

Abstract
Mitogen‐activated protein 3 kinase 7 (MAP3K7) encodes the ubiquitously expressed transforming growth factor β‐activated kinase 1, which plays a crucial role in many cellular processes. Mutationsin the MAP3K7 gene have been linked to two distinct disorders: frontometaphyseal dysplasia type 2 (FMD2) and cardiospondylocarpofacial syndrome (CSCF). The fact that different mutations can induce two distinct phenotypes suggests a phenotype/genotype correlation, but no side‐by‐side comparison has been done thus far to confirm this. Here, we significantly expand the cohort and the description of clinical phenotypes for patients with CSCF and FMD2 who carry mutations in MAP3K7. Our findings support that in contrast to FMD2‐causing mutations, CSCF‐causing mutations in MAP3K7 have a loss‐of‐function effect. Additionally, patients with pathogenic mutations in MAP3K7 are at risk for (severe) cardiac disease, have symptoms associated with connective tissue disease, and we show overlap in clinical phenotypes of CSCF with Noonan syndrome (NS). Together, we confirm a molecular fingerprint of FMD2‐ versus CSCF‐causing MAP3K7 mutations and conclude that mutations in MAP3K7 should be considered in the differential diagnosis of patients with syndromic congenital cardiac defects and/or cardiomyopathy, syndromic connective tissue disorders, and in the differential diagnosis of NS.
Keywords: cardiospondylocarpofacial syndrome, frontometaphyseal dysplasia type 2, MAP3K7, Noonan syndrome
1. INTRODUCTION
The mitogen‐activated protein 3 kinase 7 (MAP3K7) gene (MIM# 602614) encodes transforming growth factor β (TGF‐β)‐activated kinase 1 (TAK1), which plays a vital role in innate and adaptive immunity by regulating inflammatory responses and regulating cell differentiation, cell survival, and apoptosis (Dai et al., 2012; Yu et al., 2014) through interaction with TAK1‐binding proteins (TAB1, TAB2, and TAB3) (Xu & Lei, 2020). Mutations in MAP3K7 have been associated with two autosomal dominant conditions: frontometaphyseal dysplasia type 2 (FMD2; MIM #617137) caused by recurrent gain‐of‐function mutations (Wade et al., 2016) and cardiospondylocarpofacial (CSCF; MIM #157800) syndrome caused by individual mutations most often considered to induce loss‐of‐function (Le Goff et al., 2016).
FMD is a progressive skeletal dysplasia characterized by sclerosis of the skull, joint contractures, and undermodeled long bones, scoliosis, and prominent supraorbital ridges. Other features include hypertelorism, down‐slanting palpebral fissures, broad nasal bridge, full cheeks, micrognathia, hydronephrosis, cleft palate, hearing loss, ulnar deviation of the hands, camptodactyly, wrist contractures, long fingers, structural cardiac defect, and keloid scars (Costantini et al., 2018). FMD is caused by gain‐of‐function mutations in FLNA, TAB2, and MAP3K7 (Giuliano et al., 2005; Robertson, 2004; Wade et al., 2016, 2017). Most patients have normal intellect, although intellectual disability (ID) has been reported (Basart et al., 2015; Wade et al., 2016). In total, 19 FMD2 patients carrying mutations in MAP3K7 have been described in the literature (Costantini et al., 2018; Wade et al., 2017). A recurrent variant was identified in 16 patients (c.1454C>T (p.(Pro485Leu))), and the other three described mutations causing the FMD2 phenotype are c.208G>C (p.(Glu70Gln)), c.299T>A (p.(Val100Glu)), and c.502G>C (p.(Gly168Arg)). This recurrent variant leads to a severe skeletal phenotype, keloid scarring, and (in 3 out of 16 patients) ID. The three patients with the infrequent missense mutations show a milder phenotype consistent with FMD, without ID (Costantini et al., 2018).
CSCF was first described in 1966 as a new syndrome and autosomal dominant inheritance was suggested (Forney et al., 1966). The initial report describes a mother and two of her four daughters with congenital mitral valve insufficiency, skeletal malformations (fusion of cervical vertebrae/tarsal bones/carpal bones, phalangeal shortening), conductive deafness, and short stature. The hearing loss resulted from the fusion of the stapes to the round window and was improved by surgical correction. In 2010, two additional patients with an overlapping phenotype were reported to introduce the name CSCF for the first time (Sousa et al., 2010). However, it was not until 2016 that the gene MAP3K7 was identified as the cause of CSCF by Le Goff et al. (2016) who described six patients with CSCF phenotype and a causative heterozygous missense variant in the gene: three sporadic cases and one family (a father and two sons). Morlino et al. (2018) summarize these patients in their publication, adding a 12th novel patient with CSCF due to a de novo variant in the MAP3K7 gene (Morlino et al., 2018). In 2020, the same group published functional studies on patient fibroblasts demonstrating that CSCF is caused by loss‐of‐function mutations in the gene through the TGF‐β pathway (Micale et al., 2020). CSCF has also been proposed to show clinical overlap with the Noonan syndrome (NS; MIM# 163950) gestalt (Minatogawa et al., 2022; Morlino et al., 2018; Sousa et al., 2010), but functional assays to test whether this belongs to the group of RASopathies, has not been provided yet. Interestingly, ID has been described in FMD2, but not in CSCF.
Here, we expand the cohort of patients with CSCF and FMD2 carrying mutations in MAP3K7, describing 14 novel patients with CSCF and 2 novel patients with FMD2 (one with the recurrent variant and severe ID). On the basis of the phenotype of the patients in our cohort, we confirm that both CSCF and FMD2 are connective tissue diseases, as suggested previously (Minatogawa et al., 2022; Morlino et al., 2018; Mortier et al., 2019). We describe severe congenital left‐sided cardiac defects in CSCF patients and we aim to confirm the overlap of CSCF patients with the NS phenotype. A similar overlap has also been suggested for patients carrying mutations in TAB2, which is an activator for MAP3K7 (Engwerda et al., 2021). Additionally, we provide for the first time a side‐by‐side comparison of the biochemical effects of missense mutations in MAP3K7 for CSCF and FMD2, confirming a molecular fingerprint to distinguish between the two disorders.
2. METHODS
2.1. Patients
Sixteen patients with novel mutations in the MAP3K7 gene were included in this clinical observation with functional laboratory analysis of the mutations. Patients were identified in the clinical setting with trio whole‐exome sequencing (WES). Patients from five countries (seven medical centers in total) were identified using Genematcher (Sobreira et al., 2015). Written consent was obtained from adult patients and parents of pediatric patients for participation in the study and for publishing photographs.
2.2. Clinical observations
A cross‐sectional clinical assessment (medical history, physical examination, variant classification, and (if consented) photography) was performed by the treating physician to complete a detailed chart of phenotypic features. If available, radiologic and cardiac analysis outcomes were collected from the medical file. All data were entered into a pseudoanonymized digital database. The descriptive data were analyzed and the results are presented in the manuscript text and Table 1.
Table 1.
Patient Characteristic and Clinical Features
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|
| Variant | c.143 | c.143 | c.248G> | c.329 | c.329G> | 586 | 589 | 607+2 |
| G>A’ | G>A’ | A’ | G>A’ | A’ | A>G’ | G>A’ | DELT* | |
| Protein Change | p.(Gly48Glu) | p.(Gly48Glu) | p.(Arg83His) | p.(Gly110Asp) | p.(Gly110Asp) | p.(Met196Val) | p.(Ala1977Thr) | p.(E202_G203insGKWQXY) (Splicing Effect: Use Of A Later Donor Site) |
| Variant classification ACMG** | 3 | 4 | 3 | 4 | 5 | 3 | 3 | 4 |
| Inheritance | De Novo | De Novo | De Novo | De Novo | De Novo | De Novo | De Novo | De Novo |
| Phenotype | CSCF | CSCF | CSCF | CSCF | CSCF | CSCF | CSCF | CSCF |
| Age | 5 | 0 | 3 | 6 | 15 | 8 | 1 | 12 |
| In Years | (8 Months) | |||||||
| Gender | Female | Male | Female | Female | Female | Male | Female | Female |
| Ancestry | Caucasian | American/Caucasian | Hispanic | Caucasian | Caucasian | Caucasian/Asian | Middle‐East | Caucasian |
| Country Of Participation | Holland | USA | Holland | Holland | Holland | Holland | Holland | England |
| Postnatal measurements | ||||||||
| Weeks Of Gestation | 39 3/7 | 37 | 38 | 38 6/7 | 40 | 38 2/7 | 39 | 41 |
| Weight, Gr (SDS) | 2740 (‐2) | 3.5 () | 2850 (0) | 3052 (0) | 2340 (<‐2.5) | 3060 (‐0.5) | 3200 (0) | 3044 (‐1) |
| Length, Cm (SDS) | 47 (‐1,4) | 49 (‐4.0) | 46 (‐1.9) | 48 (0) | ||||
| Head Circumference, Cm (SDS) | 33 (‐1,6) | 34 (‐1.7) | 35.5 (+0.7) | 35 (0 SD) | ||||
| Measurements Last Evaluation | ||||||||
| Weight, Kg | 14.2 | 8.9 | 12 | 17 | 36.6 | 21,2 | 9,95 | 29.6 |
| Length, Cm (SDS) | 102 (‐2.7) | 66 (‐1.68) | 86,7 (‐3) | 104 (‐3,1) | 153,2 (‐2,1) | 122 (‐2,1) | 73.8 (‐1.4) | 143 (<‐2.5) |
| Head Circumference, Cm (SDS) | 48 (‐1.7) | 43.6 (‐0.74) | 49,2 (0) | 50 (‐0,6) | 53,4 (‐0.9) | 52 (‐0,3) | 48.2 (1.1) | 51,5 (‐1,4) |
| Facial | ||||||||
| Low Posterior Hair Line | Yes | No | No | Yes | No | No | ||
| Hypotonic Face | Yes | Yes | No | No | Yes | Yes | No | No |
| Full Cheeks | Yes | Yes | Yes | Yes | No | Yes | Yes | No |
| Low‐Set Ears | No | Yes | No | Yes | No | Yes | Yes | |
| Posteriorly Rotated Ears | Yes | Yes | No | Yes | Yes | Yes | No | Yes |
| Hypertelorism | Yes | Yes | No | Yes | No | Yes | Yes | Yes |
| Triangular Face | Yes | No | No | No | No | No | Yes | No |
| Strabismus | Yes | No | No | No | Yes | Yes | No | No |
| Ptosis | Yes | Yes | No | No | Yes | Yes | No | Yes |
| Downslanting Palpebral Fissures | No | No | No | No | No | No | No | Yes |
| Upslanting Palpebral Fissures | Yes | Yes | No | No | No | Yes | No | No |
| Epicanthal Folds | Yes | Yes | Yes | No | No | Yes | No | Yes |
| Peri‐Orbital Fullness | Yes | Yes | No | No | No | Yes | Yes | No |
| Anteverted Nares | Yes | Yes | Yes | No | Yes | No | Yes | |
| Round Tipped Nose | Yes | Yes | Yes | Yes | Yes | Yes | Yes | |
| Long Philtrum | Yes | Yes | Yes | No | Yes | Yes | Yes | |
| High Arched Palate | No | Yes | Yes | No | Yes | |||
| Micrognathia | No | Yes | Yes | No | No | Yes | No | No |
| Webbed Neck | Yes | Yes | No | No | No | No | No | |
| Cardiac Phenotype | ||||||||
| Congenital Heart Defect | Yes | No | No | Yes | Yes | No | Yes | |
| Ventricular Septal Defects | No | No | No | No | No | No | ||
| Atrial Septal Defects | No | No | No | No | No | No | Yes | |
| Cardiomyopathy | No | Yes | No | Yes | No | No | No | |
| Other | Dilated Cardiomyopathy with right ventricular dysfunction. He is on the waiting list for Heart Transplantation | Dilated Cardiomyopathy | Bowing Mitral Valve without Regurgitation, PDA | |||||
| Skeletal Phenotype | ||||||||
| Joint Laxity | Yes | No | Yes | Yes | Yes | Yes | Yes | |
| Scoliosis | No | No | No | No | Yes (35 Degrees) | No | No | Yes |
| Vertebral Abnormalities | Yes | No | Yes | Yes | ||||
| Carpal Fusion | Yes | No | No | No | No | Yes | ||
| Flexion Contractures | No | No | No | No | No | |||
| Brachydactyly | Yes | No | No | Yes | Yes | Yes | Yes | |
| Camptodactyly | No | No | No | No | No | |||
| Pectus Excavatum | Yes | No | No | No | No | No | No | |
| Neurologic Phenotype | ||||||||
| Hypotonia | Yes | No | Yes | No | Yes | Yes | ||
| Atrophic Adductor Pollicis | Yes | No | Yes | Yes | ||||
| Muscle Hypoplasia (Other) | No | No | ||||||
| Intellectual Disability | No | No | No | No | No | No | ||
| Behavior Disorders | No | Yes (Autism) | No | No | ||||
| MRI Brain | Left Hemispheric Ischemic Lesions At Insulo‐Parieto‐ And Temporal Region That Corresponds To MCA Territory. Thin Corpus Callosum, Chiari I Malformation | Wide Virchow‐Robin Space, Some Hyperintensity, In Particular Occipital In The White Matter. Slightly Broadened Front Horns. | Normal At Age 3 Weeks | |||||
| Miscellaneous | ||||||||
| Hearing Loss | No | No | No | Yes (Conductive) | No | Yes (Sensorineural) | ||
| Feeding Difficulties In Infancy | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes |
| Cryptorchidism | ‐ | No | ‐ | ‐ | ‐ | Yes | ‐ | ‐ |
| Widely Spaced Nipples | Yes | Yes | No | Yes | Yes | Yes | ||
| Other | Hypogamma‐globulinemia, Eczema | Hirsutism And Deep Sacral Dimple | Rhizomelic Shortening | Class 3 malocclusion jaw |
| Patient | 9 | 10 (Father P9) | 11 | 12 | 13 | 14 | 15 | 16 | Total |
|---|---|---|---|---|---|---|---|---|---|
| Variant | 616 | 616 | 617 | 713 | 721 | 721 | 337 | 1535 | |
| T>G’ | T>G’ | A>G’ | G>A’ | T>G’ | T>G’ | T>G’ | C>T’ | ||
| Protein Change | p.(Tyr206Asp) | p.(Tyr206Asp) | p.(Tyr206Cys) | p.(Arg238Gln) | p.(Trp241Gly) | p.(Trp241Gly) | p.(Tyr113Asp) | p.(Pro485Leu) | |
| Variant classification ACMG** | 4 | 4 | 4 | 4 | 5 | 5 | 3 | 4 | |
| Inheritance | Paternal | Na | De Novo | De Novo | De Novo | Not maternal; | De Novo | De Novo | 1 Inherited (1 father not tested) |
| Phenotype | CSCF | CSCF | CSCF | CSCF | CSCF | CSCF | FMD2 | FMD2 | 2/15 FMD2 |
| Age | 0 | 35 | 9 | 7 | 2 | 36 | 18 | 4 | Median 6 |
| In Years | (2 Months) | Iqr 10 | |||||||
| Gender | Female | Male | Male | Female | Female | Female | Male | Female | 10/15 Female |
| Ancestry | Caucasian/Asian | Caucasian | Caucasian | Caucasian | Caucasian | Chinese | Caucasian | Caucasian | 10/15 Caucasian |
| Country Of Participation | Holland | Holland | USA | Switzerland | England | USA | Holland | Italy | 8/15 Holland |
| Postnatal measurements | |||||||||
| Weeks Of Gestation | 37 5/7 | 40 2/7 | 38 4/7 | 36 2/7 | Term | 34 1/7 | 35 2/7 | 3 Born Premature | |
| Weight, Gr (SDS) | 2800 (‐1) | 3730 (‐0.5) | 2570 (‐1.9) | 2003 (‐2.5) | 2700 (‐2 SDS) | 1655 (<‐2.5) | 1910 (‐2.5) | 4 SGA | |
| Length, Cm (SDS) | 45 (‐1) | 50 (‐1) | 48 (‐1.4) | 41,9 (‐2.2) | 42 (‐1) | Overall Normal | |||
| Head Circumference, Cm (SDS) | 34 (+1) | 35 (+1.9) | 34.5 (+2) | 32,5 (+1) | 28,5 (‐2.5) | Broad Range | |||
| Measurements Last Evaluation | |||||||||
| Weight, Kg | 4,2 | 18 | 21 | 10.6 | 37.8 | 47,8 | 12.5 | ||
| Length, Cm (SDS) | 53 (‐1,6) | 162,5 (‐3) | 115.2 (‐3.2) | 120.9 (‐0.8) | 81.3 (‐1.3) | 146.3 (<‐2.5) | 165,1 (‐2,2) | 88 (‐3,1) | 11/16 Short Stature |
| Head Circumference, Cm (SDS) | 37 (‐1) | 55,5 (‐1,1) | 50 (‐1.8) | 53.6 (1.3) | 48 (0.3) | 45 (<‐2.5) | 1/14 Microcephalic, FMD2 patient | ||
| Facial | |||||||||
| Low Posterior Hair Line | No | No | No | No | 2/10 | ||||
| Hypotonic Face | No | No | Yes | No | No | Yes | 6/14 | ||
| Full Cheeks | Yes | No | Yes | Yes | No | Yes | Yes | 11/15 | |
| Low‐Set Ears | Yes | No | No | No | No | 5/12 | |||
| Posteriorly Rotated Ears | No | No | No | No | No | No | 6/14 | ||
| Hypertelorism | Yes | Yes | No | No | Yes | Yes | 10/14 | ||
| Triangular Face | No | Yes | No | No | No | 3/13 | |||
| Strabismus | No | No | No | No | 3/12 | ||||
| Ptosis | No | Yes | Yes | No | Yes | No | Yes | 9/15 | |
| Downslanting Palpebral Fissures | No | Yes | Yes | No | No | Yes | 4/14 | ||
| Upslanting Palpebral Fissures | No | No | No | No | No | No | 3/14 | ||
| Epicanthal Folds | No | No | Yes | Yes | Yes | 8/13 | |||
| Peri‐Orbital Fullness | Yes | Yes | No | Yes | 7/12 | ||||
| Anteverted Nares | No | No | Yes | Yes | No | No | 7/13 | ||
| Round Tipped Nose | No | Yes | Yes | Yes | No | 10/12 | |||
| Long Philtrum | No | Yes | No | No | 7/11 | ||||
| High Arched Palate | Yes | No | Yes | Yes | 6/9 | ||||
| Micrognathia | No | No | No | No | Yes | Yes | 5/14 | ||
| Webbed Neck | No | No | Yes | No | No | 3/12 | |||
| Cardiac Phenotype | |||||||||
| Congenital Heart Defect | Yes | No | Yes | Yes | No | No | Yes | Yes | 9/15 |
| Ventricular Septal Defects | No | No | No | No | Yes | Yes | 2/12 | ||
| Atrial Septal Defects | No | No | No | No | No | No | 1/13 | ||
| Cardiomyopathy | Yes | No | No | No | Yes | No | No | 4/14 | |
| Other | Hypertrophic Cardiomyopathy | Aortic Atresia, Hypoplastic Left Heart Syndrome | Mitral Valve Prolapse | Dilated cardiomyopathy, SVT with ablation age 35, mid‐moderate tricuspid regurgitation | PDA | PDA, Bicuspid Aortic Valve With Stenosis, PLSCV, Single Coronary Sinus, Ectopic Atrial Tachycardia | |||
| Skeletal Phenotype | |||||||||
| Joint Laxity | Yes | No | Yes | 8/11 | |||||
| Scoliosis | No | Yes | Yes | 4/11 | |||||
| Vertebral Abnormalities | Yes | No | Yes | 5/7 | |||||
| Carpal Fusion | No | No | No | 2/8 | |||||
| Flexion Contractures | No | No | Yes | Yes | 2/9 (Both Fmd2) | ||||
| Brachydactyly | Yes | No | Yes | 7/10 | |||||
| Camptodactyly | No | Yes | Yes | 2/8 (Both Fmd2) | |||||
| Pectus Excavatum | No | Yes | Yes | No | 3/11 | ||||
| Neurologic Phenotype | |||||||||
| Hypotonia | Yes | Yes | Yes | No | Yes | 8/11 | |||
| Atrophic Adductor Pollicis | No | No | Yes | 4/7 | |||||
| Muscle Hypoplasia (Other) | Yes (Right Pectoralis) | Yes (Hands) | Yes | 3/5 | |||||
| Intellectual Disability | No | No | No | No | Yes (Severe) | 1/11 (Fmd2) | |||
| Behavior Disorders | Yes (Autism) | Yes (Autism, Phobias, Pica) | No | 3/7 | |||||
| MRI Brain | Scattered Small Foci Of Restricted Diffusion | Cerebral Atrophy, Reduced Corpus Callosum, And Chiari 1 Malformation | |||||||
| In The Periventricular White Matter Including Lesions | |||||||||
| In Bilateral Parietal White Matter And A Single Lesion Within The Left | |||||||||
| Frontal White Matter. Most Suggestive Of White | |||||||||
| Matter Hypoxic Ischemic Injury. | |||||||||
| Miscellaneous | |||||||||
| Hearing Loss | No | Yes (Conductive) | Yes (Conductive) | Yes | Yes (Mixed) | No | 6/12 | ||
| Feeding Difficulties In Infancy | Yes | Yes | Yes | No | Yes | 11/13 | |||
| Cryptorchidism | ‐ | No | No | ‐ | ‐ | Yes | ‐ | 2/5 | |
| Widely Spaced Nipples | No | Yes | Yes | Yes | 8/10 | ||||
| Other | Short Arms | Urethral meatus slit‐like orifice positioned inferiorly on the glans penis | Myopia ‐11 diopter | Ambiguous Genitalia | Severe Eczema |
‐ = Not Applicable; Empty= Not Available, CSCF = Cardiospondylocarpofacial Syndrome, FMD2 = Frontometaphyseal Dysplasia Type 2, IQR = Interquartile Range, SGA = Small for Gestational Age, PDA = Patent Ductus Arteriosus, PLSCV = Persistent Left Superior Vena Cava
‘Complying with HGSV format and reference sequence NM_145331.2.
Complying with HGSV format and reference sequence NC_000006.12 (NM_003188.3).
Richard et al. 2015. Genet Med. doi: 10.1038/gim.2015.30
2.3. Ethical considerations
The study was conducted according to the principles of the Declaration of Helsinki (10th version, October 2013, see for the most recent version: www.wma.net (World Medical Association, 2013) and in accordance with the Dutch Medical Research Involving Human Subjects Act (WMO). Approval from the medical ethics comity of the Erasmus Medical Center was given for conducting our retrospective case‐series study on these patients.
2.4. Laboratory assessments
2.4.1. Constructs
The MAP3K7 WT (NM_003188.4) complementary DNA (cDNA) was obtained from the human brain library by PCR (Phusion High Fidelity; Thermo Fisher Scientific) (all primers used can be found in Supporting Information: Tables 1 and 2). To be able to clone this sequence into our dual promotor expression vector (Proietti Onori et al., 2018), the fragment was tagged with restriction enzymes AscI; PacI by PCR (Phusion High Fidelity; Thermo Fisher Scientific). The point mutations were introduced with site‐directed mutagenesis (Phusion High Fidelity; Thermo Fisher Scientific). For the in vivo experiments, the “empty vector” serves as a control, consisting of the dual promotor expression vector only (no gene insertion in multiple cloning sites).
The TAB1 wt (NM_006116.3) cDNA was obtained from the human brain library by PCR (Phusion High Fidelity; Thermo Fisher Scientific). This was then also cloned in our dual promotor expression vector, tagging the fragment with restriction enzymes AscI; PacI by PCR (Phusion High Fidelity; Thermo Fisher Scientific).
2.4.2. HEK293T transfection
To assess the functional effects of the different MAP3K7 mutations, HEK293T cells were cultured in DMEM/10% fetal calf serum/1% penicillin/streptomycin in six‐wells plates and transfected with the following constructs: Control vector; MAP3K7 WT ; MAP3K7 Gly48Glu ; MAP3K7 Glu70Gln ; MAP3K7 Arg83His ; MAP3K7 Val100Glu ; MAP3K7 Gly110Asp ; MAP3K7 Tyr113Asp ; MAP3K7 Gly168Arg ; MAP3K7 Met196Val ; MAP3K7 Tyr206Cys ; MAP3K7 Tyr206Asp ; MAP3K7 Trp241Gly ; MAP3K7 Pro485Leu ; TAB1 WT . In single construct transfections, 3 μg construct per well was transfected. When in cotransfection of the different MAP3K7 constructs with TAB1 WT , 1.5 μg/construct/well was transfected. Constructs for cotransfection were mixed before actual transfection, allowing both constructs to be transfected at the same time. At subconfluency (60%–70%), cells were transfected with polyethylenimine (PEI) according to the manufacturer's instructions (Sigma‐Aldrich). To avoid the toxicity of prolonged exposure to PEI, the medium was refreshed after 4–6 h. Transfected cells were used to prepare protein lysates for Western blot.
2.4.3. Western blot
The cells were harvested in ice‐cold PBS, 48–72 h after transfection. Lysate samples were prepared by homogenizing the harvested HEK293Tcells in lysate buffer (10 mM Tris‐HCl 6.8, 2.5% SDS, 2 mM EDTA) containing protease inhibitor cocktail 2 (#P5726; Sigma) and 3 (#P0044; Sigma‐Aldrich) and protease inhibitor (#P8340; Sigma‐Aldrich). Protein concentrations were determined using the BCA kit (Pierce). Final working protein concentrations were adjusted to 1 mg/ml. Western blots were probed with primary antibodies against MAP3K7 (sc‐7967, 1:1000; Santa Cruz), phospho‐MAP3K7 (Thr187; #4536, 1:1000; Cell Signaling), extracellular signal‐regulated kinase (ERK)1/2 (#9102, 1:2000; Cell Signaling), phospho‐ERK1/2 (#9101, 1:2000; Cell Signaling), actin (MAB1501R, 1:20,000; Chemicon), RFP (#600401379, 1:2000; Rockland), nuclear factor‐κB (NFκB) (sc‐514451, 1:1000; Santa Cruz), phospho‐NFκB (sc‐136548, 1:1000, Santa Cruz), glyceraldehyde 3‐phosphate dehydrogenase (2118S, 1:2000; Cell Signaling), and TAB1 (67020‐1‐Ig, 1:10.000; Proteintech) and secondary antibodies (goat anti‐mouse [#926‐32210] and goat anti‐rabbit [#926‐68021], all 1:15,000, LI‐COR). Blots were quantified using LI‐COR Odyssey Scanner and Odyssey 3.0 software.
2.5. Statistical analysis
All data were assumed to be normally distributed. Statistical difference between the conditions for the in vitro overexpression experiments was determined using one‐way analysis of variance (ANOVA) followed by Bonferroni's post hoc test for multiple comparisons. For the Western blot analysis, two‐tailed unpaired t‐test was used (dual comparison). Neuronal migration was analyzed based on the proportion of electroporated cells targeted to the cortical plate at P1 (defined as the most proximal 40% of the dorsoventral distance between the pia and ventricle (first 4 of 10 equally‐spaced bins)) or to layer 2/3 of the somatosensory cortex, defined as the proximal 30% of the dorsoventral distance between the proximal 10% of the cortex (assumed to represent layer 1) and ventricle (bin 2–4 of equally‐spaced bins). For neuronal migration analyses, we used a minimum of three targeted pups per condition.
3. RESULTS
A combined overview of the mutations in our cohort and reported in the literature are shown in Figure 1. Patient characteristics and detailed phenotypic descriptions are shown in Table 1. If a specific feature was not clearly reported as positive or negative for a patient, this feature was considered missing data for the respective patient. For this reason, the total number of patients per feature may differ in our results.
Figure 1.
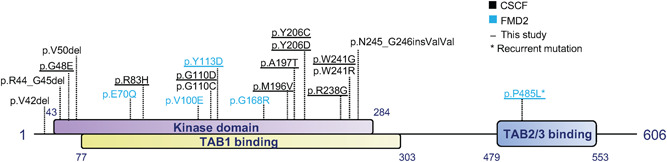
Schematic overview of MAP3K7 showing the localization of the different mutations. Schematic overview of MAP3K7 showing the kinase and TAB1 binding domain at the N‐terminal and the TAB2/3 binding domain at the C‐terminal of MAP3K7. In black, the CSCF‐related MAP3K7 mutations are indicated, and in blue, the FMD2‐related MAP3K7 mutations. The underlined mutations are the novel mutations identified in our cohort. MAP3K7Pro485Leu is the most recurrent FMD2‐causing variant. CSCF, cardiospondylocarpofacial syndrome; FMD2, frontometaphyseal dysplasia type 2; MAP3K7, mitogen‐activated protein 3 kinase 7.
3.1. Clinical phenotype of CSCF
Of the 14 patients with the CSCF phenotype, 12 carried de novo mutations in MAP3K7, one female inherited the variant from her father and one female did not inherit the variant from her mother, but the father was unavailable for testing. The clinical phenotype and patient characteristics are presented in detail in Table 1. Two adult CSCF patients were included. Figure 2 shows photographs of the patients with CSCF who consented to publication.
Figure 2.
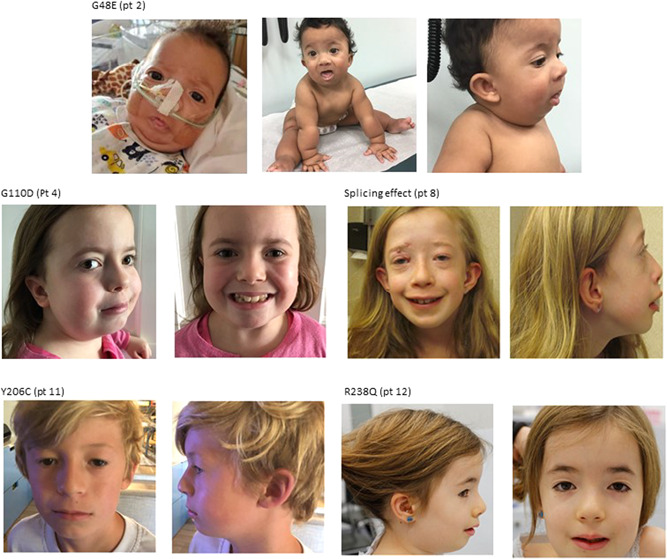
Light photographs of patients with CSCF. Five patients with CSCF: Patients show hypertelorism, full eyelids, full cheeks, round nasal tip, low set ears, and almond‐shaped eyes. CSCF, cardiospondylocarpofacial syndrome.
3.1.1. Dysmorphic features
The most frequently recorded dysmorphic features are hypotonic face (6/12 patients), full cheeks (9/13), low set ears (5/10), posteriorly rotated ears (7/12), hypertelorism (8/12), ptosis (8/13), epicanthal folds (6/12), periorbital fullness (7/11), anteverted nares (7/11), round‐tipped nose (9/11), long philtrum (7/9), and widely spaced nipples (7/9). Short stature (<−2 SDS for age and gender) was observed in 9/14 CSCF patients, ranging from −2.1 SDS to −4 SDS. Other less frequently reported features are listed in Table 1.
3.1.2. Cardiac features
Interestingly, CSCF patients showed a wide range of cardiac anomalies. Congenital heart defects that mostly seem to be left‐sided are reported in 7/13 patients ranging from mitral valve prolapse to hypoplastic left heart syndrome spectrum (c.617A>G (p.(Tyr206Cys)) variant, Patient 11; Table 1). Cardiomyopathy was observed in 4/12 patients (one hypertrophic and three dilated), the most severe in a neonate with the c.143G>A (p.(Gly48Glu)) variant (Patient 2), who is on the waiting list for heart transplantation.
3.1.3. Musculoskeletal features
Carpal fusion was assessed in seven patients and found in two of them. Brachydactyly was frequently observed (6/9). Pectus excavatum was seen in 3/11 patients. Joint laxity was a consistent feature in the pediatric patients and reported in 8/10 patients. Scoliosis was reported in two patients and for one of the patients, this was described as a Hobbs curve of 35°. Vertebral anomalies were identified in 4/6 patients and involved fusion of vertebrae (also of the odontoid process and anterior arch of C1), small intervertebral distance (thoracic), and loss of height of vertebrae and dysmorphic vertebrae. No contractures were described in our CSCF patients.
3.1.4. Neurologic features
No ID was described in any of the CSCF patients. Autism spectrum disorder, however, was present in 3/7 patients. Other neurologic features that drew attention are hypotonia (7/10) and hypoplasia of muscles; right pectoralis hypoplasia in one patient and hypoplasia of the adductor pollicis (3/6).
3.1.5. Hearing loss
Five out of 11 patients are reported to have hearing loss: One sensorineural with defective inner ear formations, and the other four conductive/mixed.
3.1.6. Other features
One male patient had cryptorchidism. Ten out of 11 patients had feeding difficulties at a young age, frequently needing nasogastric tube feeding for a period of time.
3.2. Clinical phenotype FMD2
Only two patients in this cohort have FMD2. Both patients have a de novo variant (one having the recurrent variant). The clinical phenotype and patient characteristics are presented in Table 1. More detailed information on these two cases can be found in Figure 3 and the Supporting Information.
Figure 3.
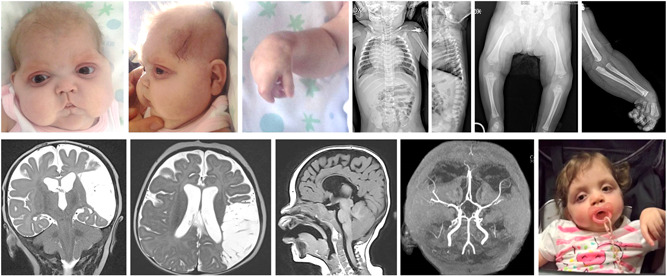
Clinical description and light photography/radiological pictures of the patient with the recurrent P485L variant causing FMD2 (Patient 16). Cranio‐facial dysmorphisms at 2 months (Panels 1–3): bitemporal narrowing, hypotelorism, down‐slanting palpebral fissures, short nose with flat nasal bridge, and depressed root, long and deeply grooved filter, microretrognathia. Radiological findings (Panels 4–7) at 7 months: a partial fusion of cervical vertebrae (C2–C3) and reduced bone length in relation to age. Magnetic resonance imaging at 7 months: T2‐weighted hyperintensity of the presumed vascular lesion in the coronal and transverse planes (Panels 8–9); T1 weighed scans in the sagittal plane: cerebral atrophy, reduced corpus callosum, and Chiari 1 malformation (Panel 10). Cranial time‐of‐flight magnetic resonance angiogram (3 T) at 7 months: flow reduction of the M2 branch of the left middle cerebral artery (Panel 11). Facial appearance at 3 years old (Panel 12).
3.2.1. Dysmorphic features
The reported dysmorphic features in our two patients are consistent with the FMD2 patients reported in medical literature, including full cheeks, hypertelorism, epicanthal folds, and micrognathia (Figure 3).
3.2.2. Cardiac features
Both FMD2 patients had a persistent ductus arteriosus, which was part of a more complex cardiac defect (ventricular septal defect, pulmonary atresia, bicuspid aortic valve with mild stenosis, progressive aortic root dilatation [> 3 z‐score], persistent left superior vena cava draining into the coronary sinus, and ectopic atrial tachycardia) in the patient born at 35 weeks of gestation with the recurrent c.1535C>T (p.(Pro485Leu)) variant. The congenital heart defect in this patient was a major reason for failure to thrive and recurrent admissions in her early months of life.
3.2.3. Musculoskeletal features
Both FMD2 patients had scoliosis and short stature (−2.2 SDS and −3.1 SDS). Both patients show camptodactyly of their fingers, flexion contractures (elbow with dislocation of the radial head), and carpal fusion. The older male patient has joint laxity. The younger female patient shows flared metaphyses, broad thumbs/fingers osteosclerosis, and partially fused with dysmorphic cervical vertebrae.
3.2.4. Neurologic features
The patient with the recurrent variant has a severe ID. Her brain magnetic resonance imaging shows cerebral atrophy, thin corpus callosum, and Chiari 1 malformation. She also has hypotonia and is suspected to possibly have muscular atrophy. The other patient has normal intellect and tone, as well as muscular hypoplasia of his hands.
3.2.5. Hearing loss
The adult patient (with the nonrecurrent variant) was shown to have mixed conductive/sensorineural hearing loss. The pediatric female patient with the recurrent variant was reported to also have hearing loss (type not specified). No temporal bone imaging is available for these patients.
3.2.6. Other features
The adult patient (with the nonrecurrent variant) had cryptorchidism and was consulted for ambiguous genitalia as a newborn. The young female patient (with the recurrent variant) had congenital stridor due to laryngo–trachea–bronchomalacia that improved with time and without the need for tracheostomy. Feeding problems are managed by PEG. Both patients showed excessive keloid scarring.
3.3. Functional assessment of the missense mutations identified in MAP3K7
The differences in clinical features seen in FMD2 versus CSCF patients are thought to be caused by different underlying molecular mechanisms. MAP3K7 mutations giving rise to FMD2 are most often recurrent, gain‐of‐function mutations (Wade et al., 2016), whereas CSCF is frequently caused by nonrecurrent missense mutations, where for some mutations, a loss‐of‐function effect was shown, but the functional effect of other mutations remains unknown. Our cohort, in combination with already published patients, allowed us to assess whether there is indeed a molecular fingerprint that can distinguish between FMD2 and CSCF. Since our cohort has only two FMD2 patients, we included the previously published FMD2‐causing missense mutations in MAP3K7 (Wade et al., 2016).
In searching for a molecular fingerprint, we first assessed the expression levels of the different MAP3K7 mutations upon overexpression in HEK‐293T cells. Whereas the mutations causing FMD2 showed similar or even a significantly higher expression level compared to MAP3K7WT, all CSCF mutations showed significantly lower expression levels compared to MAP3K7WT (FMD2 mutations: one‐way ANOVA F[5,25] = 6.16, p= 0.0008; MAP3K7WT vs. MAP3K7Glu70Gln: p = 0.39; MAP3K7WT vs. MAP3K7Val100Glu: p = 0.087; MAP3K7WT vs. MAP3K7Tyr113Asp: p = 0.42; MAP3K7WT vs. MAP3K7Gly168Arg: p= 0.40; MAP3K7WT vs. MAP3K7Pro485Leu: p = 0.0079, Dunnett's multiple comparison test; CSCF mutations: one‐way ANOVA F [7,23] = 6.39, p = 0.0003; MAP3K7WT vs. MAP3K7Gly48Glu: p = 0.004; MAP3K7WT vs. MAP3K7Arg83His: p = 0.0017; MAP3K7WT vs. MAP3K7Gly110Asp: p = 0.041; MAP3K7WT vs. MAP3K7Met196Val: p = 0.0013; MAP3K7WT vs. MAP3K7Tyr206Cys: p = 0.01; MAP3K7WT vs. MAP3K7Tyr206Asp: p = 0.0025; MAP3K7WT vs. MAP3K7Trp241Gly: p < 0.0001, Dunnett's multiple comparison test; Figure 4a). With the exception of MAP3K7Trp241Gly, reduced expression levels of the CSCF mutations could be normalized to control levels upon cotransfection with TAB1, one of the TAK1‐binding proteins known to enhance TAK1 function (Xu & Lei, 2020) (one‐way ANOVA F[7,54] = 4.3, p < 0.001; MAP3K7WT vs. MAP3K7Gly48Glu: p < 0.0001; MAP3K7WT vs. MAP3K7Arg83His: p = 0.86; MAP3K7WT vs. MAP3K7Gly110Asp: p = 0.9; MAP3K7WT vs. MAP3K7Met196Val: p = 0.25; MAP3K7WT vs. AP3K7Tyr206Cys: p0.9; MAP3K7WT vs. MAP3K7Tyr206Asp: p = 0.06; MAP3K7WT vs. MAP3K7Trp241Gly: p = 0.02, Dunnett's multiple comparison test; Figure 4b), indicating that these mutations cause instability of the MAP3K7 protein in the absence of TAB1.
Figure 4.
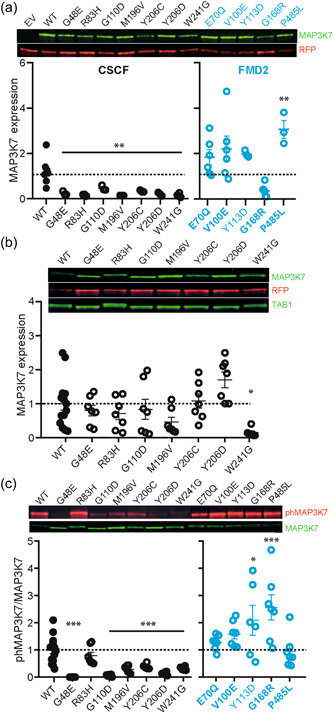
Differential expression and autophosphorylation of mutations in MAP3K7 causing CSCF or FMD2. (a) Western blot analysis shows reduced expression levels of CSCF‐causing MAP3K7 mutations (black circles), compared to MAP3K7WT, but normal or even increased expression levels of FMD2‐causing MAP3K7 mutations (blue circles), compared to MAP3K7WT, when overexpressed in HEK‐293T cells. N = 3 for all CSCF‐causing mutations, except MAP3K7Trp241Gly (N = 6). N = 6 for all FMD2‐causing mutations, except MAP3K7Tyr113Asp and MAP3K7Gly168Arg (N = 3). WT N = 7. (b) Western blot analysis showing that expression levels of CSCF‐causing MAP3K7 mutations are normalized upon TAB1 coexpression, except for MAP3K7Trp241Gly. N = 7 for all conditions, except for WT (N = 14) and MAP3K7Trp241Gly (N = 6). (c) Western blot analysis of the autophosphorylation of MAP3K7 at Thr187 shows that except for MAP3K7Arg83His, all CSCF‐causing MAP3K7 mutations show reduced pThr187 compared to MAP3K7WT, whereas FMD2‐causing mutations show normal or increased levels of pThr187 compared to MAP3K7WT. N = 7 for all conditions, except for WT (N = 14) and MAP3K7Tyr206Cys and MAP3K7Tyr113Asp (N = 6). Error bars represent the standard error of the mean. *p < 0.05; **p < 0.01; ***p < 0.0001. Mutations in bold indicate the known mutations. CSCF, cardiospondylocarpofacial syndrome; EV, empty vector; FMD2, frontometaphyseal dysplasia type 2; MAP3K7, mitogen‐activated protein 3 kinase 7; RFP, red fluorescent protein; WT, wild‐type.
Coexpression of MAP3K7 with TAB1 has been shown to result in MAP3K7 autophosphorylation at Thr187, as well as a slower migration of the TAB1 band on Western blot (Sakurai et al., 2000). Therefore, we used this assay as a readout for the kinase activity of MAP3K7. Consistent with the literature, coexpression of MAP3K7WT with TAB1 resulted in slower migration of the MAP3K7, as well as the TAB1 band on Western blot (Figure 4b), which corresponded with MAP3K7 autophosphorylation of MAP3K7 at Thr187 (Figure 4c). In support of previous findings that FMD2 is caused mainly by gain‐of‐function mutations, coexpression of the FMD2‐related MAP3K7 mutations with TAB1, all resulted in equal or (a trend towards) increased levels of autophosphorylation of MAP3K7 at Thr187 compared to MAP3K7WT (one‐way ANOVA F[5,42] = 5.43, p = 0.0006; MAP3K7WT vs. MAP3K7Glu70Gln: p = 0.9; MAP3K7WT vs. MAP3K7Val100Glu: p = 0.4; MAP3K7WT vs. MAP3K7Tyr113Asp: p = 0.03; MAP3K7WT vs. MAP3K7Gly168Arg: p = 0.0004; MAP3K7WT vs. MAP3K7Pro485Leu: p = 0.9, Dunnett's multiple comparison test [Figure 4c]). In contrast, upon cotransfection with TAB1, the majority of the CSCF‐related MAP3K7 mutations showed significantly reduced pThr187 autophosphorylation levels compared to MAP3K7WT (one‐way ANOVA F[7,54] = 15.9, p < 0.0001; MAP3K7WT vs. MAP3K7Gly48Glu: p < 0.0001; MAP3K7WT vs. MAP3K7Gly110Asp: p<.0001; MAP3K7WT vs. MAP3K7Met196Val: p < 0.0001; MAP3K7WT vs. MAP3K7Tyr206Cys: p = 0.0002; MAP3K7WT vs. MAP3K7Tyr206Asp: p < 0.0001; MAP3K7WT vs. MAP3K7Trp241Gly: p < 0.0001, Dunnett's multiple comparison test [Figure 4c]). Interestingly, one CSCF‐related MAP3K7 variant (MAP3K7R83H), did not show reduced pThr187 autophosphorylation levels compared to MAP3K7WT (MAP3K7WT vs. MAP3K7Arg83His: p = 0.48, Dunnett's multiple comparison test (Figure 4c)).
MAP3K7 is known to have several downstream substrates, through which it affects different pathways in the cell (Aashaq et al., 2019; Xu & Lei, 2020). Finding that the stability and the autophosphorylation levels seem to distinguish between CSCF and FMD2‐related MAP3K7 mutations, we next sought to understand whether there is a difference in substrate regulation between the CSCF and FMD2‐related MAP3K7 mutations. Consistent with the reduced autophosphorylation, we found that the CSCF‐related MAP3K7 mutations resulted in reduced phosphorylated NF‐kB compared to MAP3K7WT, again with the exception of MAP3K7Arg83His (one‐way ANOVA F[7,51] = 23.73, p < 0.0001; MAP3K7WT vs. MAP3K7Gly48Glu: p < 0.0001; MAP3K7WT vs. MAP3K7Arg83His: p = 0.66; MAP3K7WT vs. MAP3K7Gly110Asp: p<0.0001; MAP3K7WT vs. MAP3K7Met196Val: p < 0.0001; MAP3K7WT vs. MAP3K7Tyr206Cys: p < 0.0001; MAP3K7WT vs. MAP3K7Tyr206Asp: p < 0.0001; MAP3K7WT vs. MAP3K7Trp241Gly: p < 0.0001, Dunnett's multiple comparison test [Figure 5a]).
Figure 5.
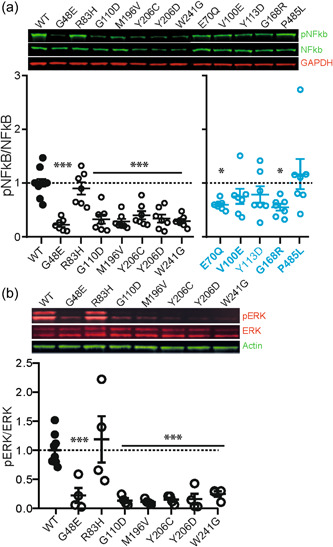
Western blot analysis of downstream targets of MAP3K7. (a) Western blot analysis reveals reduced phosphorylation of NF‐kB upon overexpression of most CSCF‐causing (black circles) and some FMD2‐causing (blue circles) MAP3K7 mutations in HEK‐293T cells, compared to MAP3K7WT. N = 7 for all conditions except for WT (N = 10). (b) Western blot analysis of phospho‐ERK1/2 (pERK) reveals reduced phosphorylation of ERK1/2 upon overexpression of most CSCF‐causing MAP3K7 mutations compared to MAP3K7WT, except for MAP3K7Arg83His. N = 4 for all conditions, except for WT (N = 8). Error bars represent standard error of the mean. *p < 0.05; ***p < 0.0001. Mutations in bold indicate the known mutations. CSCF, cardiospondylocarpofacial syndrome; ERK, extracellular signal‐regulated kinase; FMD2, frontometaphyseal dysplasia type 2; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; MAP3K7, mitogen‐activated protein 3 kinase 7; NFκB, nuclear factor‐κB;pNFκB, phospho‐NFκB; WT, wild‐type.
As for the FMD2‐related MAP3K7 mutations, the results were less straightforward. Overall, the mutations did not cause increased levels of phosphorylated NF‐kB, instead, most mutations showed a trend or significant reduction in phosphorylated NF‐kB (one‐way ANOVA F[5,49] = 4.11, p = 0.0034; MAP3K7WT vs. MAP3K7Glu70Gln: p = 0.04; MAP3K7WT vs. MAP3K7Val100Glu: p = 0.4; MAP3K7WT vs. MAP3K7Tyr113Asp: p = 0.54; MAP3K7WT vs. MAP3K7Gly168Arg: p = 0.018; MAP3K7WT vs. MAP3K7Pro485Leu: p = 0.74, Dunnett's multiple comparison test [Figure 5a]). These results indicate that whereas stability and autophosphorylation of MAP3K7 at Thr187 can potentially be used as a molecular fingerprint for distinguishing FMD2 and CSCF mutations, not all downstream pathways of MAP3K7 are necessarily differentially affected.
The majority of the CSCF patients showed clinical features in part similar to NS, where the RAS/mitogen‐activated protein kinase (MAPK) pathway is hyperactivated (Jorge et al., 2009; van der Burgt, 2007). We, therefore, assessed whether the CSCF‐related MAP3K7 mutations upregulated the RAS/MAPK pathway. However, in contrast to what is normally seen in NS, we found that most of the CSCF‐related MAP3K7 mutations resulted in reduced phosho‐ERK levels compared to MAP3K7WT (one‐way ANOVA F[7,24] = 6.78, p = 0.0002; MAP3K7WT vs. MAP3K7Gly48Glu: p = 0.016; MAP3K7WT vs. MAP3K7Gly110Asp: p = 0.007; MAP3K7WT vs. MAP3K7Met196Val: p = 0.005; MAP3K7WT vs. MAP3K7Tyr206Cys: p = 0.008; MAP3K7WT vs. MAP3K7Tyr206Asp: p = 0.009; MAP3K7WT vs. MAP3K7Trp241Gly: p = 0.021, Dunnett's multiple comparison test [Figure 5b]). Consistent with our previous results, only MAP3K7Arg83His behaved similarly to MAP3K7WT (MAP3K7WT vs. MAP3K7Arg83His: p = 0.9, Dunnett's multiple comparison test [Figure 5b]).
4. DISCUSSION
In this study, we significantly expand the published cohort of patients with mutations in the MAP3K7 gene, causing either CSCF or FMD2, and assess for the first time the genotype–phenotype correlation for MAP3K7 mutations.
4.1. Cardiospondylocarpofacial syndrome
Using WES in five independent Dutch pediatric patients (from two separate medical centers) suspected to have the RASopathy NS, mutations of unknown significance were found in MAP3K7. Further clinical assessment (detailed physical examination and radiologic assessment of the skeleton) of these patients confirmed the diagnosis of CSCF. To further characterize genotype/phenotype correlations in these patients, we studied 13 children (12 CSCF, 1 FMD2) and 3 adults (1 FMD2) with mutations in MAP3K7. Features of our CSCF patients overlapping NS included short stature (n = 9; 64%), congenital cardiac abnormalities (n = 7; 54%), cardiomyopathy (n = 4; 33%), posteriorly rotated and/or low set ears (n = 7; 58%), hypertelorism (n = 8; 67%), ptosis (n = 8; 62%), widely spaced nipples (n = 7; 78%), and epicanthal folds (n = 6; 50%). Downslanting palpebral fissures (n = 3; 23%), triangular face (n = 4; 33%), short or webbed neck (n = 3; 27%), and scoliosis (n = 2; 22%) were less often present. Distinguishing features of the CSCF patients are the wide nose with bulbous tip (n = 9; 82%), periorbital fullness (n = 7; 64%), and the full cheeks (n = 9; 69%). Information about the neurological phenotype was available for seven patients, who showed normal intelligence; three had autism spectrum disorder. Hypotonia was found in seven patients (70%), and joint laxity in eight (80%). The resemblance of CSCF to NS, which was also proposed in the literature (Minatogawa et al., 2022; Morlino et al., 2018; Sousa et al., 2010), made us wonder if CSCF should also be considered a RASopathy; however, the RAS/MAPK pathway is downregulated instead of upregulated, suggesting an alternative explanation for the resemblance of CSCF to NS. Despite these molecular results, CSCF should be considered in the differential diagnosis of NS patients based on the described clinical features.
4.2. Frontometaphyseal dysplasia type 2
Both our FMD2 patients showed phenotypes compatible with previously reported patients (Wade et al., 2017). Interestingly, our adult male FMD2 patient initially presented with ambiguous genitalia and he has a mild skeletal phenotype with normal/high intelligence. The pediatric patient with the recurrent variant for FMD2 has a severe cardiac, neurological, and skeletal phenotype. Distinguishing features of the FMD2 patients in comparison to NS and CSCF were prominent supraorbital ridges, flexion contractures of the elbows, ulnar deviation of the hands, interphalangeal joint contractures, camptodactyly, and keloid scarring (all present in both patients). Overlapping features with NS were short stature, congenital cardiac defects, hypertelorism, epicanthal folds, ptosis (n = 1), and failure to thrive in infancy. Neither patient had a triangular face, short or webbed neck. FMD2 patients have more severe dysmorphism than CSCF patients (Figure 3).
Regarding the occurrence of left‐sided heart lesions and the potential progressive aspect in our patients of aortic dilatation, these features go along with previous reports that genes which belong to the TGF‐β cascade are potentially prone to this subcategory of cardiac involvement (Baban et al., 2018; Caulfield et al., 2018; Cheng et al., 2017; Engwerda et al., 2021; Hanson et al., 2022; Yin et al., 2022). It might be wise to establish a specific screening program through serial electrocardiography and echocardiography detect progressive aortic dilatation or arrhythmias.
4.3. Functional analysis
We show that missense mutations in MAP3K7 causing CSCF to reduce MAP3K7 protein stability and autophosphorylation at Thr187, whereas MAP3K7 mutations causing FMD2 show normal or enhanced MAP3K7 protein expression and autophosphorylation levels. The only exception is the MAP3K7Arg83His variant, which is found in a patient with the clinical phenotype of CSCF. Despite the lower expression levels of this variant in absence of TAB1, this variant behaves similar to MAP3K7WT in the other molecular assays, suggesting a milder effect on protein function compared to the other CSCF mutations. Interestingly, the patient does show a clear CSCF phenotype, indicating that the full genotype/phenotype correlation remains to be uncovered.
When assessing one of the downstream targets, NF‐kB, no difference was observed between the mutations causing CSCF or FMD2, indicating that further research is required to assess what downstream targets are differentially affected, to further elucidate the precise molecular mechanism underlying CSCF and FMD2.
5. OVERALL CONCLUSION
Our cohort in combination with the MAP3K7 mutations causing FMD2 described in literature allowed for the first time to search for genotype/phenotype correlations, that is, side‐by‐side comparison of the different missense mutations causing either FMD2 or CSCF. Confirming previous reports, we found that the FMD2‐causing mutations in MAP3K7 produce a gain‐of‐function effect on MAP3K7 and cause a distinct phenotype with a higher rate of skeletal anomalies. Additionally, we now further support that the CSCF‐causing mutations in MAP3K7 have a loss‐of‐function effect and lead to more soft connective tissue manifestations than the FMD2 phenotype. This is most clearly seen when looking at the expression and pThr187 levels of MAP3K7. Interestingly, when assessing one of the downstream targets of MAP3K7, NF‐kB (Xu & Lei, 2020), this distinction between the FMD2‐ or CSCF‐causing mutations is not evident, suggesting other molecular pathways are involved in the pathogenesis of these two disorders. Indeed, we have only assessed the effect of this pathway in the coexpression of TAB1 and MAP3K7. Possibly there is a more distinct phenotype when assessing the function of MAP3K7 when in complex with TAB2/3. Additionally, other downstream pathways remain to be studied.
In this paper, we significantly expanded the cohort of patients with either CSCF or FMD2 and performed for the first time a side‐by‐side comparison of the different missense mutations related to either FMD2 or CSCF. We have shown that both clinical phenotypes, despite being largely distinct from each other, exhibit a clear overlap with other syndromic connective tissue disorders with respect to bone deformities, short stature, hypermobile joints, contractures, and cardiac anomalies. This fits what is known about MAP3K7's effect on the TGF‐β pathway (Le Goff et al., 2016; Yu et al., 2014). Additionally, we have identified that cardiac anomalies, which can be severe, are common in patients with pathogenic MAP3K7 mutations. The cardiac phenotype ranged from left‐sided congenital heart defects to cardiomyopathies (both hypertrophic and dilated) and arrhythmias. The latter subgroup is infrequent in NS (Gelb et al., 2015; Pierpont & Digilio, 2018). In conclusion, we show that expression and autophosphorylation levels of MAP3K7 can serve as a molecular fingerprint to distinguish between FMD2‐ or CSCF‐causing MAP3K7 mutations. Additionally, we show that MAP3K7 mutations should be considered in the differential diagnosis of patients with syndromic congenital cardiac (valve) anomalies and/or cardiomyopathy. Finally, we confirm in our cohort the association with syndromic connective tissue disorders as well as NS.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
The authors would like to thank patients for their willingness to share their information to further our understanding of cardiospondylocarpofacial syndrome and frontometaphyseal dysplasia type 2. They would also like to thank Dr. Ineke van der Burgt (Division of Human Genetics, Radboud Medical Center, Radboud University, Nijmegen, The Netherlands) for her valuable insights and help in setting up the study. Eric W. Klee, Joel M. Rosado, and Lisa A. Schimmenti are supported by the Center for Individualized Medicine, Mayo Clinic. Part of the work of Anwar Baban, Marcello Niceta, and Marco Tartaglia was supported by funding from the Italian Ministry of Health (CCR‐2017‐23669081, RCR‐2020‐23670068_001 to Marco Tartaglia) and Italian Ministry of Research (FOE 2019, 2020 Sviluppo di protocolli innovativi e applicazione di nuovi strumenti‐omici nei pazienti orfani di diagnosi to Marco Tartaglia). Geeske M. van Woerden was funded by NWO‐VIDI (016.Vidi.188.014)
van Woerden, G. M. , Senden, R. , de Konink, C. , Trezza, R. A. , Baban, A. , Bassetti, J. A. , van Bever, Y. , Bird, L. M. , van Bon, B. W. , Brooks, A. S. , Guan, Q. , Klee, E. W. , Marcelis, C. , Rosado, J. M. , Schimmenti, L. A. , Shikany, A. R. , Terhal, P. A. , Nicole Weaver, K. , Wessels, M. W. , … Demirdas, S. (2022). The MAP3K7 gene: Further delineation of clinical characteristics and genotype/phenotype correlations. Human Mutation, 43, 1377–1395. 10.1002/humu.24425
Contributor Information
Geeske M. van Woerden, Email: g.vanwoerden@erasmusmc.nl.
Serwet Demirdas, Email: s.demirdas@erasmusmc.nl.
REFERENCES
- Aashaq, S. , Batool, A. , & Andrabi, K. I. (2019). TAK1 mediates convergence of cellular signals for death and survival. Apoptosis: An International Journal on Programmed Cell Death, 24(1–2), 3–20. 10.1007/s10495-018-1490-7 [DOI] [PubMed] [Google Scholar]
- Baban, A. , Magliozzi, M. , Loeys, B. , Adorisio, R. , Alesi, V. , Secinaro, A. , Corica, B. , Vricella, L. , Dietz, H. C. , Drago, F. , Novelli, A. , & Amodeo, A. (2018). First evidence of maternally inherited mosaicism in TGFBR1 and subtle primary myocardial changes in Loeys‐Dietz syndrome: A case report. BMC Medical Genetics, 19(1), 170–177. 10.1186/s12881-018-0661-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basart, H. , van de Kar, A. , Adès, L. , Cho, T.‐J. , Carter, E. , Maas, S. M. , Wilson, L. C. , van der Horst, C. M. , Wade, E. M. , Robertson, S. P. , & Hennekam, R. C. (2015). Frontometaphyseal dysplasia and keloid formation without FLNA mutations. American Journal of Medical Genetics, Part A, 167(6), 1215–1222. 10.1002/ajmg.a.37044 [DOI] [PubMed] [Google Scholar]
- Caulfield, T. R. , Richter, J. E. , Brown, E. E. , Mohammad, A. N. , Judge, D. P. , & Atwal, P. S. (2018). Protein molecular modeling techniques investigating novel TAB2 variant R347X causing cardiomyopathy and congenital heart defects in multigenerational family. Molecular Genetics & Genomic Medicine, 6(4), 666–672. 10.1002/mgg3.401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, A. , Dinulos, M. , Neufeld‐Kaiser, W. , Rosenfeld, J. , Kyriss, M. , Madan‐Khetarpal, S. , Risheg, H. , Byers, P. H. , & Liu, Y. J. (2017). 6q25.1 (TAB2) microdeletion syndrome: Congenital heart defects and cardiomyopathy. American Journal of Medical Genetics, Part A, 173(7), 1848–1857. 10.1002/ajmg.a.38254 [DOI] [PubMed] [Google Scholar]
- Costantini, A. , Wallgren‐Pettersson, C. , & Mäkitie, O. (2018). Expansion of the clinical spectrum of frontometaphyseal dysplasia 2 caused by the recurrent mutation p.Pro485Leu in MAP3K7. European Journal of Medical Genetics, 61(10), 612–615. 10.1016/j.ejmg.2018.04.004 [DOI] [PubMed] [Google Scholar]
- Dai, L. , Aye Thu, C. , Liu, X.‐Y. , Xi, J. , & Cheung, P. C. F. (2012). TAK1, more than just innate immunity. IUBMB Life, 64(10), 825–834. 10.1002/iub.1078 [DOI] [PubMed] [Google Scholar]
- Engwerda, A. , Leenders, E. , Frentz, B. , Terhal, P. A. , Löhner, K. , de Vries, B. , Dijkhuizen, T. , Vos, Y. J. , Rinne, T. , van den Berg, M. P. , Roofthooft, M. , Deelen, P. , van Ravenswaaij‐Arts, C. , & Kerstjens‐Frederikse, W. S. (2021). TAB2 deletions and variants cause a highly recognisable syndrome with mitral valve disease, cardiomyopathy, short stature and hypermobility. European Journal of Human Genetics, 29(11), 1669–1676. 10.1038/s41431-021-00948-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forney, W. R. , Robinson, S. J. , & Pascoe, D. J. (1966). Congenital heart disease, deafness, and skeletal malformations: A new syndrome? The Journal of Pediatrics, 68(1), 14–26. 10.1016/s0022-3476(66)80418-3 [DOI] [PubMed] [Google Scholar]
- Gelb, B. D. , Roberts, A. E. , & Tartaglia, M. (2015). Cardiomyopathies in Noonan syndrome and the other RASopathies. Progress in Pediatric Cardiology, 39(1), 13–19. 10.1016/j.ppedcard.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano, F. , Collignon, P. , Paquis‐Flucklinger, V. , Bardot, J. , & Philip, N. (2005). A new three‐generational family with frontometaphyseal dysplasia, male‐to‐female transmission, and a previously reported FLNA mutation. American Journal of Medical Genetics, Part A, 132A(2), 222. 10.1002/ajmg.a.30396 [DOI] [PubMed] [Google Scholar]
- Hanson, J. , Brezavar, D. , Hughes, S. , Amudhavalli, S. , Fleming, E. , Zhou, D. , Alaimo, J. T. , & Bonnen, P. E. (2022). TAB2 variants cause cardiovascular heart disease, connective tissue disorder, and developmental delay. Clinical Genetics, 101(2), 214–220. 10.1111/cge.14085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorge, A. A. L. , Malaquias, A. C. , Arnhold, I. J. P. , & Mendonca, B. B. (2009). Noonan syndrome and related disorders: A review of clinical features and mutations in genes of the RAS/MAPK pathway. Hormone Research, 71(4), 185–193. 10.1159/000201106 [DOI] [PubMed] [Google Scholar]
- Le Goff, C. , Rogers, C. , Le Goff, W. , Pinto, G. , Bonnet, D. , Chrabieh, M. , Alibeu, O. , Nistchke, P. , Munnich, A. , Picard, C. , & Cormier‐Daire, V. (2016). Heterozygous mutations in MAP3K7, encoding TGF‐β‐activated kinase 1, cause cardiospondylocarpofacial syndrome. American Journal of Human Genetics, 99(2), 407–413. 10.1016/j.ajhg.2016.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micale, L. , Morlino, S. , Biagini, T. , Carbone, A. , Fusco, C. , Ritelli, M. , Giambra, V. , Zoppi, N. , Nardella, G. , Notarangelo, A. , Schirizzi, A. , Mazzoccoli, G. , Grammatico, P. , Wade, E. M. , Mazza, T. , Colombi, M. , & Castori, M. (2020). Insights into the molecular pathogenesis of cardiospondylocarpofacial syndrome: MAP3K7 c.737‐7A>G variant alters the TGFβ‐mediated α‐SMA cytoskeleton assembly and autophagy. Biochimica Et Biophysica Acta, Molecular Basis of Disease, 1866(6), 165742. 10.1016/j.bbadis.2020.165742 [DOI] [PubMed] [Google Scholar]
- Minatogawa, M. , Miyake, N. , Tsukahara, Y. , Tanabe, Y. , Uchiyama, T. , Matsumoto, N. , & Kosho, T. (2022). Expanding the phenotypic spectrum of cardiospondylocarpofacial syndrome: From a detailed clinical and radiological observation of a boy with a novel missense variant in MAP3K7. American Journal of Medical Genetics, Part A, 188(1), 350–356. 10.1002/ajmg.a.62516 [DOI] [PubMed] [Google Scholar]
- Morlino, S. , Castori, M. , Dordoni, C. , Cinquina, V. , Santoro, G. , Grammatico, P. , Venturini, M. , Colombi, M. , & Ritelli, M. (2018). A novel MAP3K7 splice mutation causes cardiospondylocarpofacial syndrome with features of hereditary connective tissue disorder. European Journal of Human Genetics, 26, 1–5. 10.1038/s41431-017-0079-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortier, G. R. , Cohn, D. H. , Cormier‐Daire, V. , Hall, C. , Krakow, D. , Mundlos, S. , Nishimura, G. , Robertson, S. , Sangiorgi, L. , Savarirayan, R. , Sillence, D. , Superti‐Furga, A. , Unger, S. , & Warman, M. L . (2019). Nosology and classification of genetic skeletal disorders: 2019 revision. American Journal of Medical Genetics Part A, 179(12), 2393–2419. 10.1002/ajmg.a.61366 [DOI] [PubMed] [Google Scholar]
- Pierpont, M. E. , & Digilio, M. C. (2018). Cardiovascular disease in Noonan syndrome. Current Opinion in Pediatrics, 30(5), 601–608. 10.1097/MOP.0000000000000669 [DOI] [PubMed] [Google Scholar]
- Proietti Onori, M. , Koopal, B. , Everman, D. B. , Worthington, J. D. , Jones, J. R. , Ploeg, M. A. , Mientjes, E. , van Bon, B. W. , Kleefstra, T. , Schulman, H. , Kushner, S. A. , Küry, S. , Elgersma, Y. , & van Woerden, G. M. (2018). The intellectual disability‐associated CAMK2G p.Arg292Pro mutation acts as a pathogenic gain‐of‐function. Human Mutation, 39(12), 2008–2024. 10.1002/humu.23647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. ACMG Standards and Guidelines, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, S. P. (2004). Molecular pathology of filamin A: Diverse phenotypes, many functions. Clinical Dysmorphology, 13(3), 123–131. 10.1097/01.mcd.0000130235.95356.40 [DOI] [PubMed] [Google Scholar]
- Sakurai, H. , Miyoshi, H. , Mizukami, J. , & Sugita, T. (2000). Phosphorylation‐dependent activation of TAK1 mitogen‐activated protein kinase kinase kinase by TAB1. FEBS Letters, 474(2–3), 141–145. 10.1016/s0014-5793(00)01588-x [DOI] [PubMed] [Google Scholar]
- Sobreira, N. , Schiettecatte, F. , Valle, D. , & Hamosh, A. (2015). GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa, S. B. , Baujat, G. , Abadie, V. , Bonnet, D. , Sidi, D. , Munnich, A. , Krakow, D. , & Cormier‐Daire, V. (2010). Postnatal growth retardation, facial dysmorphism, spondylocarpal synostosis, cardiac defect, and inner ear malformation (cardiospondylocarpofacial syndrome?)—A distinct syndrome? American Journal of Medical Genetics, Part A, 152A(3), 539–546. 10.1002/ajmg.a.33277 [DOI] [PubMed] [Google Scholar]
- van der Burgt, I. (2007). Noonan syndrome. Orphanet Journal of Rare Diseases, 2(1), 4–6. 10.1186/1750-1172-2-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade, E. M. , Daniel, P. B. , Jenkins, Z. A. , McInerney‐Leo, A. , Leo, P. , Morgan, T. , Addor, M. C. , Adès, L. C. , Bertola, D. , Bohring, A. , Carter, E. , Cho, T. J. , Duba, H. C. , Fletcher, E. , Kim, C. A. , Krakow, D. , Morava, E. , Neuhann, T. , Superti‐Furga, A. , … Robertson, S. P. (2016). Mutations in MAP3K7 that alter the activity of the TAK1 signaling complex cause frontometaphyseal dysplasia. American Journal of Human Genetics, 99(2), 392–406. 10.1016/j.ajhg.2016.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade, E. M. , Jenkins, Z. A. , Daniel, P. B. , Morgan, T. , Addor, M. C. , Adés, L. C. , Bertola, D. , Bohring, A. , Carter, E. , Cho, T. J. , de Geus, C. M. , Duba, H. C. , Fletcher, E. , Hadzsiev, K. , Hennekam, R. , Kim, C. A. , Krakow, D. , Morava, E. , Neuhann, T. , … Robertson, S. P. (2017). Autosomal dominant frontometaphyseal dysplasia: Delineation of the clinical phenotype. American Journal of Medical Genetics, Part A, 173, 1739–1746. 10.1002/ajmg.a.38267 [DOI] [PubMed] [Google Scholar]
- Xu, Y.‐R. , & Lei, C.‐Q. (2020). TAK1‐TABs complex: A central signalosome in inflammatory responses. Frontiers in Immunology, 11, 608976. 10.3389/fimmu.2020.608976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, H. , Guo, X. , Chen, Y. , Zeng, Y. , Mo, X. , Hong, S. , He, H. , Li, J. , Steinmetz, R. , & Liu, Q. (2022). TAB2 deficiency induces dilated cardiomyopathy by promoting RIPK1‐dependent apoptosis and necroptosis. The Journal of Clinical Investigation, 132(4), e152297. 10.1172/JCI152297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J. , Zhang, F. , Wang, S. , Zhang, Y. , Fan, M. , & Xu, Z. (2014). TAK1 is activated by TGF‐β signaling and controls axonal growth during brain development. Journal of Molecular Cell Biology, 6(4), 349–351. 10.1093/jmcb/mju030 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.