

Abstract
Leigh syndrome is a mitochondrial disease characterized by neurodegeneration, neuroinflammation, and early death. Mice lacking NDUFS4, a mitochondrial complex I subunit (Ndufs4 KO mice), have been established as a good animal model for studying human pathology associated with Leigh syndrome. As the disease progresses, there is an increase in neurodegeneration and neuroinflammation, thereby leading to deteriorating neurological symptoms, including motor deficits, breathing alterations, and eventually, death of the animal. However, despite the magnitude of neuroinflammation associated with brain lesions, the role of neuroinflammatory pathways and their main cellular components have not been addressed directly as relevant players in the disease pathology. Here, we investigate the role of microglial cells, the main immune cells of the CNS, in Leigh‐like syndrome pathology, by pharmacologically depleting them using the colony‐stimulating factor 1 receptor antagonist PLX3397. Microglial depletion extended lifespan and delayed motor symptoms in Ndufs4 KO mice, likely by preventing neuronal loss. Next, we investigated the role of the major cytokine interleukin‐6 (IL‐6) in the disease progression. IL‐6 deficiency partially rescued breathing abnormalities and modulated gliosis but did not extend the lifespan or rescue motor decline in Ndufs4 KO mice. The present results show that microglial accumulation is pathogenic, in a process independent of IL‐6, and hints toward a contributing role of neuroinflammation in the disease of Ndufs4 KO mice and potentially in patients with Leigh syndrome.
Keywords: IL‐6, Leigh syndrome, microglia, Ndufs4 KO, neuroinflammation
Main Points
Microglial depletion extends lifespan, delays motor symptoms, and protects from neuronal loss in Ndufs4 KO mice, demonstrating a causative role of neuroinflammation in the disease.
IL‐6 deficiency partially rescues breathing abnormalities.
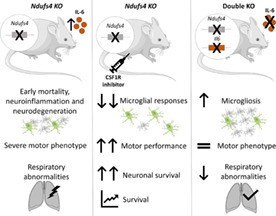
1. INTRODUCTION
Primary mitochondrial diseases are a group of rare genetic disorders characterized by mutations in nuclear or mitochondrial DNA genes that lead to dysfunction in the energetic mitochondrial machinery by affecting oxidative phosphorylation (Gorman et al., 2016). Due to the wide range of existing mutations, these disorders are highly heterogeneous and present a plethora of clinical symptoms and different disease onsets, which can vary between birth and adulthood. The most affected tissues are those that require high energy, such as muscles and the central nervous system (CNS) (Gorman et al., 2016). Leigh syndrome (LS) is the most common early‐onset mitochondrial disease. Also known as subacute necrotizing encephalomyelopathy, LS is characterized by progressive focal neurodegeneration in specific brain regions caused by numerous mutations (Lake et al., 2016; Lee et al., 2020). The time of death from such complications usually occurs around 3 years of age, and the neurological symptoms include developmental delay and regression, seizures, ataxia, dystonia, or hypotonia. Currently, there is no effective treatment available for patients with LS (Lake et al., 2016; Lee et al., 2020).
Mutations in the nuclear gene NDUFS4 (NADH dehydrogenase ubiquinone iron–sulfur protein 4), which encodes a subunit of complex I of the mitochondrial respiratory chain, have been described to produce LS (Fassone & Rahman, 2012). In this context, the Ndufs4 KO mouse recapitulates most aspects of human pathology associated with LS and has therefore been accepted as an LS mouse model (Quintana et al., 2010). Ndufs4 KO mice develop progressive encephalopathy beginning at postnatal day P30, which finally leads to the death of the animal at around P55. During the disease, Ndufs4 KO mice manifest growth retardation, decreased body weight, ataxia, paw paralysis, seizures, and respiratory abnormalities (Johnson et al., 2013; Kruse et al., 2008; Quintana et al., 2012). The main symptoms have been shown to have a neurological origin since Cre‐driven Ndufs4 deletion in neurons and glia (Nes‐KO) essentially demonstrated the same phenotype as systemic Ndufs4 KO mice (Quintana et al., 2010). Neuroinflammation, mainly microgliosis and astrogliosis, has been established as a hallmark of the disease in regions where neurodegeneration occurs in the late stages of pathology in Ndufs4 KO mice (Quintana et al., 2010). This is consistent with observations in human patients with LS (Arii & Tanabe, 2000; Lake et al., 2016). Surprisingly, the role of neuroimmune processes has not been studied per se but as a secondary event associated with neurodegeneration. It is now well described that mitochondrial defects or alterations can be major drivers of inflammation and disease progression in a wide range of pathologies, including neurodegenerative diseases (Luna‐sánchez et al., 2021; West & Shadel, 2017; Zhong et al., 2019). Similarly, several immune‐related processes have been found to be upregulated in both whole‐body and brain‐specific Ndufs4 deficient mice (McElroy et al., 2020; Perry et al., 2021).
In this study, we evaluated the contributions of microglia, which are specialized immune cells of the CNS (Bennett et al., 2016; Li & Barres, 2018), to the Ndufs4 KO mouse pathology. We used a pharmacological approach that involved chronic administration of an antagonist of the colony‐stimulating factor 1 receptor (CSF1R), PLX3397, to deplete mouse microglia from the CNS parenchyma (Elmore et al., 2014; Green et al., 2020). We also studied the role of the major cytokine, interleukin‐6 (IL‐6), which has been shown to play a significant role in neuroinflammation (Erta et al., 2012; Spooren et al., 2011) and many other scenarios (Jones & Jenkins, 2018). Moreover, recent studies have depicted increased IL‐6 expression (Balsa et al., 2020) and upregulation of the IL‐6/JAK/STAT signaling pathway in the brain of Ndufs4 KO mice compared with controls (McElroy et al., 2020). Our results strongly suggest that microglial cells constitute a major subset of cells contributing to neuronal death in Ndufs4 KO mice. In addition, IL‐6 was found to play a minor role, thereby necessitating further evaluation of the role of other cytokines.
2. MATERIAL AND METHODS
2.1. Animals
All mice used in this study were fed ad libitum and housed in a 12‐h light–dark cycle under specific pathogen‐free conditions at constant temperature (22 ± 2°C). All experiments were performed as per the approval of the Ethics Committee on Animal Experiments of the Universitat Autònoma de Barcelona (Ref. 3971 and 4155).
2.2. Mice generation
The parental mice strains used in this study were Ndufs4 +/− (Kruse et al., 2008) and Il6 −/− mice; the latter were generated in our laboratory (Sanchis, Fernández‐Gayol, Comes, et al., 2020). Both mouse lines were backcrossed with a C57BL/6 background for at least 10 generations. Both male and female mice were used in the present study.
For experiments with PLX3397, male and female Ndufs4 +/− mice were bred with each other to obtain WT (wild‐type) and Ndufs4 KO mice. Three rounds of breeding were required to generate double‐deficient mice lacking IL‐6 and NDUFS4. (i) Firstly, IL‐6 deficient mice (Il6 −/− ) were crossed with heterozygous Ndufs4 (Ndufs4 +/−) mice. (ii) Il6 +/−/Ndufs4 +/− offspring were crossed with Il6 −/− mice. (iii) Finally, the resulting Il6 −/−/Ndufs4 +/− and Il6 +/−/Ndufs4+/− mice were bred to obtain the four genotypes in the current study: control (Il6 +/−/Ndufs4 +/−), referred to as WT in the text; Il6 KO (Il6 −/−/Ndufs4 +/−), Ndufs4 KO (Il6 +/−/Ndufs4 −/−), and double KO (Il6 −/−/Ndufs4 −/−).
2.3. Genotyping
All mice were genotyped by PCR using DNA extracted from their tails. For DNA extraction, the tails were boiled in 100 μl of 50 mM NaOH solution for 7 min. The following primers were used:5′‐AGCCTGTTCTCATACCTCGG‐3′, 5′‐GTCCTCTATGAGGGTACAGAG‐3′, and 5′‐GGTGCATACTTATACTACTAGTAG‐3′ for Ndufs4 genotyping; 5′‐GAGACTGTGAGAGAGGAGTGTG‐3′, 5′‐CATCTTATCTGGGCTGACCCTAG‐3′, and 5′‐TCTCTGCTGGGATCTAGGGCC‐3′ for IL‐6 deficiency (Sanchis, Fernández‐Gayol, Comes, et al., 2020).
2.4. Microglial depletion with the colony‐stimulating factor 1 receptor (CSF1R) inhibitor, PLX3397
WT and Ndufs4 KO mice were injected with vehicle solution (Veh) or 40 mg/kg of PLX3397 (MedChem, HY‐16749) intraperitoneally 6 days a week beginning on postnatal days P24–P25. PLX3397 was dissolved in a solution of 20% DMSO, 40% PEG‐300, 5% Tween‐80, and 35% saline (vol/vol).
2.5. Disease staging and clinical evaluation
Disease staging of NDUFS4‐deficient mice was performed according to previously described guidelines (Quintana et al., 2010). In brief, disease progression in Ndufs4 KO mice can be subdivided into three stages: early‐stage (P0–P29), mid‐stage (P30–41), and late‐stage (over P41) of the disease in which clinical signs increase in severity (Kruse et al., 2008; Quintana et al., 2010). The mice were weighed and examined daily for clinical signs (dehydration, ataxia, and gait/postural alterations). Paw clasping behavior and body twisting were examined by suspending the animals by the tail for 20 s. Mice were humanely euthanized after a 20% loss of maximum body weight.
2.6. Behavioral tests
Mice were tested at the early, mid, and late stages at postnatal days P27–29, P38–39, and P47–49, respectively. All mice were tested after 30 min of habituation in the testing room.
2.6.1. Rotarod
The rotarod test was used to monitor motor coordination. The task consisted of placing the animals on a rotating bar that accelerated from 4 rpm to 40 rpm (Harvard Apparatus, Holliston, MA, USA) at a lapse of 300 s until the animal fell to the ground. The latency time to fall from the rod was also measured. At every disease stage, mice underwent two consecutive training trials. The test was performed thrice, and the mean of the trials was calculated.
2.6.2. Open field
The mice were placed in a methacrylate white box (56 [W], 36.5 [D], 31 [H] cm) and allowed to explore the apparatus freely for 10 min. The measured parameters include the total ambulation distance (horizontal activity) and the number of rearings (vertical activity; the number of times the mouse stood on its rear limbs). Rotation behavior was assessed in the corresponding late‐stage open‐field test and measured as the presence or absence of the behavior. The activity of the mice was recorded through videography. The videos were analyzed using EthoVision XT tracking software (Noldus Information Technology bv., Wageningen, The Netherlands).
2.6.3. Whole‐body plethysmography
Unrestrained whole‐body plethysmography was used to assess the ventilatory function. The detailed protocol has been described previously (Prada‐Dacasa et al., 2020). Briefly, the mice were allowed to acclimatize to the plethysmograph chamber (EMMS, catalog number: PLY310) for 45 min, followed by 15 min of the experimental period. Tidal volume normalized per body weight (μL·g−1) and respiratory frequency (breaths·min−1) have been reported in this study.
2.7. Tissue preparation
Mice were euthanized by decapitation at P47–P48 for the PLX3397 experiments and P38 and P55 for mid and late stages, respectively, in the double deficiency experiments. Brains extracted for histology were fixed in 4% paraformaldehyde (PFA) solution at 4°C for 24 h and then transferred to cryoprotective 30% sucrose in PBS (Phosphate‐buffered saline) (0.01 M pH 7.4) solution for 48 h. Later, the olfactory bulbs (OB) were separated from the rest of the brain, creating a coronal cut in the prefrontal cortex. Brains were frozen by immersion in isobutane at −30°C, whereas OBs were frozen in dry ice and embedded in an OCT medium. For multiplex analysis, brain areas were dissected, flash‐frozen in liquid nitrogen, and kept at −80°C until further use.
2.8. Multiplex
Multiplex analysis was previously optimized and validated in our laboratory (Sanchis, Fernández‐Gayol, Vizueta, et al., 2020). In short, both OBs and half cerebellums were mechanically homogenized using an MM‐400 mixer mill (Retsch GmbH, Haan, Germany), followed by ultrasonic homogenization using Sartorius‐LABSONIC P (Sartorius AG, Göttingen, Germany). Samples were homogenized in 100 μl (OB) and 200 μl (cerebellum) of ice‐cold protein extraction buffer containing 25 mM HEPES, 10% IGEPAL, 0.1 M MgCl2, 1.3 mM EDTA (pH 8.0), and 0.1 M EGTA (pH 8.0). Homogenization buffer was supplemented with a 1% protease inhibitor cocktail and 0.1 M phenylmethylsulphonyl fluoride (Sigma‐Aldrich P8340 and P7626, respectively). The homogenate was centrifuged at 12,000 g for 5 min. The supernatant was collected to determine the protein concentration using the Pierce™ BCA Protein Assay (Thermofisher 23.227). A protein concentration of 5–12 μg/μl of the tissue lysate was used to detect the abundance of IL‐6, TNF‐α, and IL‐1β using the Mouse High Sensitivity T Cell Magnetic Bead Panel (Millipore, catalog number MHSTCMAG‐70 K) according to the manufacturer's instructions. Data were obtained using a Luminex MAGPIX instrument system (Luminex Corporation) and analyzed using the xPONENT software v4.2 (Luminex Corporation). All results were normalized to the protein concentration in the lysate. Results between the blank and the first standard of the standard curve were considered as 0. IL‐1β levels were unquantifiable as the levels seemed to be below the detection threshold of the technique. Both sexes were included in this experiment and analyzed together.
2.9. Histology
Coronal sections of the brain were prepared using a cryostat at 20 μm from the bregma, 0.62 to −2.46 mm, and from −5.34 to −6.64 mm following Franklin and Paxinos mouse brain atlas (Paxinos & Franklin, 2013). The sections were preserved in an anti‐freezing solution at −20°C until staining. OBs were also cut at 20 μm, directly mounted on Superfrost slides (Thermo Fisher Scientific), dried at room temperature (RT) (20–26°C), and stored at −80°C until staining. For free‐floating brain sections, four slices were selected for each animal with duplicates for each of the following regions: cortex and hippocampus (between bregma −1.94 and − 2.30 mm), VN, and cerebellum (between bregma −5.88 and − 6.12 mm), and MCN (between bregma −6.24 and − 6.48 mm). For immunostaining, sections were blocked in 1% BSA and 0.2% Triton X‐100 in PBS (0.01 M pH 7.4) solution for 1 h at RT. Slides were then incubated with the chosen primary antibody overnight at 4°C and diluted in blocking solution (1:1000 for rabbit anti‐GFAP, Dako Z0334; 1:1000 for rabbit anti‐IBA‐1, Wako 019‐19741; 1:1000 for rabbit anti‐TMEM119, ab209064; 1:500 for chicken anti‐NeuN, Millipore ABN91). The following day, sections were left for 1 h at RT before incubation with specific secondary antibodies (1:600 goat anti‐rabbit Alexa Fluor 568 Thermofisher A11011; 1:600 goat anti‐chicken Alexa Fluor 488 Thermofisher A11039). The slides were kept away from light for 1 h at RT. Finally, sections were washed, and free‐floating brain slices were mounted on Superfrost slides (Thermo Fisher Scientific). The slides were then dried and mounted with DAPI Fluoromount (Southern Biotech). For immunohistochemical analysis of CD3+ cells, mounted brain sections were incubated with citrate buffer (10 mM sodium citrate, 0.05% Tween, pH 6) for 20 min at 96°C for antigen retrieval and incubated with endogenous peroxidase blocking solution (70% methanol and 3% hydrogen peroxide) for 15 min. Slices were then blocked as described previously, incubated with primary antibody overnight at 4°C, and diluted in blocking solution (1:100 for rabbit antihuman CD3, Dako A0452). The next day, sections were incubated for 1 h at RT with secondary antibody (1:200 biotinylated goat anti‐rabbit, Vector BA‐1000) and then with horseradish peroxidase‐coupled streptavidin (1:600, Vector SA‐5004). The immunoreactivity was visualized using 0.5 mg/ml 3,3‐diaminobenzidine and 0.033% hydrogen peroxide for 10 min at RT. Finally, the slides were dehydrated and covered with DPX. A positive control (IL‐6 overexpressing mice) with CD3+ cell infiltrates was used to correctly detect CD3+ cells (Giralt et al., 2013).
2.10. In situ hybridization using RNAscope
In situ hybridization (ISH) using RNAscope Multiplex Fluorescent kit (Advanced Cell Diagnostics) was used to detect gad2 (GABAergic neurons) and Slc17a6 (glutamatergic neurons) transcripts. Briefly, 48–49‐day old mice were euthanized, and their brains were embedded in OCT medium and frozen with dry ice. Fourteen‐micrometer brain sections containing the VN region were obtained, directly mounted on slides, and kept at −80°C until use. First, sections (between bregma −5.88 and − 6.00 mm) were fixed in cold PBS containing 4% PFA for 30 min at RT. Following two quick washes in PBS, the brain slices were dehydrated in 50% (5 min), 70% (5 min), and 2 × 100% (5 min each) ethanol and treated with protease IV solution at RT for 30 min. Protease was washed away with two PBS washes (2 min each). Target and negative control probes were applied directly to the sections and incubated at 40°C for 2 h in a HybEZ oven. Next, the slides were incubated with preamplifier and amplifier probes (AMP1, 40°C for 30 min; AMP2, 40°C for 15 min; AMP3, 40°C for 30 min). The slides were then incubated with fluorescent labels (AMP4‐Alt C). Finally, the brain sections were mounted using DAPI Fluoromount.
2.11. Microscopy and image quantification
Immunofluorescence images were captured using a Nikon Eclipse 90i microscope coupled to a Nikon Digital Camera DXM1200F using the ACT‐1 v2.70 capture software (Nikon, Tokyo, Japan). Whole Cornus Ammonis 1 (CA1), VN, OB, and MCN were photographed at ×20 magnification, while pictures of the whole cortex and cerebellum slices were taken at ×10 magnification. Images were captured under specific detector sensitivity conditions for each area. The samples were quantified using ImageJ software. For GFAP, IBA‐1, and TMEM119, immunofluorescence quantification of the integrated density normalized by the measured area was performed. The total number of IBA‐1+ cells was manually counted. Microglial phenotypes were visually discriminated. In situ hybridization images were captured using a Zeiss LSM700 confocal microscope (Carl Zeiss AG) at ×20 magnification. Positive gad2 and Slc17a6 neurons were counted and relativized by area.
2.12. Statistics
All graphics are shown as mean ± SEM, except for tidal volume and respiratory frequency (box and whisker plot). The plots were created using GraphPad 8 software (GraphPad Software, Inc.). Statistical analyses were performed using the Statistical Package for Social Sciences (SPSS) 19 (IBM). Survival was analyzed using log‐rank survival tests, with genotype as a factor. For repeated measures (weight), a generalized estimating equation (GEE) analysis was used. A generalized linear model (GzLM) was used to analyze the results of rotarod, open‐field, tibial length, respiration parameters, immunofluorescence quantification, and in situ hybridization. A chi‐square test was used for the frequency analysis (rotation behavior). For the PLX3397 experiments, both male and female mice were included in similar proportions and analyzed together (no sex effect was detected). Regarding double deficiency experiments, male and female mice were separately analyzed since IL‐6 has been described to exert sex‐dependent effects in some neurodegenerative diseases (Miller et al., 2010; Sanchis, Fernández‐Gayol, Comes, et al., 2020). For tidal volume, respiratory frequency, and multiplex results, the sexes were analyzed together owing to a lower number of experimental animals and similar results for both sexes in these variables. The major factors included in the statistical analysis for GzLM and GEE were genotype (WT vs. KO) and treatment (vehicle vs. PLX3397) in the case of PLX3397 experiments and IL‐6 (WT vs. KO) and Ndufs4 (WT vs. KO) in the case of double deficiency experiments. In some cases, the WT/Il6 KO or Ndufs4 KO/Double KO groups were analyzed separately, including genotype and age as factors (as specified in the text). For comparisons between two groups (i.e., clasping), Student's t test was used. One‐way ANOVA followed by Tukey's multiple comparison test was used for the dosage pilot study analysis. Statistical significance was defined as p ≤ .05.
3. RESULTS
3.1. Microglia depletion increases lifespan and improves the motor performance of Ndufs4 KO mice
Resembling Leigh syndrome pathology (Lake et al., 2016; Lee et al., 2020), Ndufs4 KO mice develop a fatal, early‐onset progressive encephalopathy, characterized by respiratory deficiencies, a strong decline in motor function, and neurodegeneration and neuroinflammation in several brain areas (Johnson et al., 2013; Kruse et al., 2008; Quintana et al., 2010). However, the role of neuroinflammation is poorly understood. Here, we induced neuroimmune disruption by pharmacologically depleting microglial cells using the well‐known inhibitor of CSF1R, PLX3397 (Elmore et al., 2014). To avoid off‐target effects and prevent the disruption of other cellular neuroimmune components, we considered it convenient to administer PLX3397 at a dosage causing partial microglial depletion but still capable of exerting a significant effect on disease progression. In WT mice, daily injection of PLX3397 caused dose‐dependent microglial depletion (Figure S1A). A daily dose of 40 mg/kg could reduce ~70% of the IBA‐1+ cells across the whole brain after 14 days of treatment; thus, we used this dose for further experiments.
As expected, Ndufs4 KO mice exhibited early mortality (Figure 1a), progressive motor decline (Figure 1b–e), and altered respiration (Figure 1f). They also showed lower body weight (Figure S1B) and growth (Figure S3B). Microglial depletion significantly rescued several abnormalities. Thus, it prominently increased the survival rate (Figure 1a) and decreased progressive deterioration of balance and motor coordination in the rotarod (Figure 1b). While vehicle‐treated Ndufs4 KO mice were mostly incapable of performing the rotarod test by ~2 months (late stage), PLX3397‐treated mice showed improved motor performance at all stages. However, this was statistically significant only at the corresponding mid (P38–P39) and late stages (P47–P48) of the disease (Ndufs4 KO mice treated with vehicle versus PLX3397, p = .042 and p = .009, respectively). This presumably reflects the effects on motor coordination rather than general activity since microglia depletion did not rescue ambulation in the open‐field test (Figure 1c). Microglia depletion also reduced the frequency of rotation behavior (mice circling on themselves, possibly due to vestibular lesions) in Ndufs4 KO mice at the P47–48 stage (p = .014) (Figure 1d). This also delayed the onset of clasping/twisting behavior in NDUFS4‐deficient mice (Figure 1e) at the P47–48 stage (p ≤ .001). PLX3397 administration showed a clear trend of a reduction in total body weight of male and female WT mice but did not modify the decreased body weight of Ndufs4 KO mice (Figure S1B).
FIGURE 1.
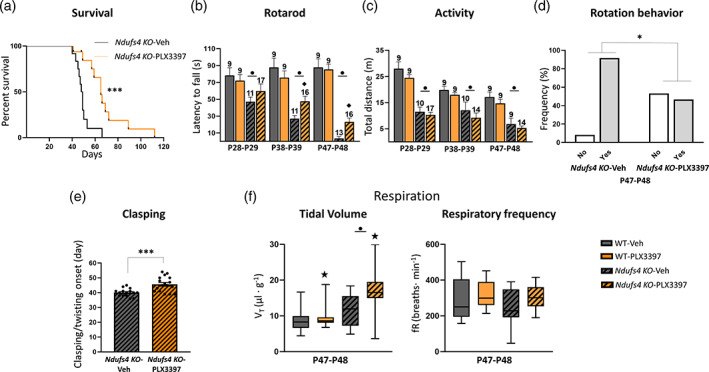
Inhibition of the CSF1R extends lifespan and delays motor symptoms in Ndufs4 KO mice. (a) Kaplan–Meier survival curve for Ndufs4 KO mice treated with vehicle (n = 12) or PLX3397 (n = 16). (b–e) Effects of PLX3397 over the motor phenotype of Ndufs4 KO. In order: Latency to fall in the accelerating rotarod, total distance traveled in the open‐field test, rotation behavior in vehicle and PLX3397 treated Ndufs4 KO mice at the late stage open‐field, and clasping onset in vehicle and PLX3397 treated Ndufs4 KO mice. (f) Tidal volume and respiratory frequency of WT and Ndufs4 KO mice treated with vehicle or PLX3397 at P47–P48, WT‐Veh (n = 12), WT‐PLX3397 (n = 12), Ndufs4 KO‐Veh (n = 10), Ndufs4 KO‐PLX3397 (n = 13). Long‐rank test for survival, GzLM statistical tests for rotarod, open‐field, tidal volume, and respiratory frequency, chi‐square test for rotation behavior, and Student's t‐test for clasping. Numbers within bars represent the “n” of the group. ● p ≤ .05 (WT vs. Ndufs4 KO), ★ treatment effect p ≤ .05 (Veh vs. PLX3397), ♦ p ≤ .05 interaction between both factors, * p = .05–.01, *** p ≤ .001. WT (wild‐type), Veh (vehicle)
Patients with LS normally present with respiratory abnormalities that are strongly associated with mortality (Arii & Tanabe, 2000). Constitutive Ndufs4 KO mice also show abnormal breathing patterns, which increase in severity as pathology progresses (Quintana et al., 2012). Respiratory rhythm generation and control include numerous CNS regions, such as the VN, cerebellum, and different brainstem nuclei. Respiratory abnormalities in Ndufs4 KO mice are partially associated with mitochondrial dysfunction due to NDUFS4 loss in the VN and abnormal responses in the pre‐Bötzinger complex of the brainstem (Quintana et al., 2012). In line with these findings, using whole‐body plethysmography (Prada‐Dacasa et al., 2020), we observed an increased tidal volume (volume of air inspired by the animal in one breath) in Ndufs4 KO mice at P47–P48 (Figure 1F, left), but no changes in respiratory frequency (Figure 1F, right). Microglial depletion significantly increased the tidal volume, while a similar trend was observed for respiratory frequency (PLX3397 effect, p = .09).
3.2. PLX3397 treatment protects against IBA‐1+ cell accumulation and proliferation in brain regions associated with neurodegeneration in Ndufs4 KO mice
Ndufs4 KO mice present with a characteristic fatal encephalopathy in which neurodegeneration and neuroinflammation occur in specific brain regions. These two processes are mainly observed in the VN, OB, brainstem, cerebellum, and hippocampus (Johnson et al., 2013; Miller et al., 2020; Quintana et al., 2010; Shil et al., 2021). This neurodegenerative and neuroinflammatory process is the main cause of the Ndufs4 KO phenotype since brain‐specific NDUFS4 deficiency (using Nes‐Cre mice) results in an almost identical phenotype to that of systemic Ndufs4 KO (Quintana et al., 2010). Furthermore, conditional deletion of Ndufs4 in different neuron subtypes was accompanied by neuroinflammation and contributed differently to the pathology (Bolea et al., 2019). Here, we wanted to examine whether microglial depletion could deplete or reduce microglia in Ndufs4 KO mice and prevent IBA‐1+ cell accumulation in the known neurodegenerative areas found in late‐stage mice. As expected, prominent gliosis (as revealed by IBA‐1 and GFAP immunostaining) was observed in the VN, OB, and cerebellum, where massive neurodegeneration was described in vehicle‐treated Ndufs4 KO mice (Figure 2a,b). In addition, GFAP (but not IBA‐1) immunostaining clearly increased in the cortex and hippocampus (Figure 2a). Treatment with PLX3397 caused a robust decrease in IBA‐1 fluorescence intensity in all the studied brain areas. WT animals showed microglial depletion like that observed in the pilot dosage study (Figure S1A), which confirmed the reproducibility of the experiments. Ndufs4 KO mice also showed dramatic microglial depletion both in areas where microgliosis is known to occur (VN, OB, and cerebellum) and in those lacking it (cortex and hippocampus). In comparison, massive cell accumulation hindered adequate cellular individualization and, thereby, a precise cell count in the first three areas. This was possible in the cortex and hippocampus, where PLX3397 administration reduced IBA‐1+ cells by ~70% and 60%, respectively. Different results depending on the brain area were observed regarding GFAP immunoreactivity due to PLX3397 administration (Figure 2A,B). Thus, in the VN, PLX3397 did not affect GFAP immunofluorescence in either WT or Ndufs4 KO mice, whereas in the cerebellum, a reduction was observed in both the genotypes. In contrast, within the OB, cortex, and hippocampus, PLX3397 treatment increased GFAP immunofluorescence. Taken together, the GFAP immunofluorescence results suggest that astrocytes respond to the microglial reduction in a region‐dependent manner. These findings are consistent with those of previous reports (Najafi et al., 2018).
FIGURE 2.
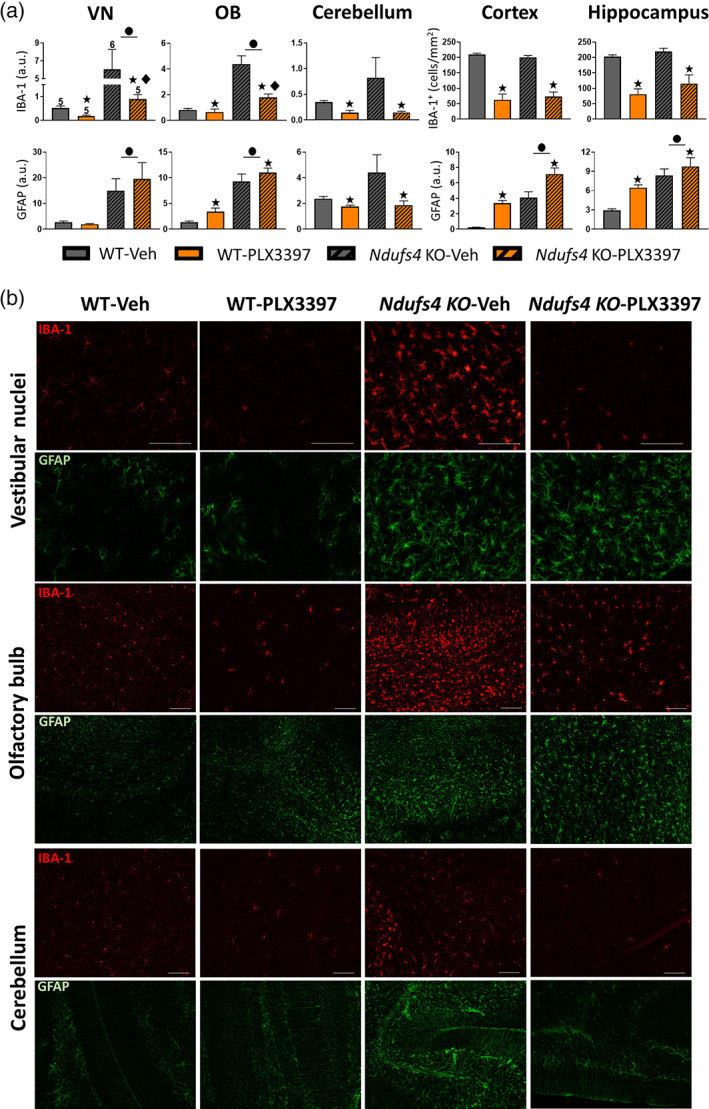
Inhibition of the CSF1R prevents IBA‐1+ cell accumulation in neurodegeneration‐related areas of Ndufs4 KO mice brains. (a) IBA‐1 fluorescence intensity or total IBA‐1+ positive cell and GFAP fluorescence intensity in VN, OB, cerebellum, cortex, and hippocampus of WT and Ndufs4 KO mice treated with vehicle or PLX3397 at P47–48 days of age. GzLM statistical tests. The experimental “n” is the same for all graphs and stated in VN IBA‐1. (b) Representative images of IBA‐1 and GFAP immunofluorescence in VN, OB, and cerebellum. ● p ≤ .05 (WT vs. Ndufs4 KO), ★ treatment effect p ≤ .05 (Veh vs. PLX3397), ♦ p ≤ .05 interaction between both factors. Scale bar 100 μm. VN (vestibular nuclei), OB (olfactory bulb)
3.3. Microglial response promotes both glutamatergic and GABAergic neuronal loss in Ndufs4 KO mice
After showing that PLX3397 treatment could prevent microglial accumulation in major neurodegenerative areas of Ndufs4 KO, we investigated whether a reduction in the microglial response could prevent neuronal loss in Ndufs4 KO mice in two areas with massive neuroinflammation, the OB and the VN. By quantifying the area occupied by NeuN immunostaining, we observed that microglial depletion partially protected against neuronal loss in the granular cell layer of the olfactory bulb (GrOB) (Figure 3a). In the whole VN, in contrast, microglial depletion did not prevent neuronal loss (Figure 3b). However, a more thorough inspection indicated that although extensive microgliosis occurred in the whole VN of Ndufs4 KO mice, it was especially dramatic between the magnocellular part of the medial vestibular nucleus (MVeMC) and the lateral vestibular nucleus (LVe), where extensive bilateral symmetrical lesions appeared (Figure 3d) (Quintana et al., 2010). Analysis of neuronal loss in this area indicated that microglial depletion tended to rescue neuronal loss (Figure 3c). Since NeuN is a general neuronal marker, we sought to investigate if specific neuronal populations were affected by microglial depletion in the VN. To this end, we used ISH to assess specific transcripts as markers of glutamatergic (Slc17a6) and GABAergic (Gad2) neurons (Figures 3e,f, and S2). Microglial depletion rescued both glutamatergic and GABAergic neuronal loss in the VN (Figure S2A). When the VN was divided into non‐lesioned (MVePC, SuVe, and the medial part of the MVeMC) (Figure 3e, left and S2B) and lesioned areas (lateral part of MVeMC and LVe) (Figure 3e, right and f), microglial depletion rescued neuronal losses in Ndufs4 KO mice, mainly in the lesion‐associated area (Ndufs4 KO mice vehicle versus PLX3397, p = .002). This reduced neuronal loss can be attributed to both glutamatergic (Figure 3e, middle) and GABAergic neuronal preservation (Figure 3e, bottom). Collectively, these results indicate that microglial cells promote neuronal loss in Ndufs4 KO mice independent of the neuronal subtype.
FIGURE 3.
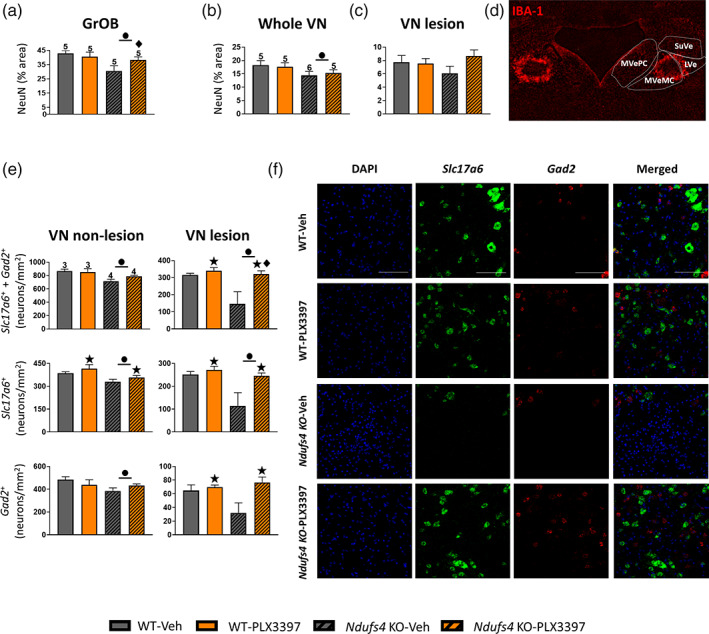
Microglia mediates neuronal loss in Ndufs4 KO mice. (a) Percentage of the area occupied by NeuN staining in the GrOB. (b) Percentage of the area occupied by NeuN staining in the whole VN and the area associated with the main lesion. (c) Image of IBA‐1 staining of the VN of a Ndufs4 KO mouse showing where Ndufs4 KO mice use to develop massive lesions. (d) Number of neurons of both glutamatergic and GABAergic subtypes in the non‐lesioned and the lesioned area of the VN of Ndufs4 KO mice, and the number of neurons subdivided by the neuronal subtype. (e) Representative ISH images of glutamatergic and GABAergic neurons of the different genotypes in the lesioned area of the VN. (a, b, and d) GzLM statistical tests. ● p ≤ .05 (WT vs. Ndufs4 KO), ★ treatment effect p ≤ .05 (Veh vs. PLX3397), ♦ p ≤ .05 interaction between both factors. Scale bar 100 μm. GrOB (granular cell layer of the olfactory bulb), VN (vestibular nucleus), MVePC (medial vestibular nucleus, parvicellular part), MVeMC (medial vestibular nucleus, magnocellular part), SuVe (superior vestibular nucleus), LVe (lateral vestibular nucleus)
3.4. Lack of evidence of infiltrating CD3 + cells in motor‐related neurodegenerative regions of Ndufs4 KO mice
Some reports using PLX3397 at concentrations like those used in this study have described that besides depleting microglia, it may also alter the populations of other immune cells (Han et al., 2020; Lei et al., 2020). Thus, we investigated whether Ndufs4 KO mice presented T lymphocyte infiltrates in the most prominent neurodegenerative areas and whether PLX3397 treatment modified them to some extent. Interestingly, we did not find CD3+ cell infiltrates in either the cerebellum or the VN of Ndufs4 KO mice at the late stage (Figure 4a, right), but substantial CD3 infiltrates were present in the OB at this stage, which were not affected by microglial depletion (Figure 4b, left, right). A GFAP‐IL6 mouse cerebellum (a chronic model of neuroinflammation with CD3+ cell infiltrates) was used as a positive control (Giralt et al., 2013) (Figure 4a, left).
FIGURE 4.
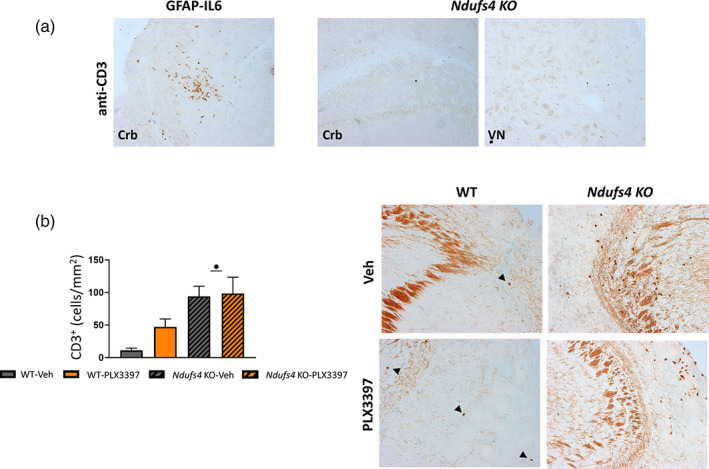
T‐cells do not infiltrate into the VN and the cerebellum of Ndufs4 KO mice, but they do infiltrate in the OB at the late stage of the disease (a) image showing the positive control used in the CD3 immunohistochemistry (GFAP‐IL6) together with representative images of the cerebellum and the VN of Ndufs4 KO mice showing no CD3+ cells infiltration in these regions at P48–P49. (b) Number of CD3+ cells in the OB of the different genotypes at P48–P49 together with representative images. Black arrows indicate CD3+ cells. GzLM statistical test. ● p ≤ .05 (WT vs. Ndufs4 KO)
3.5. IL‐6 levels are increased in Ndufs4 KO mice brains, but IL‐6 deficiency does not protect against Ndufs4 KO clinical signs or neuropathology
Once we demonstrated that the microglial response is noxious in Ndufs4 KO mice, we next investigated one of the potential candidates implicated in this process, the cytokine IL‐6. IL‐6 is highly induced in chronic neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and multiple sclerosis, with potentially detrimental roles (Erta et al., 2012; Spooren et al., 2011). To study putative IL‐6 contributions to the Ndufs4 KO mouse phenotype, we generated a mouse line with combined IL‐6 and NDUFS4 deficiencies and their respective littermate controls. First, we investigated IL‐6 protein levels by multiplex analysis in two of the main neurodegenerative regions of Ndufs4 KO mice, the OB and cerebellum (Quintana et al., 2010). IL‐6 levels were significantly increased in Ndufs4 KO mice in both brain areas at the late stage of the disease compared with WT mice (WT vs. Ndufs4 KO, p = .005 and p = .001 for OB and cerebellum, respectively) (Figure 5A, left). This observation is consistent with previous studies (Balsa et al., 2020; McElroy et al., 2020). The lack of IL‐6 was confirmed in the IL‐6 deficient groups. Moreover, we investigated whether other pro‐inflammatory cytokines could also be involved and if IL‐6 deficiency could modify their regulation. In line with the described neuroinflammation in Ndufs4 KO mice, we also observed a significant increase in TNF‐α in the OB of Ndufs4 KO and Double KO mice at the late stage compared with their littermates (Ndufs4 effect, p ≤ .001), and the same trend was observed in the cerebellum (Ndufs4 effect, p = .13) (Figure 5a, right). Interestingly, despite the massive neuroinflammation present in NDUFS4‐deficient mice at late stages of pathology (Johnson et al., 2013; Quintana et al., 2010), IL‐1β levels were undetectable in any genotype or region (not shown).
FIGURE 5.
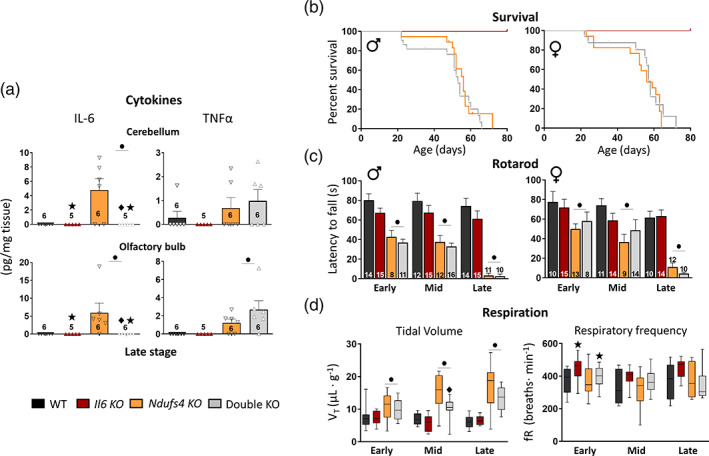
IL‐6 levels are increased in Ndufs4 KO mice brains, but IL‐6 deficiency does not modify disease progression. (a) IL‐6 and TNF‐α levels in the olfactory bulb and cerebellum at the late stage. (b) Kaplan–Meier survival curve for WT (n = 14 males, n = 11 females), Il6 KO (n = 15 males, n = 13 females), Ndufs4 KO (n = 18 males, n = 17 females), and double KO (n = 22 males, n = 16 females) mice. (c) Latency to fall in the accelerating rotarod. (d) Whole‐body plethysmography for assessing breathing alterations. For both tidal volume and respiratory frequency, WT (early, n = 9; mid, n = 9; late, n = 8), Il6 KO (early, n = 8; mid, n = 10; late, n = 9), Ndufs4 KO (early, n = 12; mid, n = 11; late, n = 7) and double KO (early, n = 12; mid, n = 17; late, n = 10). (a–d) GzLM statistical test for a, c, and d; long‐rank test for survival for b. ● Ndufs4 effect p ≤ .05 (WTs vs. Ndufs4 KOs), ★ IL‐6 effect p ≤ .05 (WTs vs. Il6 KOs), ♦ p ≤ .05 interaction between both factors
Additionally, IL‐6 deficiency in Ndufs4 KO male and female mice did not modify survival (Figure 5b) or motor coordination (Figure 5c). In addition, IL‐6 deficiency did not rescue Ndufs4 KO mice in terms of body weight and growth, total motor activity, vertical behavior, or the onset of clasping, regardless of sex (Figure S3). In contrast, IL‐6 deficiency rescued the increased respiratory tidal volume shown by Ndufs4 KO mice (Figure 5D, left), particularly at the mid‐stage of the disease. This effect could not be explained by neuronal loss or clear neuroinflammatory changes in important breathing‐related regions during the mid‐stage of the disease (Figure S4). We did not observe changes in respiratory frequency due to NDUFS4 deficiency at any stage, whereas IL‐6 deficiency tended to increase respiratory frequency, particularly in NDUFS4 non‐deficient mice (Figure 5d, right). This was statistically significant when comparing IL‐6 deficient and WT mice with genotype and age as statistical factors (WT vs. Il6 KO, p = .001).
In line with the behavioral results, double KO mice did not show substantial changes in gliosis at either the mid (Figure S5) or late‐stage of the diseases (Figures S6 and S7), in which IL‐6 deficiency increased microgliosis in Ndufs4 KO mice, particularly in male mice (Figure S7).
4. DISCUSSION
As shown above, neuroinflammation is a key manifestation of the Ndufs4 KO mice pathology. Interventions that either extend the lifespan or rescue motor decline in Ndufs4 KO mice, such as hypoxia (Jain et al., 2016; Jain et al., 2019), rapamycin treatment (Johnson et al., 2013), or doxycycline administration (Perry et al., 2021), have also been shown to reduce gliosis and immune‐related pathways in Ndufs4 KO brains. Whether this neuroimmune process is a pathological mechanism contributing to the Ndufs4 KO phenotype or simply is a consequence of neurodegeneration deserves adequate investigation in the context of complex I‐associated mitochondrial disorders. Here, we report one of the first pieces of evidence that depicts microglia as a mediator of Ndufs4 KO pathology since its reduction improved the motor phenotype and decreased the neuropathology in these mice. Importantly, these results strongly suggest that microglia may be controlling neuronal survival. Interestingly, despite VN glutamatergic neurons that have been shown to be susceptible to NDUFS4 deficiency (Bolea et al., 2019), we found that microglia promote both glutamatergic and GABAergic neuron loss, indicating that microglial‐mediated neuronal loss is not restricted to susceptible neurons. In addition, increased neuronal preservation in the GrOB can be mostly attributed to the GABAergic neuronal subpopulation since it is eminently a GABAergic neuronal region (Nagayama et al., 2014). In contrast, the VN is mainly composed of glutamatergic and GABAergic neuronal subpopulations as well as a small glycinergic subpopulation (Bagnall et al., 2007). The precise pathways by which microglia promote neuronal death remain unclear and require further investigation.
A recently published study using CSF1R inhibition with PLX3397, but at higher concentrations, which may target and eliminate a wider range of immune cell types than microglia and myeloid CNS‐associated cells, showed an increased lifespan in Ndufs4 KO mice (Stokes et al., 2022). An interesting debate in the scientific community exists regarding whether CSF1R inhibition targets microglial cells almost exclusively or if other immune cells are also being affected, which could potentially have relevant roles in CNS diseases. It has been speculated that the higher the concentration of PLX3397, the higher are the number of targets affected (i.e., circulating monocyte subpopulation, some T‐cell subpopulations, or other CNS‐associated myeloid cells) (Green & Hume, 2021; Han et al., 2020; Lei et al., 2020). Although the concentration used in this study is within the standards used in the field and is supposed to act exclusively on microglia, we cannot rule out the possibility that our dose and timing of PLX3397 also eliminate some other immune cell subpopulations. It seems reasonable to speculate that the dose‐dependent effect of PLX3397 on the Ndufs4 KO mouse phenotype may be associated with the type(s) of the immune cell population being eliminated. Henceforth, more efforts are needed to elucidate immune cell‐type‐specific contributions to this type of disorder (i.e., peripheral or central nervous system‐associated macrophages) and to mechanistically describe how this immune interplay affects Ndufs4 KO mouse pathology. Earlier, Strokes et al. claimed to obtain leukocyte depletion; however, no other immune subpopulations apart from IBA‐1+ were investigated in the context of PLX3397 administration. To look at other leukocyte subpopulations, apart from those of myeloid origin (IBA‐1+ cells), we immunohistochemically stained for CD3, a well‐described pan‐T‐cell marker, in the neurodegenerative areas of Ndufs4 KO mice. The absence of CD3+ cell accumulation in the main areas responsible for the major symptoms of Ndufs4 KO (VN and cerebellum) reinforces the idea that the pathogenic neuroimmune response present in these mice is mainly guided by innate immunity mechanisms, as observed in RNAseq data (McElroy et al., 2020; Perry et al., 2021).
IL‐6 is an important mediator of neuroinflammation and is known to have strong detrimental effects during chronic neuroinflammation. Therefore, we hypothesized that IL‐6 could potentially guide microglial activity in Ndufs4 KO mice. Collectively, our results indicate that IL‐6 is not behind the major microglial effects and that other mediators guide the response since its deficiency does not ameliorate the Ndufs4 KO mice neuropathology. However, IL‐6 deficiency partially reversed the respiratory abnormalities observed in these mice. Respiration rhythm generation and coordination require complex circuitry in which the VN, MCN, and brainstem are key components. Respiration is strongly influenced by inflammation (Peña‐Ortega, 2019). Furthermore, microglia and different inflammatory mediators can modulate respiratory rhythm (Lorea‐Hernández et al., 2016; Peña‐Ortega, 2019). For instance, IL‐1β (Lorea‐Hernández et al., 2020) and IL‐10 (Giannakopoulou et al., 2019) can modulate tidal volume and respiratory frequency in nonpathological conditions. To our knowledge, this is the first direct report showing that IL‐6 can also modulate breathing. Since we found no differences in gliosis between Ndufs4 KO and Double KO mice at the mid‐stage, we hypothesize that IL‐6 has an intrinsic capacity to affect breathing, as observed in the case of other cytokines (Giannakopoulou et al., 2019; Lorea‐Hernández et al., 2020). This could be achieved by changing neuronal firing or playing a relevant role in glia–neuron communication (Peña‐Ortega, 2019).
Regarding the strong neuroimmune response present in Ndufs4 KO mice, IL‐6 signaling is only one of the multiple neuroinflammatory mediators that can be the contributing factor. Mitochondrial defects have been known to elicit different inflammatory responses, including TLR9, inflammasome pathway, or IFN type I responses (Luna‐sánchez et al., 2021; Zhong et al., 2019). We did not find evidence of inflammasome activation because one of the main mechanisms of action of this complex is the final conversion of pro‐IL‐1β into its active form (Yang et al., 2019), and we could not detect IL‐1β in Ndufs4 KO mice. Interestingly, some studies have reported interferon‐related response activation in NDUFS4‐deficient mice (McElroy et al., 2020; Perry et al., 2021) and other mitochondrial disease mouse models (Y. Lei et al., 2021). Thus, this pathway could be a potential candidate associated with the microglial response present in Ndufs4 KO mice and deserves further attention.
Overall, our results indicate that microglial depletion using PLX3397 has a protective effect in Ndufs4 KO mice, revealing the pathogenicity of the neuroimmune response in Ndufs4 KO mice as more than just a consequence of neurodegeneration, and serves as a proof‐of‐concept to study neuroimmune‐related processes in other mitochondrial diseases that affect the brain.
AUTHOR CONTRIBUTIONS
Kevin Aguilar, Albert Quintana, Elisenda Sanz, and Juan Hidalgo conceived and designed the study. Kevin Aguilar, Gemma Comes, and Carla Canal performed research. Kevin Aguilar analyzed data. Kevin Aguilar and Juan Hidalgo wrote the initial draft of the manuscript. All authors edited, reviewed, and revised the manuscript.
CONFLICT OF INTEREST
The authors claim no competing interests.
Supporting information
Appendix S1 Supplementary Figures
ACKNOWLEDGMENTS
Kevin Aguilar and Carla Canal acknowledge the support of FPU17/02065 and PIF UAB fellowships, respectively. This study was supported by Ministerio de Ciencia, Innovación y Universidades y Fondo Europeo de Desarrollo Regional (RTI2018‐101105‐B‐I00; Juan Hidalgo) [Correction added on August, 11, 2022, after first online publication: the grant/award number for the European Regional Development Fund has been changed from RTI2018‐101105–I00 to RTI2018‐101105‐B‐I00.]. Albert Quintana and Elisenda Sanz received Ramón y Cajal fellowships (RyC‐2012‐11873; Albert Quintana, RYC2019‐028501‐I; Elisenda Sanz). Elisenda Sanz received funds from MICIU Proyectos I + D + i “Retos Investigación” (RTI2018‐101838‐J‐I00). Albert Quintana received funds from the European Research Council (Starting grant NEUROMITO, ERC‐2014‐StG‐638106), MINECO Proyectos I + D de Excelencia (SAF2014‐57981P; SAF2017‐88108‐R), MICINN Proyectos I + D + i (PID2020‐114977RB‐I00), AGAUR (2017SGR‐323) and “la Caixa” Foundation (ID 100010434), under the agreement LCF/PR/HR20/52400018. We acknowledge Prof. Ian L. Campbell for the GFAP‐IL6 mouse. We also want to thank Dr. Gemma Manich for kindly providing anti‐TMEM119.
Aguilar, K. , Comes, G. , Canal, C. , Quintana, A. , Sanz, E. , & Hidalgo, J. (2022). Microglial response promotes neurodegeneration in the Ndufs4 KO mouse model of Leigh syndrome. Glia, 70(11), 2032–2044. 10.1002/glia.24234
[Correction added on August, 11, 2022, after first online publication: the grant/award number for the European Regional Development Fund has been changed from RTI2018‐101105–I00 to RTI2018‐101105‐B‐I00.]
Funding information “la Caixa” Foundation, Grant/Award Number: ID 100010434; Agència de Gestió d'Ajuts Universitaris i de Recerca, Grant/Award Number: 2017SGR‐323; European Regional Development Fund, Grant/Award Number: RTI2018‐101105‐B‐I00; H2020 European Research Council, Grant/Award Number: ERC‐2014‐StG‐638106; Ministerio de Ciencia e Innovación, Grant/Award Number: PID2020‐114977RB‐I00; Ministerio de Ciencia, Innovación y Universidades, Grant/Award Number: RTI2018‐101838‐J‐I00; Secretaría de Estado de Investigación, Desarrollo e Innovación, Grant/Award Numbers: SAF2014‐57981P, SAF2017‐88108‐R
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Arii, J. , & Tanabe, Y. (2000). Leigh syndrome: Serial MR imaging and clinical follow‐up. American Journal of Neuroradiology, 21(8), 1502–1509. http://www.ajnr.org/content/21/8/1502 [PMC free article] [PubMed] [Google Scholar]
- Bagnall, M. W. , Stevens, R. J. , & du Lac, S. (2007). Transgenic mouse lines subdivide medial vestibular nucleus neurons into discrete, Neurochemically distinct populations. Journal of Neuroscience, 27(9), 2318–2330. 10.1523/JNEUROSCI.4322-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsa, E. , Perry, E. A. , Bennett, C. F. , Jedrychowski, M. , Gygi, S. P. , Doench, J. G. , & Puigserver, P. (2020). Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nature Communications, 11(1), 1–12. 10.1038/s41467-020-16423-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M. L. , Bennett, F. C. , Liddelow, S. A. , Ajami, B. , Zamanian, J. L. , Fernhoff, N. B. , Mulinyawe, S. B. , Bohlen, C. J. , Adil, A. , Tucker, A. , Weissman, I. L. , Chang, E. F. , Li, G. , Grant, G. A. , Hayden Gephart, M. G. , & Barres, B. A. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences, 113(12), 1738–1746. 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolea, I. , Gella, A. , Sanz, E. , Prada‐Dacasa, P. , Menardy, F. , Bard, A. M. , Machuca‐Márquez, P. , Eraso‐Pichot, A. , Mòdol‐Caballero, G. , Navarro, X. , Kalume, F. , & Quintana, A. (2019). Defined neuronal populations drive fatal phenotype in a mouse model of Leigh syndrome. eLife, 8, e47163. 10.7554/ELIFE.47163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore, M. R. P. , Najafi, A. R. , Koike, M. A. , Dagher, N. N. , Spangenberg, E. E. , Rice, R. A. , Kitazawa, M. , Matusow, B. , Nguyen, H. , West, B. L. , & Green, K. N. (2014). Colony‐stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82(2), 380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erta, M. , Quintana, A. , & Hidalgo, J. (2012). Interleukin‐6, a major cytokine in the central nervous system. International Journal of Biological Sciences, 8(9), 1254–1266. 10.7150/ijbs.4679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassone, E. , & Rahman, S. (2012). Complex I deficiency: Clinical features, biochemistry and molecular genetics. Journal of Medical Genetics, 49(9), 578–590. 10.1136/JMEDGENET-2012-101159 [DOI] [PubMed] [Google Scholar]
- Giannakopoulou, C. E. , Sotiriou, A. , Dettoraki, M. , Yang, M. , Perlikos, X. F. , Toumpanakis, D. , Prezerakos, G. , Koutsourelakis, I. , Kastis, G. A. , Vassilakopoulou, V. , Mizi, E. , Papalois, A. , Greer, J. J. , & Vassilakopoulos, T. (2019). Regulation of breathing pattern by IL‐10. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 317, 190–202. 10.1152/ajpregu.00065.2019 [DOI] [PubMed] [Google Scholar]
- Giralt, M. , Ramos, R. , Quintana, A. , Ferrer, B. , Erta, M. , Castro‐Freire, M. , Comes, G. , Sanz, E. , Unzeta, M. , Pifarré, P. , García, A. , Campbell, I. L. , & Hidalgo, J. (2013). Induction of atypical EAE mediated by transgenic production of IL‐6 in astrocytes in the absence of systemic IL‐6. Glia, 61(4), 587–600. 10.1002/glia.22457 [DOI] [PubMed] [Google Scholar]
- Gorman, G. S. , Chinnery, P. F. , DiMauro, S. , Hirano, M. , Koga, Y. , McFarland, R. , Suomalainen, A. , Thorburn, D. R. , Zeviani, M. , & Turnbull, D. M. (2016). Mitochondrial diseases. Nature Reviews. Disease Primers, 2, 16080. 10.1038/nrdp.2016.80 [DOI] [PubMed] [Google Scholar]
- Green, K. N. , Crapser, J. D. , & Hohsfield, L. A. (2020). To kill a microglia: A case for CSF1R inhibitors. Trends in Immunology, 41(9), 771–784. 10.1016/J.IT.2020.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, K. N. , & Hume, D. A. (2021). On the utility of CSF1R inhibitors. Proceedings of the National Academy of Sciences, 118(4), 1–2. 10.1073/PNAS.2019695118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, J. , Fan, Y. , Zhou, K. , Zhu, K. , Blomgren, K. , Lund, H. , Zhang, X. M. , & Harris, R. A. (2020). Underestimated peripheral effects following pharmacological and conditional genetic microglial depletion. International Journal of Molecular Sciences, 21(22), 8603. 10.3390/IJMS21228603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, I. H. , Zazzeron, L. , Goldberger, O. , Marutani, E. , Wojtkiewicz, G. R. , Ast, T. , Wang, H. , Schleifer, G. , Stepanova, A. , Brepoels, K. , Schoonjans, L. , Carmeliet, P. , Galkin, A. , Ichinose, F. , Zapol, W. M. , & Mootha, V. K. (2019). Leigh syndrome mouse model can be rescued by interventions that normalize brain Hyperoxia, but not HIF activation. Cell Metabolism, 30(4), 824–832.e3. 10.1016/j.cmet.2019.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, I. H. , Zazzeron, L. , Goli, R. , Alexa, K. , Schatzman‐Bone, S. , Dhillon, H. , Goldberger, O. , Peng, J. , Shalem, O. , Sanjana, N. E. , Zhang, F. , Goessling, W. , Zapol, W. M. , & Mootha, V. K. (2016). Hypoxia as a therapy for mitochondrial disease. Science, 352(6281), 54–61. 10.1126/SCIENCE.AAD9642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, S. C. , Yanos, M. E. , Kayser, E. B. , Quintana, A. , Sangesland, M. , Castanza, A. , Uhde, L. , Hui, J. , Wall, V. Z. , Gagnidze, A. , Oh, K. , Wasko, B. M. , Ramos, F. J. , Palmiter, R. D. , Rabinovitch, P. S. , Morgan, P. G. , Sedensky, M. M. , & Kaeberlein, M. (2013). MTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science, 342(6165), 1524–1528. 10.1126/SCIENCE.1244360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S. A. , & Jenkins, B. J. (2018). Recent insights into targeting the IL‐6 cytokine family in inflammatory diseases and cancer. Nature Reviews Immunology, 18, 773–789. 10.1038/s41577-018-0066-7 [DOI] [PubMed] [Google Scholar]
- Kruse, S. E. , Watt, W. C. , Marcinek, D. J. , Kapur, R. P. , Schenkman, K. A. , & Palmiter, R. D. (2008). Mice with mitochondrial complex I deficiency develop a fatal Encephalomyopathy. Cell Metabolism, 7(4), 312–320. 10.1016/J.CMET.2008.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake, N. J. , Compton, A. G. , Rahman, S. , & Thorburn, D. R. (2016). Leigh syndrome: One disorder, more than 75 monogenic causes. Annals of Neurology, 79, 190–203. 10.1002/ana.24551 [DOI] [PubMed] [Google Scholar]
- Lee, J. S. , Yoo, T. , Lee, M. , Lee, Y. , Jeon, E. , Kim, S. Y. , Lim, B. C. , Kim, K. J. , Choi, M. , & Chae, J. H. (2020). Genetic heterogeneity in Leigh syndrome: Highlighting treatable and novel genetic causes. Clinical Genetics, 97(4), 586–594. 10.1111/CGE.13713 [DOI] [PubMed] [Google Scholar]
- Lei, F. , Cui, N. , Zhou, C. , Chodosh, J. , Vavvas, D. G. , & Paschalis, E. I. (2020). CSF1R inhibition by a small‐molecule inhibitor is not microglia specific; affecting hematopoiesis and the function of macrophages. Proceedings of the National Academy of Sciences, 117(38), 23336–23338. 10.1073/PNAS.1922788117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, Y. , Guerra Martinez, C. , Torres‐Odio, S. , Bell, S. L. , Birdwell, C. E. , Bryant, J. D. , Tong, C. W. , Watson, R. O. , West, L. C. , & West, A. P. (2021). Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice. Science Advances, 7(22), 1–16. 10.1126/SCIADV.ABE7548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. , & Barres, B. (2018). Microglia and macrophages in brain homeostasis and diseases. Nature Reviews Immunology, 18(4), 225–242. 10.1038/nri.2017.125 [DOI] [PubMed] [Google Scholar]
- Lorea‐Hernández, J. J. , Camacho‐Hernández, N. P. , & Peña‐Ortega, F. (2020). Interleukin 1‐beta but not the interleukin‐1 receptor antagonist modulates inspiratory rhythm generation in vitro. Neuroscience Letters, 734, 134934. 10.1016/J.NEULET.2020.134934 [DOI] [PubMed] [Google Scholar]
- Lorea‐Hernández, J. J. , Morales, T. , Rivera‐Angulo, A. J. , Alcantara‐Gonzalez, D. , & Peña‐Ortega, F. (2016). Microglia modulate respiratory rhythm generation and autoresuscitation. Glia, 64(4), 603–619. 10.1002/GLIA.22951 [DOI] [PubMed] [Google Scholar]
- Luna‐sánchez, M. , Bianchi, P. , & Quintana, A. (2021). Mitochondria‐induced immune response as a trigger for neurodegeneration: A pathogen from within. International Journal of Molecular Sciences, 22(16), 8523. 10.3390/ijms22168523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy, G. S. , Reczek, C. R. , Reyfman, P. A. , Mithal, D. S. , Horbinski, C. M. , & Chandel, N. S. (2020). NAD+ regeneration rescues lifespan, but not ataxia, in a mouse model of brain mitochondrial complex I dysfunction. Cell Metabolism, 32(2), 301–308.e6. 10.1016/J.CMET.2020.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, H. C. , Louw, R. , Mereis, M. , Venter, G. , Boshoff, J.‐D. , Mienie, L. , Reenen, M. , Venter, M. , Lindeque, J. Z. , Domínguez‐Martínez, A. , Quintana, A. , & Westhuizen, F. H. (2021). Metallothionein 1 overexpression does not protect against mitochondrial disease pathology in Ndufs4 knockout mice. Molecular Neurobiology, 58(1), 243–262. 10.1007/S12035-020-02121-Y [DOI] [PubMed] [Google Scholar]
- Miller, V. M. , Lawrence, D. A. , Coccaro, G. A. , Mondal, T. K. , Andrews, K. , Dreiem, A. , & Seegal, R. F. (2010). Sex effects of Interleukin‐6 deficiency on neuroinflammation in aged C57Bl/6 mice. Brain Research, 1318, 11–22. 10.1016/J.BRAINRES.2009.12.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama, S. , Homma, R. , & Imamura, F. (2014). Neuronal organization of olfactory bulb circuits. Frontiers in Neural Circuits, 98(8), 1–19. 10.3389/fncir.2014.00098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi, A. R. , Crapser, J. , Jiang, S. , Ng, W. , Mortazavi, A. , West, B. L. , & Green, K. N. (2018). A limited capacity for microglial repopulation in the adult brain. Glia, 66(11), 2385–2396. 10.1002/glia.23477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos, G. , & Franklin, K. B. J. (2013). Paxinos and Franklin's the mouse brain in stereotaxic coordinates. Academic Press. [Google Scholar]
- Peña‐Ortega, F. (2019). Clinical and experimental aspects of breathing modulation by inflammation. Autonomic Neuroscience, 216, 72–86. 10.1016/J.AUTNEU.2018.11.002 [DOI] [PubMed] [Google Scholar]
- Perry, E. A. , Bennett, C. F. , Luo, C. , Balsa, E. , Jedrychowski, M. , O'Malley, K. E. , Latorre‐Muro, P. , Ladley, R. P. , Reda, K. , Wright, P. M. , Gygi, S. P. , Myers, A. G. , & Puigserver, P. (2021). Tetracyclines promote survival and fitness in mitochondrial disease models. Nature Metabolism, 3(1), 33–42. 10.1038/s42255-020-00334-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada‐Dacasa, P. , Urpi, A. , Sánchez‐Benito, L. , Bianchi, P. , & Quintana, A. (2020). Measuring breathing patterns in mice using whole‐body plethysmography. Bio‐Protocol, 10(17), 3741. 10.21769/BIOPROTOC.3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana, A. , Kruse, S. E. , Kapur, R. P. , Sanz, E. , & Palmiter, R. D. (2010). Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proceedings of the National Academy of Sciences, 107(24), 10996–11001. 10.1073/PNAS.1006214107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana, A. , Zanella, S. , Koch, H. , Kruse, S. E. , Lee, D. , Ramirez, J. M. , & Palmiter, R. D. (2012). Fatal breathing dysfunction in a mouse model of Leigh syndrome. The Journal of Clinical Investigation, 122(7), 2359–2368. 10.1172/JCI62923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis, P. , Fernández‐Gayol, O. , Comes, G. , Aguilar, K. , Escrig, A. , Giralt, M. , Palmiter, R. D. , & Hidalgo, J. (2020). A new mouse model to study restoration of interleukin‐6 (IL‐6) expression in a Cre‐dependent manner: Microglial IL‐6 regulation of experimental autoimmune encephalomyelitis. Journal of Neuroinflammation, 17(1), 1–17. 10.1186/S12974-020-01969-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchis, P. , Fernández‐Gayol, O. , Vizueta, J. , Comes, G. , Canal, C. , Escrig, A. , Molinero, A. , Giralt, M. , & Hidalgo, J. (2020). Microglial cell‐derived interleukin‐6 influences behavior and inflammatory response in the brain following traumatic brain injury. Glia, 68(5), 999–1016. 10.1002/GLIA.23758 [DOI] [PubMed] [Google Scholar]
- Shil, S. K. , Kagawa, Y. , Umaru, B. A. , Nanto‐Hara, F. , Miyazaki, H. , Yamamoto, Y. , Kobayashi, S. , Suzuki, C. , Abe, T. , & Owada, Y. (2021). Ndufs4 ablation decreases synaptophysin expression in hippocampus. Scientific Reports, 11(1), 10969. 10.1038/s41598-021-90127-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spooren, A. , Kolmus, K. , Laureys, G. , Clinckers, R. , de Keyser, J. , Haegeman, G. , & Gerlo, S. (2011). Interleukin‐6, a mental cytokine. Brain Research Reviews, 67, 157–183. 10.1016/j.brainresrev.2011.01.002 [DOI] [PubMed] [Google Scholar]
- Stokes, J. C. , Bornstein, R. L. , James, K. , Park, K. Y. , Spencer, K. A. , Vo, K. , Snell, J. C. , Johnson, B. M. , Morgan, P. G. , Sedensky, M. M. , Baertsch, N. A. , & Johnson, S. C. (2022). Leukocytes mediate disease pathogenesis in the Ndufs4(KO) mouse model of Leigh syndrome. JCI Insight, 7(5), e156522. 10.1172/jci.insight.156522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, A. P. , & Shadel, G. S. (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nature Reviews Immunology, 17, 363–375. 10.1038/nri.2017.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y. , Wang, H. , Kouadir, M. , Song, H. , & Shi, F. (2019). Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death & Disease, 10(2), 1–11. 10.1038/s41419-019-1413-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, F. , Liang, S. , & Zhong, Z. (2019). Emerging role of mitochondrial DNA as a major driver of inflammation and disease progression. Trends in Immunology, 40, 1120–1133. 10.1016/j.it.2019.10.008 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplementary Figures
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.