GABAA as well as to GABAB receptors and functions as a neuromodulator by regulating the inhibiting neurotransmitter release. 1 , 2 , 3 , 4 GHB exhibits a dose‐dependent effect. Low doses induce euphoria, loss of inhibition, amnesia, and drowsiness. Manifestations of an overdose can include sedation, coma, hypothermia, bradycardia, respiratory depression, and death. 3 , 5 Besides GHB's natural occurrence, it is further used as a therapeutic drug. Applications of GHB include the treatment of narcolepsy exhibiting cataplexy in form of the sodium salt (sodium oxybate) as well as the treatment of alcohol withdrawal. 6
GHB is of particular interest in forensic cases due to its abuse as a recreational drug, sports performing enhancing drug as well as its implication in drug‐facilitated sexual assault (DFSA). The detection period of exogenous administered GHB is 6 h in blood and 12 h in urine due to its complete and rapid metabolism, which makes it difficult to differentiate between endogenous and (low) exogenous levels of GHB. 7 , 8 , 9 , 10 GHB undergoes hepatic metabolism and is oxidized by GHB dehydrogenase which produces succinic semialdehyde. Succinic semialdehyde can be further oxidized to succinic acid, which then can be introduced into the Krebs cycle. Furthermore, GHB is easily biosynthesized in the body from its synthetic precursors, gamma‐butyrolactone (GBL), and 1,4‐butanediol (1,4‐BD). Both substances are sold as solvents for industrial use, which facilitates the use of GHB as a knock‐out drug. 11 Due to GHBs short half‐life, finding a suitable biomarker to extend the detection window has been of great interest. Especially phase‐II‐metabolites have been applied successfully in extending the detection window of, for example, alcohol. Alcohol is usually only detectable for 4–6 h in breath, 10–12 h in blood, and up to 24 h in urine. Ethyl glucuronide (EtG) and ethyl sulfate (EtS), two products of an alternative pathway of alcohol metabolism, have been used as markers in forensic analysis, due to their slightly longer half‐life. 12 , 13 Likewise, GHB‐β‐O‐glucuronide and GHB‐4‐sulfate have been investigated as possible phase‐II‐metabolite conjugates. These potential markers did not prove to be suitable due to their large interindividual variability in test subjects. 14 , 15 , 16 In recent years, an untargeted metabolomics study has been conducted to identify new potential biomarkers. GHB conjugates with carnitine, the amino acids glutamate, glycine and taurine, as well as ribose have been identified as markers of interest; however, reference substances were not available to verify the identity. 9 , 10 In a different study, GHB‐related fatty acids—for example, palmitoyl‐GHB—have been detected, which could possibly also indicate an exogenous GHB intake. 17 Forensic toxicology still requires a biomarker capable of distinguishing between endogenous and exogenous GHB and having a longer detection window. Due to the lack of reference compounds, targeted methods on GHB conjugates are scarce in literature. The availability of targeted screenings could finally help forensic toxicologists to evaluate significant concentration levels of GHB conjugates/metabolites and potentially enlarge the detection window for GHB intake. The aim of this work was the development of an easy and small‐scale synthesis of GHB conjugates and metabolites for application by forensic toxicology laboratories.
2. MATERIALS AND METHODS
2.1. Chemicals and reagents
GBL, triethylamine, imidazole, dry methylene chloride (DCM), trifluoroacetic acid (TFA), tert‐butyldimethylsilyl chloride (TBDMSCl), and fatty acid chlorides were obtained from Sigma‐Aldrich (Buchs Switzerland) in the highest analytical grade. Stearoyl‐chloride was purchased in purity of 90% (Sigma‐Aldrich, Buchs, Switzerland). Tert‐butyl 4‐hydroxybutanoate was obtained from Toronto Research Chemicals (Toronto, Canada). DCM, dimethylformamide (DMF), tetrahydrofuran (THF), cyclohexane, and ethyl acetate (EtOAc) were purchased from Sigma‐Aldrich and VWR (Darmstadt, Germany), respectively. Protected amino acids were obtained from IRIS Biotech (Marktredwitz Germany) and were all in L‐configuration. Oxyma® and diisopropylcarbodiimide (DIC) were purchased from Apollo scientific (Stockport, United Kingdom). Thin layer chromatography (TLC) silica gel 60 TLC plates on aluminum sheets with fluorescent indicator UV254 were obtained from VWR (Darmstadt, Germany). Pure water was obtained from an in‐house water purification system from Labtec (Villmergen, Switzerland). Acetonitrile (ACN), methanol (MeOH), and water of HPLC grade were purchased from Fluka (Buchs, Switzerland). Deuterated solvents were from Cambridge Isotope Laboratories (Tewksbury, USA).
2.2. Chemical synthesis
2.2.1. Large‐scale synthesis of 4‐((tert‐butyldimethlysilyl)oxy)butanoic acid (TBDMS‐GHB)
Synthesis of 4‐((tert‐butyldimethylsilyl)oxy)butanoic acid (TBDMS‐GHB) was performed as described previously with minor modifications. 18 In short, to a mixture of sodium hydroxide (NaOH, 12.3 g, 0.31 mol, 1.1 equivalents) in dry MeOH (280 ml) at 0°C (dry MeOH was added to the NaOH pellets at 0°C, internal temperature raised to approximately 20°C), GBL (21.4 ml, 0.28 mol, 1.0 equivalent) was added dropwise. The reaction mixture was stirred at room temperature for 4 h (no reaction control), before the solvent was removed under reduced pressure. A white solid was formed. 1H‐nuclear magnetic resonance (NMR) in D2O indicated the content of MeOH to be appproximately 2%. To a mixture of the resulting sodium salt in dry DMF (560 ml) at 0°C, imidazole (62.6 g, 0.92 mol, 3.3 equivalents) was added in a single portion and TBDMSCl (92.4 g, 0.61 mmol, 2.2 equivalents) in equal portions. The reaction mixture was stirred at room temperature for 18 h (no reaction control), before it was slowly quenched with water (1600 ml) and extracted with pentane (3 × 450 ml). The combined organic layers were dried over anhydrous magnesium sulfate (MgSO4), and pentane was removed under reduced pressure. To a solution of the crude bisprotected product in MeOH (280 ml) and THF (280 ml), a solution of K2CO3 (77.1 g, 0.56 mmol, 2.0 equivalents) in water (920 ml) was added at room temperature. The reaction was stirred at room temperature for 16 h, then acidified with 1 M HCl to appproximately pH = 2 (1200 ml), before it was extracted with diethyl ether (4 × 400 ml). The combined organic layers were dried over anhydrous MgSO4, and the solvent was removed under reduced pressure. The resulting oil was co‐evaporated with toluene (3 × 200 ml) to remove residual THF and tert‐butyldimethylsilanol. The final product TBDMS‐GHB was obtained as a pale‐yellow oil (44.4 g, yield: 73%).
2.2.2. Synthesis of fatty acid‐GHB‐OtBu
To a mixture of tert‐butyl 4‐hydroxybutanoate (160 mg, 1 mmol, 1.0 equivalent) in dry DCM (10 ml) at 0°C, 125 μl trimethylamine was added. Reaction was done in preheated glassware and under a nitrogen atmosphere. Fatty acid chloride was added dropwise (1.1 equivalents). The reaction mixtures were stirred at 0°C for an hour before the ice bath was removed. The solutions were allowed to warm to room temperature and were stirred for further 24 h (no reaction control). Mixtures were quenched with water, and 15 ml of DCM was added. Organic phases were extracted with saturated NaHCO3 (3*25 ml) and subsequently washed with brine (3*25 ml). Organic layers were dried over anhydrous sodium sulfate (Na2SO4), and DCM was removed under reduced pressure. Crude was dissolved in cyclohexane (1 ml) and subsequently purified by silica gel chromatography (40 times excess of silica gel related to the crude, 12 g) using cyclohexane‐EtOAc (9:1) as eluent (100 ml). A clear pale‐yellow residue was obtained after removal of the solvent.
2.2.3. Synthesis of fatty acid‐GHB‐OH
Purified tert‐butyl protected fatty acid GHB precursors were dissolved in 5 ml TFA/DCM (20% final [v/v]) 19 The mixture was stirred overnight at room temperature. Reactions were monitored by TLC by UV detection (λ = 254 nm) or stained with potassium permanganate solution. Reactions were quenched with 15 ml water. Organic layers were washed with brine (3*25 ml) and dried over anhydrous Na2SO4. After evaporation of the solvent under reduced pressure, a white solid was obtained without further purification.
2.2.4. Synthesis of GHB‐amino acid‐OtBu
DIC (188.8 mg, 1.5 mmol), ethyl cyanohydroxyiminoacetate (213.8 mg, 1.5 mmol), and TBDMS‐GHB from the aforementioned synthesis (332.7 mg, 1.5 mmol) were stirred for 5 min at 0°C in DMF (20 ml). Amino acid‐OtBu (1.0 mmol) and diisopropylethylamine (189.3 mg, 1.5 mmol) were added. After 1 h at 0°C, the reaction was stirred at room temperature overnight. The reaction was quenched with water (50 ml) and EtOAc (50 ml) subsequently. The EtOAc phase was extracted with 10 mM HCl (3 × 25 ml), saturated NaHCO3 (3 × 25 ml), water (25 ml), and brine (3 × 25 ml), then dried with anhydrous sodium sulfate. Afterwards, the solution was concentrated in vacuo to obtain a viscous and clear yellow liquid. Crude was purified by silica gel column (40 times excess of silica gel related to the crude) using cyclohexane/EtOAc (4:1) as eluent.
2.2.5. Synthesis of GHB‐amino acid‐OH
Purified GHB‐amino acid precursor (GHB‐amino acid‐OtBu) was dissolved in 5 ml TFA/DCM (20% final [v/v]) as previously described in publications. 19 , 20 The mixture was stirred overnight at room temperature. Reactions were monitored by TLC by UV detection (λ = 254 nm) or stained with potassium permanganate solution. Reactions were quenched with 10 ml water. DCM layer was separated, and the aqueous phase was extracted three times with chloroform/isopropanol (3:1, 10 ml). Organic layers were combined and washed with brine (3*10 ml) and dried over anhydrous Na2SO4. After evaporation of the organic solvent under reduced pressure, a white clear oil was obtained. All chemical illustrations were done with ChemDraw professional (Perkin Elmer, Version 19.0.0.22).
2.3. Analytical measurements
2.3.1. Infrared (IR)
IR spectra were recorded on a PerkinElmer FT‐IR Spectrometer Spectrum 100 (Schwerzenbach, Switzerland) in the range of 4000–600 cm−1 with 16 scans per measurement. Background was recorded before the measurement and automatically subtracted. The used software was Perkin Elmer Spectrum (version 10.5.0).
2.3.2. NMR
1H and 13C NMR spectra were recorded using a Bruker Avance III 400 MHz spectrometer (Ettlingen, Germany) at 298 K using deuterated chloroform, deuterium oxide, or deuterated acetone as solvent. The solvent peaks were used as internal standards: Chloroform (δ 7.26), water (δ 4.79), or acetone (δ 2.05). Data are reported in parts per million (ppm), with multiplicity assigned as follows: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, and m = multiplet. Coupling constants J are reported in Hertz. Spectra were analyzed using the MestReNova software (Mestrelab, version 14.1.0–24,037).
2.3.3. Liquid chromatography‐high‐resolution mass spectrometry (LC‐HRMS)
HRMS measurements of synthesized GHB conjugates (1 μg/ml in water [amino acid conjugates]) or MeOH (fatty acid conjugates) were performed on a Thermo Fischer Ultimate 3000 UHPLC system (Thermo Fischer Scientific, San Jose, CA, USA) coupled to a HR quadrupole time of flight (qTOF) instrument system (TripleTOF 6600, Sciex, Concord, ON, Canada) as described in detail elsewhere. 10 , 21 , 22 Briefly, two different columns—reversed phase (RP) (Waters XSelect HSST RP‐C18 column, Waters, Baden‐Daettwil, Switzerland [150 mm × 2.1 mm, 2.5 μm particle size]) and hydrophilic interaction liquid chromatography (HILIC) (Merck SeQuant ZIC HILIC column, Merck, Darmstadt, Germany [150 mm × 2.1 mm, 3.5 μm particle size])—were used for chromatographic separation. HRMS (mass range m/z 100–1000) and MS/MS (mass range m/z 50–1000) data were acquired by information‐dependent data acquisition (IDA; top 4) in positive and negative ionization mode (resolving power 30,000 in MS and 15,000 in MS/MS).
3. RESULTS AND DISCUSSION
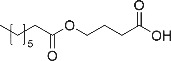
Despite recent advances in the search for new or alternative biomarkers for detection/interpretation of GHB intake, none of the possibly new markers, including amino acid or fatty acid conjugates, have been proven under real routine (forensic) conditions yet. 9 , 10 , 17 , 23 A reason for that is the lack of suitable, purified reference material necessary for (targeted) method development, validation, and routine quantification. Synthesis of reference standards is beyond the capability of analytical chemistry laboratories, while the commissioning of a custom‐made synthesis for a single compound is too expensive. Therefore, the aim of the current work was the development and application of an easy, low‐cost, and small‐scale chemical synthesis, suitable for analytical laboratories, of amino and fatty acid conjugates of GHB. Next to the general establishment of a synthesis strategy for amino acids and fatty acids conjugates in general, the synthesized compounds, partly identified in references, given in Table 1 should be synthesized. 9 , 10 , 17
TABLE 1.
Overview of synthesized GHB conjugates
| Entry | Precursor (pro) | Target compound |
|---|---|---|
| 1 Caprylic acid (C8:0) |
|
|
|
2 Capric acid (C10:0) |
|
|
|
3 Lauric acid (C12:0) |
|
|
|
4 Myristic acid (C14:0) |
|
|
|
5 Palmitic acid (C16:0) |
|
|
|
6 Stearic acid (C18:0) |
|
|
|
7 Phenylalanine (Phe) |
|
|
|
8 Glycine (Gly) |
|
|
|
9 Glutamic acid (Glu) |
|
|

















Abbreviation: GHB, gamma‐hydroxybutyric acid.
3.1. Chemical synthesis procedures
There are numerous reports described in the literature for amid coupling and esterification of primary amine and primary alcohol functional groups, respectively. 24 , 25 , 26 , 27 , 28 In this work, the following procedure was applied (Figure 1).
FIGURE 1.

Synthesis scheme of gamma‐hydroxybutyric acid (GHB)‐conjugates. n = 5, 7, 9, 11, 13, 15; R = Glu, Gly, Phe
3.1.1. Synthesis of GHB conjugates with fatty acids
The proposed scheme for synthesis of fatty acids conjugates of GHB can theoretically be applied to all fatty acids of interest. In the present study, we focused on those fatty acids with high occurrence in body fluids. Myristic acid, palmitic acid and stearic acid are the most common unsaturated acids found in human tissue and in most vegetable oils. 29 , 30 In a previous work, Kraemer et al. identified palmitoyl‐GHB (5) and GHB conjugates with myristic acid, stearic acid, and oleic acid in blood (plasma) samples after GHB intake. 17
The use of fatty acid chlorides and C‐terminal protected amino acids offered the greatest utility for use due to their commercial availability and ease of handling. Esterification was done in a 1 mmole scale with fatty acid chlorides and C‐terminal tert‐butyl protected GHB precursor. Fatty acid chlorides avoid the use of additional chemicals and activators, for example, dimethyl amino pyridine and dicyclocarbodiimide. 25 A similar synthesis approach was also proposed by Kraemer et al. 17 However, in their work, complete conversion of the acid chloride and the primary alcohol, which was used in a 1:1 ratio at room temperature without exclusion of humidity, was only assumed. However, further analysis revealed about 10% of unchanged GHB. Fatty acid conjugates were further used without purification. Furthermore, GHB was used without protection of the carboxylic acid function. Due to their reactivity, fatty acid chlorides should be protected from humidity and stored under nitrogen. Also, reactions should be done under a nitrogen atmosphere, preheated glassware, and dry solvents to guarantee highest synthetic yields. Residual water directly inactivates acid chloride, and the free carboxylic acid is formed. Our approach used preheated glassware, and the reaction was done under a nitrogen atmosphere to reduce side hydrolytic reactions with residual water. Nevertheless, yields of the first step were up to 70% for all fatty acids. Incomplete reaction required a short silica chromatography column to obtain the protected precursor in the highest purity. Silica gel chromatography was necessary to obtain purest compounds as starting material for the cleavage procedure, which was quantitative. However, size of the chromatographic set up is feasible even in analytical laboratories. For fatty acid conjugates, separation of the target compound from crude material was done using less than 100 ml of the solvent mix. Fractions containing the target compound were combined, and the solvent was evaporated under reduced pressure. Subsequently, a deprotection step was done in TFA/DCM (20% v/v), which is sufficient for the removal of C‐terminal tert‐butyl protection group. 20 Deprotection was done overnight, and TLC indicated a quantitative conversion. Reaction was quenched with water, and the organic phase was extracted three times with brine (each 15 ml). After drying over anhydrous sodium sulfate, solvents were removed under reduced pressure. All fatty acid GHB conjugates were finally obtained as a white solid. Overall yields were between 60% and 70%. In general, the complete synthesis approach comprised 2 days of synthetic work to obtain a single target compounds in high purity. In Figure 2, 1H‐NMR spectra of the precursor (lower panel) and the target compound (upper panel) are shown. Spectra clearly indicate the removal of the tert‐butyl protecting group of compound by loss of the signal at δ = 1.38 ppm (1 pro). Furthermore, in the spectra of the target compound, a clear split of methylene groups next to the carbonyl groups is observed at δ = 2.46 ppm (t, 3H) and δ = 2.29 ppm (t, 3H) in the final product, respectively. Target compound was obtained without further purification steps. Our approach yielded approximately 150 mg of each fatty acid conjugates. Purity of all fatty acid conjugates was higher than 95% (as determined by 1H‐NMR).
FIGURE 2.

Stacked view of the protected precursor (1 pro, black) and final compound (1, red). Lower panel shows the protected precursor (1 pro). Upper panel shows the target compound (1) [Colour figure can be viewed at wileyonlinelibrary.com]
HRMS and MS/MS spectra were recorded in the positive and negative ionization mode. In the negative ionization mode, a delta m/z of 28 (C2H4) based on varying length of the alkyl chain is clearly visible, as exemplified in Figure 3 for C14, C16, and C18 conjugates. In positive ionization mode, a fragment related to the GHB core structure of the molecule (m/z 87.0446) is detected (supporting information). MS/MS spectra of (5) correspond to spectra published previously. 17
FIGURE 3.

Fragmentation patterns of fatty acid gamma‐hydroxybutyric acid (GHB) conjugates (4, 5, 6) in negative ionization mode [Colour figure can be viewed at wileyonlinelibrary.com]
3.1.2. Synthesis of GHB‐amino acids conjugates
Based on previous studies, glutamic acid and glycine were selected to be synthesized as presented in Figure 1. Although phenylalanine‐GHB conjugate was not detected in previous studies, (7) was used to establish the synthesis and purification procedure. Detection of the aromatic ring system allows the simple reaction monitoring by UV. Other conjugates were stained with potassium permanganate. All precursor and target analytes were susceptible to this staining method.
Reagents for amid bond formation, for example, DIC and Oxyma®, showed reasonable stability to residual moisture and mild reaction conditions. 31 , 32 Also, for safety and toxicity reasons, the use of DIC and Oxyma® is preferred over other reagents (e.g., benzotriazoles). 32 For amid bond formation between carboxylic acid of GHB and the N‐terminus of the amino acid, the reactive hydroxyl group of GHB needs to be protected. Protection of the gamma hydroxyl group of GHB with tert‐butyldimethylsilyl group is common practice and well described in literature. 18 The preparation of the synthetic precursor TBDMS‐GHB was done without silica chromatography even in a large‐scale synthesis approach. Yield was 73%, and purity was higher than 95% (as determined by NMR).
Amidation was performed in 1 mmol scale and calculated on the protected amino acid precursor. 31 , 33 Reactions were stirred overnight at room temperature. After amide bond formation, a short silica gel chromatography using cyclohexane and EtOAc as solvent was performed. Protected amino acid conjugates and fatty acid‐GHB‐OtBu showed different lipophilicity. Therefore, chromatographic conditions for the purification of protected amino acid‐GHB precursor were more polar as reported above. For silica gel chromatography, crude was dissolved in 0.7 ml cyclohexane/EtOAc (4:1). Solution was transferred to a silica gel column, which was equilibrated in cyclohexane/EtOAc (4:1). For purification of the amino acid conjugates compared with fatty acid conjugates, a higher content of EtOAc was used. Care must be taken because of the acid character of the silanol groups, which are able to cleave the labile TBDMS‐protecting. The addition of 2% trimethylamine to the solvent is recommended to protect acid labile TBDMS‐group. Again, fractions containing the target compound were combined and the solvent was removed under reduced pressure. Identity of the protected precursor was confirmed by IR and 1H/13C‐NMR. Mass spectrometric detection was not performed on the level of the precursor. Cleavage of the labile TBDMS‐protecting group was observed in the heated ESI source. The final step was done in TFA/DCM (20% TFA). 20 Deprotection was performed overnight, and TLC indicated a quantitative conversion. The reaction was quenched by the addition of 10 ml water. For amino acid conjugates, the extraction method for the target analytes was modified due to higher polarity of the analytes. The organic phase was separated, and the aqueous phase was washed two times with diethyl ether (10 ml each). Afterwards, the aqueous phase was extracted with mixture of chloroform/isopropanol (3:1; 3 × 10 ml). Organic layers were combined and washed again with brine.
After drying over anhydrous sodium sulfate, solvents were removed under reduced pressure. The identity of all amino acid GHB conjugates was confirmed by 1H/13C NMR, FT‐IR, and MS. Analytical data can be found in the supporting information. Overall yields were between 10% and 40%. These lower yields were attributed to the higher polarity of target analytes and therefore lower recovery from the aqueous phase after the deprotection step. Nevertheless, the presented approach represents an interesting synthetic strategy for analytical laboratories. The overall time frame is around 2 days to obtain the final product.
In Figure 4, 1H‐NMR spectra of (8) is shown. The signal of the Cα‐hydrogen appears as a singlet at a shift of 3.98 ppm (s, 2H). Hydrocarbons related to the GHB body of the molecule are detected at 3.61 ppm (t, 2H), 2,38 (t, 2H), and 1.89–1.79 ppm (m, 2H), respectively.
FIGURE 4.

1H‐NMR in D2O (4.79 ppm) of (8), after deprotection with 20% trifluoroacetic acid (TFA) in methylene chloride. Protons in the molecule are numbered according to signals from low to high shift. The number of protons is indicated by numbers above the signal
HRMS and MS/MS spectra were recorded in positive and negative ionization modes. In Figure 5, fragmentation pattern of synthesized amino acid conjugates is shown (7–9). Fragmentation of the amide bond is the preferred fragmentation reaction of amino acid conjugates. However, for (9), further fragments can be detected.
FIGURE 5.

Fragmentation patterns of amino acid GHB conjugates (7, 8, 9) in negative ionization mode [Colour figure can be viewed at wileyonlinelibrary.com]
4. LIMITATIONS
Only three out of 20 proteinogenic amino were synthesized. However, in previous studies only (8) and (9) were found in authentic samples. In general, conjugates with glycine and taurine were predominantly found in metabolomics studies. 34 , 35 , 36 , 37 The presented protocol can be easily applied for the synthesis of other proteinogenic and nonproteinogenic amino acid conjugates as shown for Phe‐GHB. However, in contrast to fatty acids, the purification of amino acids conjugates depends to a higher degree from the individual chemistry and physico‐chemical properties of each amino acid, for example, regarding polarity or other functional groups. Two different, very promising conjugates—GHB‐taurine and GHB‐carnitine—have not been reported within this work. Synthesis was performed by the authors, but due to stability and purification issues, the synthetic strategy—which should be applicable in an analytical laboratory—was discontinued and will be part of further studies. Additionally, several fatty acid conjugates were synthesized, to show the applicability of our approach. The synthesis protocol represents an interesting starting point for the preparation of other physiological important unsaturated fatty acids GHB conjugates.
5. CONCLUSION
Easy availability of reference compounds is essential for successful analytical work in forensic and clinical laboratories. Employing a small‐scale coupling procedure GHB conjugates has been synthesized in a two‐step reaction scheme. This quick method employs commercially available starting materials and reagents and allows for an efficient synthetic preparation of GHB conjugates. The method is simple and high yielding and has been designed for use in analytical laboratories. This synthetic procedure will make GHB conjugate reference materials easily available to forensic toxicology laboratories.
Supporting information
Data S1.Supporting Information
ACKNOWLEDGEMENT
We are grateful to Ina Schmidt for laboratory support. Open access funding enabled and organized by Projekt DEAL.
Steuer C, Quattrini D, Raeber J, Waser P, Steuer AE. Easy and convenient millimole‐scale synthesis of new, potential biomarkers for gamma‐hydroxybutyric acid (GHB) intake: Feasible for analytical laboratories. Drug Test Anal. 2022;14(8):1460‐1470. doi: 10.1002/dta.3273
REFERENCES
- 1. Carter LP, Koek W, France CP. Behavioral analyses of GHB: Receptor mechanisms. Pharmacol Ther. 2009;121(1):100‐114. doi: 10.1016/j.pharmthera.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dave RA, Follman KE, Morris ME. Gamma‐hydroxybutyric acid (GHB) pharmacokinetics and pharmacodynamics: Semi‐mechanistic and physiologically relevant PK/PD model. AAPS J. 2017;19(5):1449‐1460. doi: 10.1208/s12248-017-0111-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Felmlee MA, Morse BL, Morris ME. γ‐hydroxybutyric acid: Pharmacokinetics, pharmacodynamics, and toxicology. AAPS J. 2021;23(1):22. doi: 10.1208/s12248-020-00543-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maitre M. The gamma‐hydroxybutyrate signalling system in brain: Organization and functional implications. Prog Neurobiol. 1997;51(3):337‐361. doi: 10.1016/s0301-0082(96)00064-0 [DOI] [PubMed] [Google Scholar]
- 5. Morse BL, Vijay N, Morris ME. gamma‐Hydroxybutyrate (GHB)‐induced respiratory depression: Combined receptor‐transporter inhibition therapy for treatment in GHB overdose. Mol Pharmacol. 2012;82(2):226‐235. doi: 10.1124/mol.112.078154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abanades S, Farre M, Segura M, et al. Gamma‐hydroxybutyrate (GHB) in humans: Pharmacodynamics and pharmacokinetics. Ann N Y Acad Sci. 2006;1074:559‐576. doi: 10.1196/annals.1369.065 [DOI] [PubMed] [Google Scholar]
- 7. Busardo FP, Jones AW. GHB pharmacology and toxicology: Acute intoxication, concentrations in blood and urine in forensic cases and treatment of the withdrawal syndrome. Curr Neuropharmacol. 2015;13(1):47‐70. doi: 10.2174/1570159X13666141210215423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Busardo FP, Kyriakou C, Marchei E, Pacifici R, Pedersen DS, Pichini S. Ultra‐high performance liquid chromatography tandem mass spectrometry (UHPLC‐MS/MS) for determination of GHB, precursors and metabolites in different specimens: Application to clinical and forensic cases. J Pharm Biomed Anal. 2017;137:123‐131. doi: 10.1016/j.jpba.2017.01.022 [DOI] [PubMed] [Google Scholar]
- 9. Steuer AE, Raeber J, Simbuerger F, et al. Towards Extending the Detection Window of Gamma‐Hydroxybutyric Acid‐An Untargeted Metabolomics Study in Serum and Urine Following Controlled Administration in Healthy Men. Metabolites. 2021;11(3):166‐181. doi: 10.3390/metabo11030166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steuer AE, Raeber J, Steuer C, et al. Identification of new urinary gamma‐hydroxybutyric acid markers applying untargeted metabolomics analysis following placebo‐controlled administration to humans. Drug Test Anal. 2019;11(6):813‐823. doi: 10.1002/dta.2558 [DOI] [PubMed] [Google Scholar]
- 11. Marinelli E, Beck R, Malvasi A, Lo Faro AF, Zaami S. Gamma‐hydroxybutyrate abuse: Pharmacology and poisoning and withdrawal management. Arh Hig Rada Toksikol. 2020;71(1):19‐26. doi: 10.2478/aiht-2020-71-3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andresen‐Streichert H, Muller A, Glahn A, Skopp G, Sterneck M. Alcohol Biomarkers in Clinical and Forensic Contexts. Dtsch Arztebl Int. 2018;115(18):309‐315. doi: 10.3238/arztebl.2018.0309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walsham NE, Sherwood RA. Ethyl glucuronide and ethyl sulfate. Adv Clin Chem. 2014;67:47‐71. doi: 10.1016/bs.acc.2014.09.006 [DOI] [PubMed] [Google Scholar]
- 14. Hanisch S, Stachel N, Skopp G. A potential new metabolite of gamma‐hydroxybutyrate: Sulfonated gamma‐hydroxybutyric acid. Int J Leg Med. 2016;130(2):411‐414. doi: 10.1007/s00414-015-1235-x [DOI] [PubMed] [Google Scholar]
- 15. Petersen IN, Tortzen C, Kristensen JL, Pedersen DS, Breindahl T. Identification of a new metabolite of GHB: Gamma‐hydroxybutyric acid glucuronide. J Anal Toxicol. 2013;37(5):291‐297. doi: 10.1093/jat/bkt027 [DOI] [PubMed] [Google Scholar]
- 16. Piper T, Mehling LM, Spottke A, et al. Potential of GHB phase‐II‐metabolites to complement current approaches in GHB post administration detection. Forensic Sci Int. 2017;279:157‐164. doi: 10.1016/j.forsciint.2017.08.023 [DOI] [PubMed] [Google Scholar]
- 17. Kraemer M, Broecker S, Kueting T, Madea B, Maas A. Fatty acid esters as novel metabolites of γ‐hydroxybutyric acid: A preliminary investigation. Drug Test Anal. 2022;14(4):690‐700. doi: 10.1002/dta.3213 [DOI] [PubMed] [Google Scholar]
- 18. Davies JA, Bull FM, Walker PD, et al. Total Synthesis of Kalimantacin A. Org Lett. 2020;22(16):6349‐6353. doi: 10.1021/acs.orglett.0c02190 [DOI] [PubMed] [Google Scholar]
- 19. Vineberg JG, Wang T, Zuniga ES, Ojima I. Design, Synthesis, and Biological Evaluation of Theranostic Vitamin–Linker–Taxoid Conjugates. J Med Chem. 2015;58(5):2406‐2416. doi: 10.1021/jm5019115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuzniewski CN, Glauser S, Gaugaz FZ, et al. Synthesis, Profiling, and Bioactive Conformation of trans‐Cyclopropyl Epothilones. Helv Chim Acta. 2019;102(5):e1900078. doi: 10.1002/hlca.201900078. [DOI] [Google Scholar]
- 21. Boxler MI, Schneider TD, Kraemer T, Steuer AE. Analytical considerations for (un)‐targeted metabolomic studies with special focus on forensic applications. Drug Test Anal. 2019;11(5):678‐696. doi: 10.1002/dta.2540 [DOI] [PubMed] [Google Scholar]
- 22. Steuer AE, Kaelin D, Boxler MI, et al. Comparative Untargeted Metabolomics Analysis of the Psychostimulants 3,4‐Methylenedioxy‐Methamphetamine (MDMA), Amphetamine, and the Novel Psychoactive Substance Mephedrone after Controlled Drug Administration to Humans. Metabolites. 2020;10(8):306. doi: 10.3390/metabo10080306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuting T, Schneider B, Heidbreder A, et al. Detection of gamma‐hydroxybutyric acid‐related acids in blood plasma and urine: Extending the detection window of an exogenous gamma‐hydroxybutyric acid intake? Drug Test Anal. 2021;13(9):1635‐1649. doi: 10.1002/dta.3097 [DOI] [PubMed] [Google Scholar]
- 24. Pan W‐B, Chang F‐R, Wei L‐M, Wu M‐J, Wu Y‐C. New and efficient method for esterification of carboxylic acids with simple primary and secondary alcohols using cerium (IV) ammonium nitrate (CAN). Tetrahedron Lett. 2003;44(2):331‐334. doi: 10.1016/S0040-4039(02)02578-9 [DOI] [Google Scholar]
- 25. Neises B, Steglich W. Simple Method for the Esterification of Carboxylic Acids. Angew Chem Int Ed Engl. 1978;17(7):522‐524. doi: 10.1002/anie.197805221 [DOI] [Google Scholar]
- 26. Komura K, Ozaki A, Leda N, Sugi Y. FeCl3 center dot 6H(2)O as a Versatile Catalyst for the Esterification of Steroid Alcohols with Fatty Acids. Synthesis‐Stuttgart. 2008;2008(21):3407‐3410. doi: 10.1055/s-0028-1083175 [DOI] [Google Scholar]
- 27. Gaspa S, Porcheddu A, De Luca L. Metal‐Free Oxidative Cross Esterification of Alcohols via Acyl Chloride Formation. Adv Synth Catal. 2016;358(1):154‐158. doi: 10.1002/adsc.201500912 [DOI] [Google Scholar]
- 28. Massolo E, Pirola M, Benaglia M. Amide bond formation strategies: Latest advances on a dateless transformation. Eur J Org Chem. 2020;2020(30):4641‐4651. doi: 10.1002/ejoc.202000080 [DOI] [Google Scholar]
- 29. Hodson L, Skeaff CM, Fielding BA. Fatty acid composition of adipose tissue and blood in humans and its use as a biomarker of dietary intake. Prog Lipid Res. 2008;47(5):348‐380. doi: 10.1016/j.plipres.2008.03.003 [DOI] [PubMed] [Google Scholar]
- 30. Dubois V, Breton S, Linder M, Fanni J, Parmentier M. Fatty acid profiles of 80 vegetable oils with regard to their nutritional potential. Eur J Lipid Sci Technol. 2007;109(7):710‐732. doi: 10.1002/ejlt.200700040 [DOI] [Google Scholar]
- 31. El‐Faham A, Al Marhoon Z, Abdel‐Megeed A, Albericio F. OxymaPure/DIC: An efficient reagent for the synthesis of a novel series of 4‐[2‐(2‐acetylaminophenyl)‐2‐oxo‐acetylamino] benzoyl amino acid ester derivatives. Molecules. 2013;18(12):14747‐14759. doi: 10.3390/molecules181214747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subiros‐Funosas R, Prohens R, Barbas R, El‐Faham A, Albericio F. Oxyma: An efficient additive for peptide synthesis to replace the benzotriazole‐based HOBt and HOAt with a lower risk of explosion. Chemistry. 2009;15(37):9394‐9403. doi: 10.1002/chem.200900614 [DOI] [PubMed] [Google Scholar]
- 33. von Mentzer B, Russo AF, Zhang Z, et al. A CGRP receptor antagonist peptide formulated for nasal administration to treat migraine. J Pharm Pharmacol. 2020;72(10):1352‐1360. doi: 10.1111/jphp.13317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Falany CN, Johnson MR, Barnes S, Diasio RB. Glycine and taurine conjugation of bile acids by a single enzyme. Molecular cloning and expression of human liver bile acid CoA:amino acid N‐acyltransferase. J Biol Chem. 1994;269(30):19375‐19379. doi: 10.1016/S0021-9258(17)32178-6 [DOI] [PubMed] [Google Scholar]
- 35. Ito T, Okazaki K, Nakajima D, Shibata D, Murakami S, Schaffer S. Mass spectrometry‐based metabolomics to identify taurine‐modified metabolites in heart. Amino Acids. 2018;50(1):117‐124. doi: 10.1007/s00726-017-2498-y [DOI] [PubMed] [Google Scholar]
- 36. Trottier J, Białek A, Caron P, et al. Metabolomic profiling of 17 bile acids in serum from patients with primary biliary cirrhosis and primary sclerosing cholangitis: A pilot study. Dig Liver Dis. 2012;44(4):303‐310. doi: 10.1016/j.dld.2011.10.025 [DOI] [PubMed] [Google Scholar]
- 37. Knights KM, Sykes MJ, Miners JO. Amino acid conjugation: Contribution to the metabolism and toxicity of xenobiotic carboxylic acids. Expert Opin Drug Metab Toxicol. 2007;3(2):159‐168. doi: 10.1517/17425255.3.2.159 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.Supporting Information