

Abstract
Despite several new therapeutic options for acute myeloid leukemia (AML), disease relapse remains a significant challenge. We have previously demonstrated that augmenting ceramides can counter various drug‐resistance mechanisms, leading to enhanced cell death in cancer cells and extended survival in animal models. Using a nanoscale delivery system for ceramide (ceramide nanoliposomes, CNL), we investigated the effect of CNL within a standard of care venetoclax/cytarabine (Ara‐C) regimen. We demonstrate that CNL augmented the efficacy of venetoclax/cytarabine in in vitro, ex vivo, and in vivo models of AML. CNL treatment induced non‐apoptotic cytotoxicity, and augmented cell death induced by Ara‐C and venetoclax. Mechanistically, CNL reduced both venetoclax (Mcl‐1) and cytarabine (Chk1) drug‐resistant signaling pathways. Moreover, venetoclax and Ara‐C augmented the generation of endogenous pro‐death ceramide species, which was intensified with CNL. Taken together, CNL has the potential to be utilized as an adjuvant therapy to improve outcomes, potentially extending survival, in patients with AML.
Keywords: cell death; ceramides; cytarabine; drug resistance; leukemia, myeloid, acute; models, animal; recurrence; signal transduction; standard of care; venetoclax
Abbreviations
- ANOVA
analysis of variance
- AML
acute myeloid leukemia
- Ara‐C
cytarabine
- BLI
bioluminescence imaging
- BSA
bovine serum albumin
- CERS
ceramide synthase
- CNL
ceramide nanoliposomes
- DAPI
4′,6‐diamidino‐2‐phenylindole
- FBS
fetal bovine serum
- FLT3‐ITD
FMS‐like tyrosine kinase‐3 internal tandem duplication
- HexCer
hexosyl ceramide
- LC‐MS/MS
liquid chromatography with tandem mass spectrometry
- MDS
myelodysplastic syndrome
- MTS
[3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt
- PARP
poly‐ADP ribose polymerase
- PCR
polymerase chain reaction
- PMS
phenazine methosulfate
- RIPA
radioimmune precipitation assay
- RPMI‐1640
Roswell Park Memorial Institute ‐1640 medium
- SEM
standard error of mean
- SDS
sodium dodecyl sulphate
- TBS
tris‐buffered saline
1. INTRODUCTION
The Bcl‐2 inhibitor venetoclax has received clinical adoption in combination with low dose cytarabine (Ara‐C) 1 or hypomethylating agents (azacitidine or decitabine) 2 as frontline treatment options for AML patients unfit for intensive chemotherapy, and now represent a standard of care for this disease. However, primary treatment results in only 60%–70% remission, and de novo resistance or clonal adaptation eventually leads to relapse in these combinatorial regimens. Therefore, improved treatment strategies, particularly for venetoclax‐resistant leukemia, are needed. 3
Dysfunctional ceramide metabolism contributes to leukemia pathogenesis. 4 , 5 Ceramide is a pro‐death sphingolipid metabolite 6 that may augment the efficacy of standard of care therapeutics by limiting drug‐resistance mechanisms. 7 , 8 We have engineered a C6‐ceramide nanoliposome (CNL) that solubilizes the lipid to improve pharmacokinetics and efficacy, allowing for bioactive ceramides to be used as a cancer therapeutic. 4 , 9 We have previously shown that CNL yields longer term disease control in AML with myelodysplastic syndrome (MDS)‐related changes, and in a model of de novo AML when combined with lysosome dysregulators, such as vinblastine. 4 This efficacy was also attributed to the activities of sphingolipid metabolizing enzymes that remodel short‐chain C6‐ceramide into pro‐death metabolites, such as long‐chain ceramide species or sphingosine. 4 As we have previously demonstrated that ceramide and its analogs modulate Mcl‐1 expression in AML, 10 a known resistance mechanism of venetoclax, we now examine if CNL augments venetoclax/Ara‐C responses in AML by reducing drug‐resistance mechanisms.
2. METHODS
2.1. Reagents
C6‐Ceramide and all other lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Antibodies for Mcl‐1 (#5453), Bcl‐2 (#4223), survivin (#2808), XIAP (#14334), Phospho‐Ser216 cdc25C (#4901), total cdc25C (#4688), Phospho‐Ser317 Chk1 (#12302), total Chk1 (#2360), Phospho‐Thr68 Chk2 (#2197), total Chk2 (#6334), PARP (#9542), LC3B (#3868), and alpha‐tubulin (#2125) were purchased from Cell Signaling Technology Inc (Danvers, MA). Anti‐FLAG M2 (#F1804) and beta‐actin (#A‐5441) antibodies were purchased from Millipore Sigma (Burlington, MA). Anti‐active Caspase3‐AF647 conjugated antibodies (#560626) and anti‐cytochrome C‐AF647 conjugated antibodies (#558709) were purchased from BD Pharmingen (Franklin Lakes, NJ). DAPI and Polybrene were purchased from Millipore Sigma. Venetoclax (#S8048) and Ara‐C were purchased from Selleck Chemicals (Houston, TX). CNL (C6‐ceramide nanoliposomes) and Lip‐Ghost (control nanoliposomal formulation with no C6‐ceramide) were prepared as previously described. 4
2.2. Cell lines
Human acute myeloid leukemia cell line MOLM‐14 was obtained from Dr. Mark Levis, Johns Hopkins Medical Institutions (Baltimore, MD), and OCI‐AML2 from Dr. Xiaorong Gu, Cleveland Clinic (Cleveland, OH). C1498 cells were purchased from ATCC (Gaithersburg, MD) and were transduced with viral particles containing pUltra‐LUC‐Chili (Addgene plasmid #48688) and polybrene (8 μg/ml) three times every 12 hr. Cells were allowed to expand and sorted for tdTomato expression by flow cytometry. MOLM‐13‐YFP‐Luc cells were generously gifted by Dr. Hong‐Gang Wang (Penn State University, Hershey, PA). All cell lines were authenticated prior to use. HL‐60 cells were obtained from American Tissue Culture Collection (Manassas, VA). Cells were cultured in RPMI‐1640 media (ThermoFisher Scientific, Waltham, MA) with 10% FBS (MOLM‐14, HL‐60) or 20% (OCI‐AML2) (Gemini Bio, Calabasas, CA) and 1% Penicillin–Streptomycin (ThermoFisher Scientific) at 37°C and 5% CO2.
To model venetoclax resistance, MV4‐11 cells were conditionally adapted over time to 1.0 μM venetoclax, in a manner previously described for other chemotherapy drugs. 8 The EC50 for venetoclax‐resistant cells is 4.41 μM and 15.9 nM for drug‐naïve cells.
2.3. Plasmids
Lentiviral plasmid used for cloning, pLKO5d.EFS.SpCas9.P2A.BSD, (Addgene plasmid #57821) was a gift from Dr. Benjamin Ebert (Harvard Medical School, Boston, MA, USA). Packaging plasmids, pMD2.G (Addgene plasmid #12259) and psPAX2, (Addgene plasmid #12260) were a gift from Dr. Didier Trono (School of Life Sciences, Ecole Polytechnique Fédérale de Lausanne, Lausanne, Switzerland). Plasmids pcDNA3.1‐CERS5 (isoform 1, NP_671723.1) and pcDNA3.1‐CERS6 (isoform 1, NP_001243055.1) encoding CERS5/CERS6 cDNAs were a gift from Dr. Antony Futerman (Department Biomolecular Sciences, Weizmann Institute of Science, Rehovot, Israel).
For expression of ceramide synthase 6, its coding sequence was amplified by PCR from pcDNA3.1‐CERS6 using primers 5′‐CGCGGATCCGCCACCATGGCAGGGATCTTAGCCTGGTTCTG‐3′ and 5′‐CGCGACGCGTATCATCCATGGAGCAGGAGCCAGTC‐3′. The first primer contained a BamHI restriction endonuclease site in front of CERS6 ATG while the second primer has CERS6 Stop codon replaced with MluI restriction endonuclease site. The PCR products was digested with BamHI and MluI enzymes and subcloned into pLKO5d.EFS.mCherry.P2A vector created by replacement of the SpCas9.P2A.BSD cassette of the pLKO5d.EFS.SpCas9.P2A.BSD plasmid with one containing BamHI site followed by MluI site, a FLAG epitope tag (5′‐GATTACAAGGATGACGACGATAAG‐3, DYKDDDDK) followed by in‐frame P2A self‐cleavage peptide and the fluorescent protein mCherry. The MluI site in the PCR primer above and lentiviral vector were designed in such a way that FLAG‐P2A‐mCherry coding sequence was in frame with CERS6, resulting in expression of single CERS6‐FLAG‐P2A‐mCherry mRNA. Upon translation, the P2A polypeptide undergoes self‐cleavage resulting in two separate CERS6‐FLAG and P2A‐mCherry peptides. The same strategy was used for creating CERS5 FLAG‐P2A‐mCherry construct.
2.4. Lentiviral production and cell infection
To produce lentiviral particles, plasmids encoding CERS5, CERS6, or mCherry alone were co‐transfected with packaging plasmids into Lenti‐Pac 293 TA cells (GeneCopoeia, Rockville, MD) grown on 100‐mm dishes with Fugene 6 (Roche, Branford, CT) according to manufacturer's instructions. Forty‐eight hours later, culture medium was harvested and passed through a 0.45‐micron filter to remove cell debris. Viral stocks were used undiluted right away or aliquoted and stored at −80°C.
To create MOLM‐14 cells expressing CERS5, CERS6, or mCherry, cells were dispensed on 12‐well plates (Genesee Scientific, Morrisville, NC) at a density of 500 000 per well in 0.1 ml of growth media. Lentiviral stocks were mixed with Polybrene to final concentration of 8 μg/ml and added to cells in the amount of 1 ml per well. Plates were incubated at 37°C and 5% CO2 for 15 min, sealed with Parafilm and centrifuged at 25°C for 30 min at 1220g. After centrifugation, 3 ml of fresh growth medium was added to each well. The next day, media was removed and infection repeated used 1 ml of fresh lentiviral supernatant as described above.
Two or 3 days after infection, cells demonstrated a transduction efficiency of 90% based on the mCherry signal, and were further sorted to obtain a pure population of mCherry‐positive cells. Expression of CERS5 or CERS6 was confirmed by Western blotting.
2.5. MTS cell viability assay
Cytotoxicity was measured by plating 15 000 cells/well into 96‐well plates followed by growth for 24 h in the presence of ghost liposomes, CNL or other drugs for 24 h. After 24 h, [3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt (Promega, Madison, WI)/phenazine methosulfate (MilliporeSigma) (MTS/PMS) mixture was added and adsorption was read at 560 nm using Bio‐Tek Cytation3 (Agilent, Santa Clara, CA) plate reader.
2.6. Western blotting
Whole cell lysates were prepared by dissolving cells in RIPA Buffer (Alfa Aesar, Ward Hill, MA, Cat #J62524 diluted to 1x) containing protease inhibitors (ThermoFisher Scientific A32955) and phosphatase inhibitors (ThermoFisher Scientific A32957) for 10–15 min on ice with occasional shaking. After centrifugation for 15 min at 13–14 000g at 4°C supernatants were saved and protein concentration was measured with the Bio‐Rad DC protein assay (Bio‐Rad Laboratories, Hercules, CA). Equal amount of protein (80–100 μg) was mixed with NuPAGE LDS 4x sample buffer (Invitrogen #NP0007, ThermoFisher Scientific) to achieve 1x final strength and 2‐mercaptoethanol was added to 3%.
Protein samples were heated at 70°C for 10 min and separated using NuPAGE 4%–12% Bis‐Tris precast gels (Life Technologies #NP0335, ThermoFisher Scientific) in SDS‐containing buffer and transferred to Immobilon membrane (Bio‐Rad). After 15–30 min of blocking in 1% casein blocker in TBS (ThermoFisher Scientific #37532), membranes were incubated with the primary antibody overnight at 4°C, washed and incubated with the horseradish peroxidase‐conjugated secondary antibody. Primary and secondary antibodies were diluted in 0.05% TBS/Tween‐20 containing 0.05% casein blocker. Protein bands were visualized using a chemiluminescence kit (Prometheus #GSC‐925‐D10, Genesee Scientific). Images were taken by Chemi XX6 G: BOX digital imaging system (Syngene, Frederick, MD) and quantified using GeneTools software (Syngene). Readings were normalized to actin (Figures 3A, 4A,B) or to tubulin (Figure 4C). If required, membranes were stripped with Restore Stripping Buffer (#21059, ThermoFisher Scientific) for 30 min, washed, re‐blocked and probed as described above with different antibodies.
FIGURE 3.
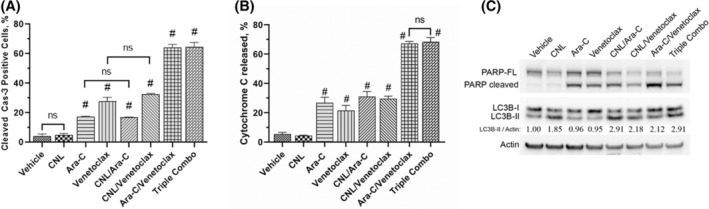
The effect of CNL treatment on formation of apoptotic‐ and autophagy‐specific markers. (A) MOLM‐14 were treated with drugs alone or in combinations (CNL 15 μM, Ara‐C 1.25 μM, venetoclax 50 nM) for 24 h, fixed and processed for active Caspase‐3 staining. Percentage of active Cas‐3 positive cells were accessed by flow cytometry; N = 3, run in triplicates, ns—non‐significant; #p < .005 versus vehicle, shown are means +/− SEM. (B) MOLM‐14 cells were treated with the drug combinations as indicated above, fixed and processed for cytochrome C release assay. Percentage of cells from which cytochrome C has been released were accessed by flow cytometry; N = 3, run in triplicates, ns‐non‐significant; shown are means +/− SEM, #p < .001 versus vehicle. (C) MOLM‐14 cells were treated with the drug combinations as indicated above, harvested and proteins extracted. Western blots were performed and quantitated via densitometry. LC3B‐II/Actin numbers: relative LC3B‐II autophagosome marker expression to vehicle normalized to actin loading. Representative Western of N = 3 replicates.
FIGURE 4.
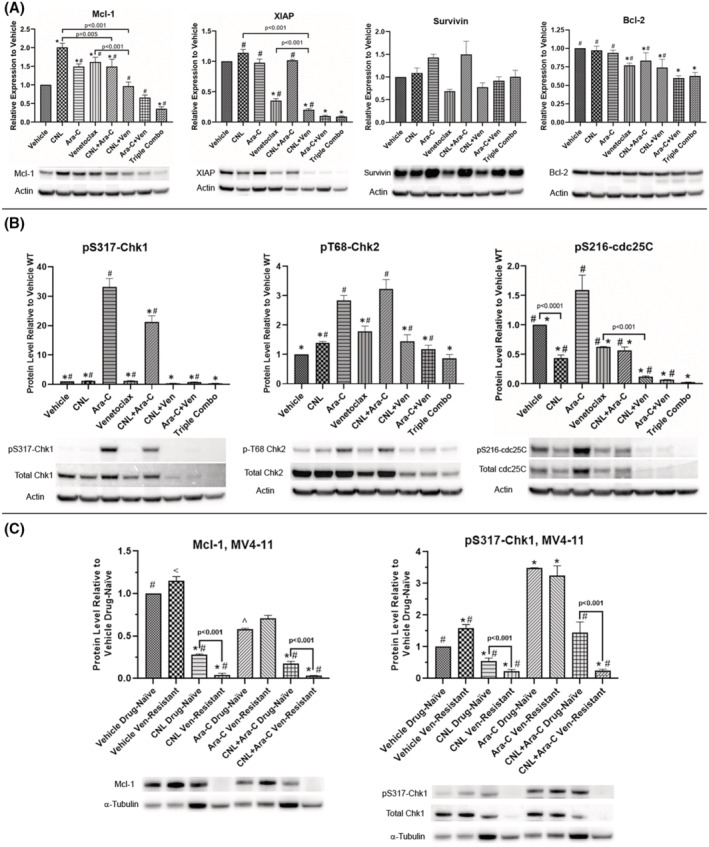
CNL reduces Ara‐C and/or venetoclax‐induced drug‐resistant mechanisms: MOLM‐14 (A, B) and MV4‐11 (C) cells made resistant to venetoclax or drug‐naïve were treated with the indicated drug combinations (MOLM‐14: CNL 15 μM, Ara‐C 1.25 μM, venetoclax 50 nM; MV4‐11: CNL 15 μM, Ara‐C 1.25 μM) for 24 h, harvested and proteins extracted. Western blots were performed and quantitated via densitometry. Loading was normalized to actin (3A, 3B) or to tubulin (3C). Shown is mean +/− SEM of relative protein expression to vehicle for proteins indicated, N = 3–5. Of note, we chose tubulin instead of actin for the MV4‐11 drug‐resistant cell line as actin levels were significantly reduced in this cell lines compared with wild type (data not shown). Even though, CNL elevated tubulin in wild type but not the drug‐resistant cell line. CNL reduced Mcl‐1 and P‐S317‐Chk1 significantly in the drug‐resistant cell line. (A) *p < .05 versus vehicle; #p < .05 versus triple. (B) *p < .005 versus Ara‐C; #p < .005 versus triple. (C) *p < .05 versus vehicle drug‐naïve; ^p = .05 versus vehicle drug‐naive; #p < .02 versus Ara‐C drug‐naive; <p = .048 versus Ara‐C drug‐naive. Representative blots are shown beneath each graph.
2.7. Flow cytometry
For experiments described in Figure 3A, cells were plated in a 24‐well plate at 1 × 106 per well in 2 ml of media with drugs added, mixed well, and 200 μl cell aliquots were transferred to 96‐well plate. Seventeen hours later, cells were fixed and stained for apoptotic markers as described in Ref [11].
For experiments in Figure 6, cells were plated in a 24‐well tray at 660 000 per well in 2 ml of media and CNL or other drugs were added. Twenty‐four hours later, 100 μl cell aliquots were transferred to 96‐well plates and mixed with 50 μl of growth media containing 9 μM DAPI. After 10 min incubation at room temperature, cells were analyzed with Attune NxT flow cytometer (ThermoFisher Scientific). Percentage of cells positive for DAPI staining was calculated using FlowJo V. 10.6.2 (Becton, Dickinson & Co, Franklin Lakes, NJ) software.
FIGURE 6.
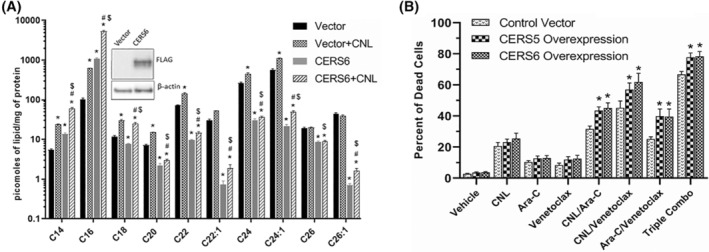
Ceramide Synthase 6 (CERS6) overexpression increases C16 ceramide generation in CNL treated MOLM‐14 cells, leading to augmented cell death in the presence of Ara‐C and/or venetoclax. (A) Ceramide levels from MOLM‐14 cells transduced with a control vector or CERS6 for overexpression and treated with vehicle or CNL 15 μM for 24 h. N = 5; shown are means +/− SEM; *p < .001 to vector, #p < .036 versus CERS6, $p < .03 versus vector+CNL. Insert shows expression of FLAG‐CERS6 and β‐actin. (B) Cell death was assessed in control or CERS5 or CERS6 overexpressing MOLM‐14 treated with the indicated drug combinations (CNL 15 μM, Ara‐C 1.25 μM, venetoclax 50 nM) for 24 h. N = 5; shown are means +/− SEM; *p < .05 between control vector and CERS5 or control vector and CERS6 overexpression in each treatment group.
For experiments in Figure 3B, cells were plated in a 24‐well plates at 0.8‐1 × 106 per well in 2 ml of media with drugs added, mixed well and 200 μl cell aliquots were transferred to 96‐well tray. Twenty‐four hours later, cells were fixed by addition of formaldehyde to 1% for 10 min at room temperature followed by neutralization with the addition of ¼ (vol/vol) of 1.7 M Tris, 1.25 M Glycine, pH 9.1 for 5–20 min. Cells were washed once with 1% BSA/0.2% Tween 20/PBS (cytoC Stain buffer) and incubated overnight at 4°C with 1:400 anti‐cytochrome C‐AF647 antibody diluted in the same buffer. The next day, cells were washed once with cytoC Stain buffer, resuspended in 150 μl of the same buffer and analyzed with Attune flow cytometer. Percentage of cells negative for cytochrome C staining was calculated using FlowJo.
2.8. Sphingolipid analyses
Samples were prepared and analyzed for sphingolipids essentially as previously described. 10 Briefly, cellular lysates corresponding typically to 400 μg of protein were subjected to lipid extractions. Extracts were subsequently analyzed by LC–MS/MS on an I‐class Acquity with a 2.1 mm × 10 cm C18 CSH 1.7 μm particle size column coupled to an in‐line TQ‐S mass spectrometer using multiple reaction monitoring.
2.9. Patient samples and colony‐forming assay
All clinical samples and information were collected at the Penn State College of Medicine under Institutional Review Board‐approved informed consent (Table 1). The ex vivo cell colony assay of primary AML cells was as described. 12 , 13
TABLE 1.
Clinical samples used in this study
| Code | Gender | Age | Dis stage | AML subtype | WBC (10e3/μl) | % Blasts | Molecular | Cytogenetics |
|---|---|---|---|---|---|---|---|---|
| 1103 | F | 41 | Dx | De Novo | 247.25 | 86% | FLT3‐ITD, NOTCH1, PTPN11 | 46,XX |
| 1172 | F | 46 | Dx | De Novo | 149.23 | 96% | FLT3‐ITD | 46,XX |
| 1241 | F | 50 | Dx | De Novo | 180.4 | 90% | NPM1, FLT3‐ITD | 46,XX |
| 1099 | M | 86 | Dx | De Novo | 141.02 | 61% | N/A | 46,XY |
| 934 | F | 72 | Dx | De Novo | 137.78 | 98% | NPM1, FLT3‐ITD | 46,XX |
| 1071 | M | 81 | Dx | De Novo | 148.89 | 98% | N/A | 47, XY,+8[18] |
Briefly, cryopreserved human AML patient cells were thawed and washed with RPMI‐1640 with 10% FBS medium before treating with 15 μM ceramide nanoliposome (CNL), 1 μM cytarabine (Ara‐C), 0.05 μM venetoclax (VEN) alone and in combinations (double or triple) for 24 h in RPMI‐1640 with 20% FBS medium. Posttreatment, the cells were washed, counted, and cultured in triplicate in 12‐well plates at a density of 0.1 × 105 to 2 × 105 cells per well in Human Methylcellulose Complete Media (HSC0003; R&D Systems, Minneapolis, MN). Plating densities were selected for each case to yield more than 20 colonies per well. Colonies were propagated for 10–14 days, and blast colonies (>20 cells/colony) were counted in a masked manner under a light microscope.
2.10. Animal studies
Human AML cell line MOLM‐13‐Luc‐YFP cells (5 × 105) were intravenously injected into 6–8‐week‐old female NOD.Cg‐Rag1tm1Mom Il2rgtm1Wjl/SzJ (NRG) (The Jackson Laboratory, Bar Harbor, ME) mice via the tail vein to form a measurable systemic leukemia. Animals (n = 5) were randomized and assigned to control and treatment groups on day 4 based on total body Bioluminescence Imaging (BLI) signals. Beginning on day 4 (post‐engraftment), mice were treated with CNL (30 mg/kg; IV; alternate day), Ara‐C (25 mg/kg; IP) venetoclax (100 mg/kg; Oral gavage) both 5 days on/2 days off then 5 days on again alone or in combination until day 16. Animals were imaged using the IVIS Lumina LT Series III imaging system (Perkin Elmer, Waltham, MA) after 7–10 min of D‐luciferin (Gold Biotechnology, St Louis MO) injection (150 mg/kg; IP) on indicated days. The photons emitted from cells, expressed as total flux (photons/s/cm2/steradian), were quantified and analyzed using the “Living image” software (Perkin Elmer). The study was terminated when all the animals reached humane endpoints or died.
For the syngeneic mouse AML model, murine leukemia C1498‐Luc cells (1 × 106) were injected via IV route into 6–8 week‐old albino C57BL/6J mice (N = 7; The Jackson Laboratories) as described previously. 13 Animals were randomized based on body weights on day 7 (post‐engraftment) and treated with CNL (31.2 mg/kg; IV; alternate day), Ara‐C (75 mg/kg; IP; alternate day) alone or in combination till day 18. The study was terminated when all animals reached humane endpoints or died.
The Institutional Animal Care and Use Committees (IACUC) of the Penn State College of Medicine approved of the studies and procedures utilizing laboratory animals.
2.11. Synergy calculations
Synergy for combination drug responses were based on the Bliss independence model using SynergyFinder software. 14 Combination drug responses were evaluated using a two‐drug format (e.g., drug A alone vs. drug B alone vs. the combination). Triple combination drug responses were also evaluated using a two‐drug format (e.g., drug A vs. drug B/C vs. triple combination). Synergy scores represent the percent difference between the predicted versus expected drug responses, and drug combinations were considered synergistic or antagonistic based on the difference between predicated versus observed responses. Scores less than 10 were considered antagonistic. Score between −10 and 10 were considered additive. Scores greater than 10 were considered synergistic.
2.12. Statistical analysis
Data shown in Figures 1A–D, 3A, and 5A–D were analyzed using one‐way ANOVA for a factorial experiment in complete randomized blocks, with “experiment” as the blocking factor. 15 Data in Figure 1E were analyzed using two‐way ANOVA, with factors “patient” and “group.” The factor “patient” was treated as a random effect. Kaplan–Meier analysis was used for data shown in Figure 2A–D. Data shown in Figure 4A–C were analyzed by one‐way ANOVA. Data shown in Figures 3B and 6B were analyzed by two‐way ANOVA. Data analysis was done using GraphPad Prism software (GraphPad, V. 9.4, San Diego, CA).
FIGURE 1.
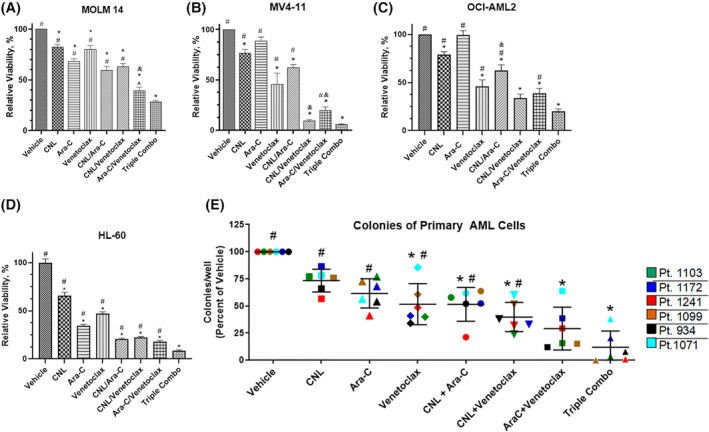
CNL augments the efficacy of cytarabine/venetoclax treatment in in vitro and ex vivo AML models. (A–D) MOLM‐14, MV4‐11, OCI‐AML2, and HL‐60 cells were treated with the indicated drug combinations (CNL 15 μM, Ara‐C 1.25 μM, venetoclax 50 nM) for 24 h (A–C) or 48 h (D) and viability was assessed by MTS assay. N = 4, shown are means +/− SEM. MOLM‐14: *p < .0005 versus vehicle; #p < .0001 versus Triple Combo (CNL/Ara‐C/venetoclax), ^p < .04 versus Triple Combo; MV4‐11: *p < .01 versus vehicle; #p < .0001 versus Triple Combo; OCI‐AML2 and HL‐60: *p < .001 to vehicle, #p < .001 versus Triple Combo. &, denotes combination treatments with synergetic effect on cell viability. (E) Cryopreserved human de novo AML patient cells (n = 6) cultured and treated for 24 h with ceramide nanoliposome (CNL, 15 μM), Ara‐C (1 μM), venetoclax (50 nM), or combinations. Posttreatment, the cells were propagated for 10–14 days, and blast colonies counted. Data are the mean ± SEM; *p < .02 versus vehicle; #p < .002 versus Triple Combo. Shown are means +/− SD.
FIGURE 5.
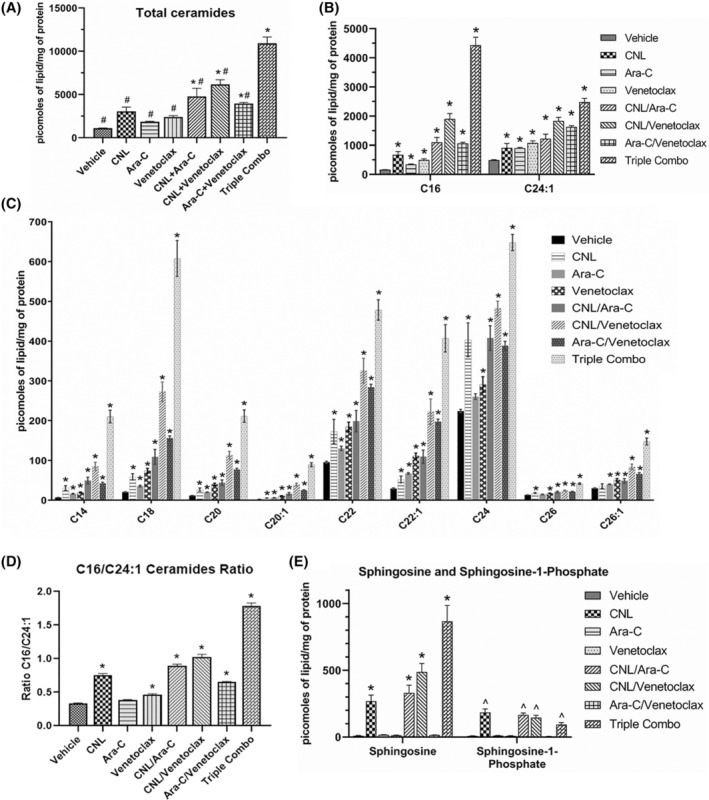
Combinatorial Ara‐C/venetoclax treatment regimen augments metabolism of CNL to long‐chain ceramide metabolites. (A‐E). MOLM‐14 cells were treated with CNL 15 μM, Ara‐C 1.25 μM, venetoclax 50 nM alone or in combinations as indicated for 24 h, prior to lipid analyses. Shown are total ceramide levels (A), individual ceramide species (A‐C), ratios of the change in long‐chain C16‐ to very‐long‐chain C24:1‐ceramide (D), and levels of sphingosine and sphingosine‐1‐phosphate (E). N = 4–5, shown are means +/− SEM *p < .02 to vehicle; #p < .001 to CNL/Ara‐C/venetoclax (A); ^p < .005 to vehicle (E).
FIGURE 2.
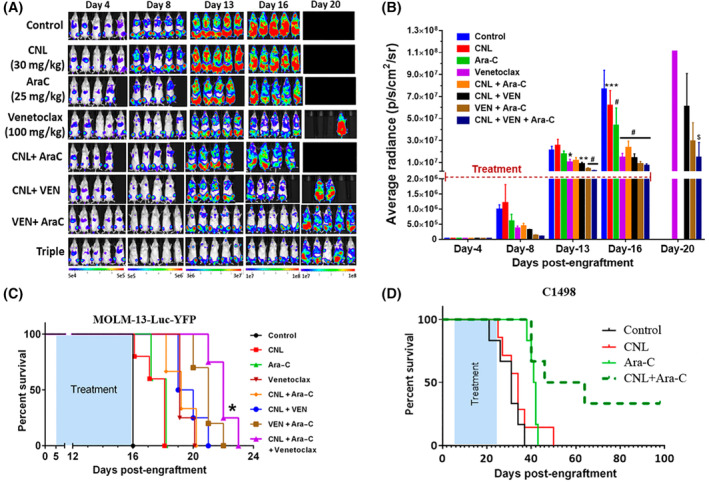
CNL augments the efficacy of cytarabine/venetoclax treatment in in vivo AML models. (A–C) Bioluminescence images (A), their quantification (B) and survival of MOLM‐13 Luc‐YFP bearing NRG mice (C) treated for 12 days with control or CNL (30 mg/kg; intravenous; every other day), Ara‐C (25 mg/kg; intraperitoneally), and venetoclax (100 mg/kg; oral gavage) 13 both 5 days on/2 days off then 5 days on again alone or in combination. N = 5/group. B: *p < .05; **p < .01; ***p < .001; #p < .0001 versus control by two‐way ANOVA (Dunnett's test); $p < .001 versus CNL + Ara‐C by two‐way ANOVA (Sidak's test). Shown are means +/− SD; C: *p < .005 between VEN/Ara‐C and CNL/VEN/Ara‐C by Kaplan–Meier analysis. CNL dose and dosage regimen was determined by previous PK and efficacy studies in solid and non‐solid tumor models 45 (D) Survival of C1498‐bearing C57Bl/6J mice treated 11 days with alternative days dosing frequency with either Control (intravenous, similar volume equivalent to CNL formulation), CNL (intravenous, 31.2 mg/kg), Ara‐C (intraperitoneally, 75 mg/kg) or CNL/Ara‐C. The CNL/Ara‐C group had median survival time of 55 days compared with 34 days for the CNL monotherapy group (p < .01), 41 days for the Ara‐C monotherapy group (p < .05), and 31 days for control group (p < .001) by Kaplan–Meier analysis.
3. RESULTS
We assessed if CNL could augment the efficacy of venetoclax and Ara‐C in AML cell lines with different mutations (Table 2), patient specimens, and in vivo models. We noted that the combinatorial treatment of CNL with Ara‐C/venetoclax at suboptimal doses induced significant loss of viability compared to single or most double drug treatments. (Figure 1A–D). For example, in four cell lines tested (MOLM‐14, OCI‐AML2, HL‐60, and MV4‐11), CNL added to Ara‐C/venetoclax treatment, significantly increased cell death compared to Ara‐C/venetoclax alone. In some cell lines, combination of CNL with Ara‐C or venetoclax had synergistic effects on cell viability, but in general was additive (Figure 1B,C and Table S1). Specifically, CNL was synergistic with venetoclax in MV4‐11 cells and Ara‐C in OCI‐AML2 cells. The Bliss synergy scores for CNL + Ara‐C/venetoclax triple combination fell just short of the synergy threshold in these two cell lines (9.55 in MV4‐11 and 9.53 in OCI‐AML2). A colony formation assay with primary AML patient samples confirmed that the combination of CNL with Ara‐C and venetoclax were significantly more effective than these drugs as single agents or in double combinations (Figure 1E). This combination was just below the cutoff for synergy at 8.79 (Table S1).
TABLE 2.
Human AML cell lines used in this study
| Cell line | Source | Mutations |
|---|---|---|
| OCI‐AML2 | peripheral blood of a 65‐year‐old male with acute myeloid leukemia | DNMT3A R635W (MLL‐MLLT4; MLL‐AF6) gene fusion |
| MOLM‐14 | peripheral blood of a 20‐year‐old male with acute myeloid leukemia | FLT3‐ITD (MLL‐MLLT3; MLL‐AF9) fusion |
| HL‐60 | Peripheral blood of the 36‐year‐old female with acute promyelocytic leukemia | N‐RAS Q61L, amplified c‐MYC |
| MV4‐11 | Blasts of a 10‐year‐old male with biphenotypic B‐myelomonocytic leukemia (AML FAB M5) | FLT3‐ITD, MLL‐AFF1 gene fusion |
We next documented if these in vitro and ex vivo results could be replicated in vivo. Immune‐compromised NRG mice were engrafted with the very aggressive MOLM‐13 cells that overexpress luciferase. MOLM‐13 cells are derived from the same patient with relapsed AML as MOLM‐14 cells. 16 Here, we demonstrate that CNL/Ara‐C/venetoclax treatment, applied for only 12 days, decreased the leukemia burden (Figure 2A,B) compared to other treatments, including Ara‐C/venetoclax at the advanced stage of disease (Figure 2B, day 20). Importantly, the CNL/Ara‐C/venetoclax regimen increased the lifespan of mice (Figure 2C) compared to single and double treatment regimens, including Ara‐C/venetoclax‐treated mice. Utilizing a separate syngeneic, venetoclax‐resistant (EC50 > 10 μM), C1498‐luc mouse AML model, single‐agent Ara‐C or CNL treatment yielded no increase in survival. However, the combinatorial short‐term 11‐day therapy resulted in a median survival of 55 days versus 31 days in the control group, with 30% of the animals on combination therapy surviving >100 days. (Figure 2D).
Given that we observed decreased cell viability with CNL alone that was exacerbated with Ara‐C/venetoclax treatment, we next probed the mechanism(s) of cell death. To assess apoptosis, we utilized caspase 3 activation as a readout and noted that CNL did not induce caspase 3 cleavage or alter Ara‐C and/or venetoclax‐induced caspase‐3 cleavage (Figure 3A). Similarly, Ara‐C and venetoclax both increased cytochrome C release, which was unaffected by CNL (Figure 3B). Furthermore, these data were confirmed by PARP cleavage as another marker for apoptosis (Figure 3C, top panel). Again, we noted that CNL treatment did not induce apoptosis, nor did it augment the apoptotic effects induced by Ara‐C and venetoclax. While CNL did not alter markers of apoptosis, this treatment did induce the formation of an autophagy‐specific marker, the phosphatidylethanolamine‐conjugated derivative of the LC3B protein, LC3B‐II (Figure 3C, middle panel). Ara‐C and/or venetoclax treatment did not induce LC3B expression and did not markedly affect CNL induction of LC3B‐II. Thus, our data suggest that the mechanisms underlying cooperativity between CNL and Ara‐C/venetoclax‐induced cell death are independent of apoptosis or autophagy.
We next assessed if CNL alters drug‐resistance survival mechanisms induced by venetoclax or Ara‐C. Venetoclax‐resistant cells have been shown to upregulate pro‐survival Mcl‐1, 17 while Ara‐C resistant cells can activate checkpoint kinase 1 (Chk‐1) to permit DNA repair. 18 While venetoclax, CNL, and Ara‐C all induced Mcl‐1 expression in MOLM‐14 cells, combination treatments reduced this response, with the triple combination reducing levels 64% below basal (Figure 4A). Combination treatments also reduced the pro‐survival protein XIAP, but not survivin levels. Venetoclax moderately decreased Bcl‐2 protein levels, but neither CNL nor Ara‐C further diminished this expression. CNL attenuated the Ara‐C‐induced protein phosphorylation of Chk‐1, but not Chk‐2 (Figure 4B). Venetoclax, by itself, reduced both the expression and Ara‐C‐induced phosphorylation of Chk1 and 2. CNL and/or venetoclax also reduced Ara‐C‐induced cdc25C phosphorylation, which is regulated by Chk1. 19 Taken together, CNL treatment reduced drug‐resistant signaling mechanism induced by venetoclax or Ara‐C.
We next investigated the effects of CNL, Ara‐C, and both agents together on a venetoclax‐resistant, MV4‐11 cell line generated by chronic treatment with venetoclax (Figure 4C). Mcl‐1 expression was increased in venetoclax‐resistant cells. CNL, in the presence or absence of Ara‐C, reduced basal and venetoclax‐dependent Mcl‐1 expression, confirming data in Figure 4A. Similarly, CNL reduced the phosphorylation and expression of basal and Ara‐C‐induced Chk1, especially in the venetoclax‐resistant cells. Even in this venetoclax‐resistant model, CNL reduced drug‐resistant and pro‐survival signaling mechanisms and these effects were most pronounced with combinatorial approaches.
To determine whether Ara‐C and/or venetoclax treatment alters endogenous sphingolipid or C6‐ceramide metabolism, we conducted lipidomic analyses (Figure 5A–E). We observed that combinatorial Ara‐C and venetoclax treatment increased total ceramide levels in MOLM‐14 cells compared to vehicle control. While significant elevations in highly abundant (Figure 5B) and less abundant (Figure 5C) ceramide species were seen with the triple combination versus control and/or the single or double combinations, the most pronounced elevations were noted for C16‐ceramide (Figure 5B). The CNL/Ara‐C/venetoclax triple regimen increased the C16/C24:1 ratio by 3.86‐fold relative to vehicle (Figure 5D). Moreover, the CNL/Ara‐C/venetoclax combination also dramatically elevated sphingosine, a pro‐death sphingolipid metabolite compared to other treatment groups. In contrast, the level of sphingosine‐1‐phosphate, a pro‐survival sphingolipid, while elevated by CNL, was significantly reduced by Ara‐C/venetoclax treatment (Figure 5E). The combination of CNL with Ara‐C, venetoclax, or Ara‐C + venetoclax largely had no effect on HexCer levels beyond those seen with single agent CNL, although decreased levels of C22‐ and C24‐HexCeramide species were observed between the combination of CNL/Ara‐C/venetoclax and CNL alone (Figure S1A,B). CNL treatment decreased sphingomyelin levels alone or in combination with Ara‐C, venetoclax, and Ara‐C + venetoclax for the most abundant SM species, C16 and C24.1 (Figure S1C).
As long‐chain ceramides, like C16‐ceramide, have been suggested to be preferentially more pro‐death than very‐long‐chain C24‐ceramide species, 20 , 21 we next investigated if C16‐ceramide generation selectively drives AML cell death. We transduced MOLM‐14 cells with ceramide synthase 6 (CERS6), which selectively produces C14/C16‐ceramide. 22 Here, CERS6 overexpression increased C14‐ and C16‐ceramides at the expense of C22/24/26 species (Figure 6A) and was exacerbated by CNL. While overexpression of CERS6 was insufficient to alter cell death in MOLM‐14 cells treated with CNL, Ara‐C, or venetoclax alone, the combination treatments of Ara‐C and venetoclax with or without CNL significantly decreased cell viability (Figure 6B). Similar results were seen by overexpressing the C16‐generating CERS5 (Figure 6B). Taken together, these data suggest that combinations of CNL with Ara‐C and venetoclax can remodel short‐chain C6‐ceramides into long‐chain, pro‐death, C16‐ceramide species; driving the increase in the C16‐ceramide to C24:1‐ceramide ratio (Figure 5D) and inducing cell death (Figure 6B).
4. DISCUSSION
Our data show that CNL augmented the efficacy of Ara‐C and/or venetoclax within in vitro human AML cell lines (Figure 1A–D), ex vivo AML patient specimens (Figure 1E), and extended the lifespans within two distinct murine models after therapy discontinuation (Figure 2A–D). While we did not observe an overt apoptotic or autophagic mechanism underlying interactions between CNL and Ara‐C and/or venetoclax (Figure 3), we did demonstrate that CNL reduced pro‐mitogenic and pro‐survival drug‐resistance proteins and kinases induced by Ara‐C and venetoclax (Figure 4) and led to augmented C16‐ceramide and sphingosine production to increase cell death (Figures 5 and 6).
While clinical adoption of venetoclax regimens has provided treatment options to previously untreatable and relapsed AML patients, incomplete remission rates and eventual treatment resistance remain a challenge. The action of CNL to significantly augment Ara‐C/venetoclax therapy, in part through suppressing resistance mechanisms, offers some promise for improving patient outcomes. Venetoclax‐dependent upregulation of anti‐apoptotic proteins such as Mcl‐1, as well as Ara‐C‐upregulation of drug‐resistant kinases that induce DNA repair and promote survival, such as Chk‐1 and cdc25C, has been described. 23 Moreover, targeting Chk1 24 , 25 and Mcl‐1 26 , 27 have been shown to improve responses to Ara‐C and venetoclax, respectively. In this regard, it is worth noting that CNL treatment of venetoclax‐resistant MV4‐11 cells induced a more profound decrease in Mcl‐1 and phospho‐Chk1 levels than in drug‐naïve MV4‐11 (Figure 4C). The mechanism for the observed cell line specific dynamics of CNL on these proteins are presently unknown, but could reflect changes in cofactors and upstream kinases that regulates these targets. Additional experiments are required to answer these intriguing observations.
Our team was the first to report that exogenous addition of ceramide through CNL could limit survivin‐dependent drug‐resistance mechanisms. 28 Substantiating these findings, our group has recently published that elevation of endogenous ceramides via acid ceramidase inhibition induced apoptosis in AML cell models via alternative Mcl‐1 splicing and degradation. 10 Moreover, the Pitson group has recently reported that inhibition of sphingosine kinase, which also elevates endogenous ceramide, downregulated venetoclax‐induced Mcl‐1 expression through inducing an integrated stress response. 29 Independent of CNL, to our knowledge, this is the first demonstration that venetoclax also suppressed Ara‐C activation of Chk1‐dependent drug resistance.
The effect of CNL to downregulate these drug‐resistance signaling mechanisms in a chronically maintained cell model of venetoclax resistance was also recapitulated by demonstrating CNL/Ara‐C therapeutic efficacy in a venetoclax‐resistant C1498 syngeneic murine model. The extended survival of C1498 bearing animals receiving Ara‐C and CNL, unusual for models of this kind, suggests that CNL given with this cytotoxic agent may yield durable remissions and is reminiscent of the efficacy observed with CNL in combination with vinblastine that we demonstrated in a MV4‐11 xenograft model. 4 In fact, as was shown in our recent work, 4 , 30 inhibitors of lysosomal function (e.g., chloroquine and vinblastine) can switch protective autophagy to a lethal mechanism. Thus, we speculated that Ara‐C or venetoclax would also exert similar effects upon CNL‐induced autophagy, given that venetoclax has been shown to induce autophagy‐associated cell death in solid tumors. 31 However, our studies assessing LC3B‐II suggest that Ara‐C or venetoclax are not exerting such an effect in AML. Regardless, these results offer the prospect of improved and more durable therapeutic responses for AML patients, and the circumvention of de‐novo and acquired venetoclax‐ and/or Ara‐C‐resistance.
AML is highly heterogeneous in disease presentation, mutational profile, and prognosis, making it a challenge to uniformly treat. 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 Although we demonstrated beneficial outcomes in several models, the influence of driver mutations and chromosomal rearrangements on therapeutic efficacy, possibly through altered sphingolipid metabolism, requires addition studies. FLT3‐ITD mutations, translocation t(8;21) and inversion (inv16) have indicated differential sphingolipid metabolism in AML subsets. 7 , 40 Given that several of our cellular models (MOLM‐13, MOLM‐14, MV4‐11) contain a poor prognosis associated FLT3‐ITD driver mutation, it is of interest that the Ogretman group has previously shown that FLT3‐ITD inhibits CERS1‐dependent C18‐ceramide formation and mitophagy in AML. 7 The roles of specific ceramide species distinguished by their fatty acid composition is gaining greater appreciation. Our work argues for a novel lipid‐centric metabolic mechanism by which Ara‐C and/or venetoclax augmented the efficacy of CNL in AML. We demonstrated that Ara‐C and venetoclax preferentially stimulated the generation of long‐chain ceramide species, such as C16‐ceramide. This was exacerbated by the addition of CNL, which in the presence of Ara‐C/venetoclax, led to augmented conversion of CNL (C6‐ceramide) preferentially to C16‐ceramide species versus very‐long‐chain C24:1 species (Figure 5). The importance of this conversion is demonstrated upon overexpression of C16‐ceramide generating ceramide synthases 5 or 6, which led to augmented cell death (Figure 6). While the pathophysiological role of individual ceramide species is still controversial, evidence is starting to accumulate for a distinct dichotomy between ceramide species. For example, generation of C24‐ceramide species by CERS2 expression stimulated, while CERS6 expression inhibited colony formation, cell proliferation, and induction of apoptosis in human cancer models. 20 Long‐chain C16‐ and C18‐ceramides, in contrast to very‐long‐chain ceramides (≥C22), may preferentially induce cell death via lethal autophagy. 41 Moreover, CERS2‐generated very‐long‐chain ceramides interfere with C16‐ceramide‐induced mitochondrial channel formation. 42 Overall, these studies suggest that ceramide ratios between long chain (e.g., C16) and very‐long chain (e.g., C24:1) may predict or regulate therapeutic efficacy and advocate for studies to apply sphingolipidomic analysis to specimens from molecularly defined AML patients and their responses to therapeutics. While similar alterations in ceramide species ratios have been used as predictive of cardiovascular risk, 43 , 44 our data provide compelling evidence that these ratios may also be relevant to cancer.
Taken together, our data offer a strong rationale for the addition of CNL to the existing standard of care Ara‐C and venetoclax regimen. The extended survival of two models of AML treated with CNL and cytotoxic agents (vinblastine for MV4‐11 and Ara‐C for C1498) suggest that these, and other CNL drug combinations, 4 may prove to offer improved remission and survivorship for AML. It could be argued that the models used by us recapitulate relapsed/refractory disease to venetoclax and further supports the putative utility of CNL administration for patients who have relapsed or become refractory to either Ara‐C or venetoclax. In fact, based upon the present data and success of CNL as a monotherapy in solid tumors (NCT0283461), with no observed dose‐limiting toxicities at doses fivefold over presumed efficacious doses, the initial phase of a Ceramide/Ara‐C/venetoclax (CAV) trial for relapsed/refractory AML patients has now received FDA IND approval (142902).
AUTHOR CONTRIBUTIONS
AK, TD, DF, TL, AS, DC, JS, TF, and MK designed the experiments; AK, TD, JS, PCP, AC, UG, AS, and TF performed the experiments; MCC and SFT provided cells; AK, TD, AS, DF, TL, DC, JS, JU, MRC, TF, and MK analyzed the data; AK, TD, DF, TL, DC, TF, AS, and MK drafted the manuscript.
FUNDING INFORMATION
This work was supported by NCI Award P01‐CA171983 and a Cancer Center Support Grant P30‐CA044579. P.C.P was funded by the National Cancer Institute of the National Institutes of Health (F99‐CA245802). J.U. was supported in part by F31‐CA271809 and by the Robert R. Wagner Fellowship at the University of Virginia.
DISCLOSURES
Penn State Research Foundation has licensed CNL to Keystone Nano, Inc (PA). MK is CTO and cofounder of Keystone Nano. TPL is a member of the SAB of Keystone Nano, Bioniz Therapeutics, Kymera Therapeutics, and Dren Bio. Other authors declare no competing financial interests.
Supporting information
Table S1
ACKNOWLEDGMENTS
The authors thank Drs. Benjamin Ebert, Didier Trono, and Anthony Futerman for gifts of plasmids, The University of Virginia Flow Cytometry Core, and the Bioluminescence Imaging Core at the Penn State University College of Medicine.
Khokhlatchev AV, Sharma A, Deering TG, et al. Ceramide nanoliposomes augment the efficacy of venetoclax and cytarabine in models of acute myeloid leukemia. The FASEB Journal. 2022;36:e22514. doi: 10.1096/fj.202200765R
Andrei V. Khokhlatchev and Arati Sharma contributed equally.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in the methods of this article. Further information and requests for reagents are available from the corresponding author, Dr. Todd Fox, upon reasonable request.
REFERENCES
- 1. Wei AH, Montesinos P, Ivanov V, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo‐controlled trial. Blood. 2020;135(24):2137‐2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment‐naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791‐803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barth BM, Wang W, Toran PT, et al. Sphingolipid metabolism determines the therapeutic efficacy of nanoliposomal ceramide in acute myeloid leukemia. Blood Adv. 2019;3(17):2598‐2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ricci C, Onida F, Ghidoni R. Sphingolipid players in the leukemia arena. Biochim Biophys Acta. 2006;1758(12):2121‐2132. [DOI] [PubMed] [Google Scholar]
- 6. Morad SAF, Cabot MC. Ceramide‐orchestrated signalling in cancer cells. Nat Rev Cancer. 2013;13(1):51‐65. [DOI] [PubMed] [Google Scholar]
- 7. Dany M, Gencer S, Nganga R, et al. Targeting FLT3‐ITD signaling mediates ceramide‐dependent mitophagy and attenuates drug resistance in AML. Blood. 2016;128(15):1944‐1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kao L‐P, Morad SAF, Davis TS, et al. Chemotherapy selection pressure alters sphingolipid composition and mitochondrial bioenergetics in resistant HL‐60 cells. J Lipid Res. 2019;60(9):1590‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stover TC, Sharma A, Robertson GP, Kester M. Systemic delivery of liposomal short‐chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin Cancer Res. 2005;11(9):3465‐3474. [DOI] [PubMed] [Google Scholar]
- 10. Pearson JM, Tan S‐F, Sharma A, et al. Ceramide analogue SACLAC modulates sphingolipid levels and MCL‐1 splicing to induce apoptosis in acute myeloid leukemia. Mol Cancer Res. 2020;18(3):352‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jayappa KD, Portell CA, Gordon VL, et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv. 2017;1(14):933‐946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Annageldiyev C, Tan S‐F, Thakur S, et al. The PI3K/AKT pathway inhibitor ISC‐4 induces apoptosis and inhibits growth of leukemia in preclinical models of acute myeloid leukemia. Front Oncol. 2020;10:393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Annageldiyev C, Gowda K, Patel T, et al. The novel Isatin analog KS99 targets stemness markers in acute myeloid leukemia. Haematologica. 2020;105(3):687‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng S, Wang W, Aldahdooh J, et al. SynergyFinder plus: toward better interpretation and annotation of drug combination screening datasets. Genomics Proteomics Bioinformatics. 2022. Online ahead of print; S1672‐02229(22)00008‐0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montgomery D. Design and analysis of experiments. 3rd ed. John Wiley and Sons; 1991. p. Chapter 14. [Google Scholar]
- 16. Matsuo Y, MacLeod RA, Uphoff CC, et al. Two acute monocytic leukemia (AML‐M5a) cell lines (MOLM‐13 and MOLM‐14) with interclonal phenotypic heterogeneity showing MLL‐AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia. 1997;11(9):1469‐1477. [DOI] [PubMed] [Google Scholar]
- 17. Bose P, Gandhi V, Konopleva M. Pathways and mechanisms of venetoclax resistance. Leuk Lymphoma. 2017;58(9):1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sampath D, Cortes J, Estrov Z, et al. Pharmacodynamics of cytarabine alone and in combination with 7‐hydroxystaurosporine (UCN‐01) in AML blasts in vitro and during a clinical trial. Blood. 2006;107(6):2517‐2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu K, Zheng M, Lu R, et al. The role of CDC25C in cell cycle regulation and clinical cancer therapy: a systematic review. Cancer Cell Int. 2020;20:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hartmann D, Lucks J, Fuchs S, et al. Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth. Int J Biochem Cell Biol. 2012;44(4):620‐628. [DOI] [PubMed] [Google Scholar]
- 21. Sheridan M, Ogretmen B. The role of ceramide metabolism and signaling in the regulation of mitophagy and cancer therapy. Cancers (Basel). 2021;13(10):2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cingolani F, Futerman AH, Casas J. Ceramide synthases in biomedical research. Chem Phys Lipids. 2016;197:25‐32. [DOI] [PubMed] [Google Scholar]
- 23. Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. 2014;134(5):1013‐1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vincelette ND, Ding H, Huehls AM, et al. Effect of CHK1 inhibition on CPX‐351 cytotoxicity in vitro and ex vivo. Sci Rep. 2019;9(1):3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. David L, Fernandez‐Vidal A, Bertoli S, et al. CHK1 as a therapeutic target to bypass chemoresistance in AML. Sci Signal. 2016;9(445):ra90. [DOI] [PubMed] [Google Scholar]
- 26. Liu F, Zhao Q, Su Y, et al. Cotargeting of Bcl‐2 and mcl‐1 shows promising antileukemic activity against AML cells including those with acquired cytarabine resistance. Exp Hematol. 2022;105:39‐49. [DOI] [PubMed] [Google Scholar]
- 27. Zhou F‐J, Zeng C‐X, Kuang W, et al. Metformin exerts a synergistic effect with venetoclax by downregulating mcl‐1 protein in acute myeloid leukemia. J Cancer. 2021;12(22):6727‐6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu X, Ryland L, Yang J, et al. Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK‐LGL leukemia. Blood. 2010;116(20):4192‐4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lewis AC, Pope VS, Tea MN, et al. Ceramide‐induced integrated stress response overcomes Bcl‐2 inhibitor resistance in acute myeloid leukemia. Blood. 2022;139(26):3737‐3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shaw JJP, Boyer TL, Venner E, et al. Inhibition of lysosomal function mitigates protective mitophagy and augments ceramide nanoliposome‐induced cell death in head and neck squamous cell carcinoma. Mol Cancer Ther. 2020;19(12):2621‐2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alhoshani A, Alatawi FO, Al‐Anazi FE, et al. BCL‐2 inhibitor Venetoclax induces autophagy‐associated cell death, cell cycle arrest, and apoptosis in human breast cancer cells. Onco Targets Ther. 2020;13:13357‐13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Kouchkovsky I, Abdul‐Hay M. Acute myeloid leukemia: a comprehensive review and 2016 update. Blood Cancer J. 2016;6(7):e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997‐2002. Cancer Causes Control. 2008;19(4):379‐390. [DOI] [PubMed] [Google Scholar]
- 34. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5‐29. [DOI] [PubMed] [Google Scholar]
- 35. Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yamada O, Kawauchi K. The role of the JAK‐STAT pathway and related signal cascades in telomerase activation during the development of hematologic malignancies. JAK‐Stat. 2013;2(4):e25256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Steensma DP, McClure RF, Karp JE, et al. JAK2 V617F is a rare finding in de novo acute myeloid leukemia, but STAT3 activation is common and remains unexplained. Leukemia. 2006;20(6):971‐978. [DOI] [PubMed] [Google Scholar]
- 38. Kantarjian H, Kadia T, DiNardo C, et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 2021;11(2):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stefanko A, Thiede C, Ehninger G, Simons K, Grzybek M. Lipidomic approach for stratification of acute myeloid leukemia patients. PLoS One. 2017;12(2):e0168781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang Z, Wen L, Zhu F, et al. Overexpression of ceramide synthase 1 increases C18‐ceramide and leads to lethal autophagy in human glioma. Oncotarget. 2017;8(61):104022‐104036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stiban J, Perera M. Very long chain ceramides interfere with C16‐ceramide‐induced channel formation: a plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim Biophys Acta. 2015;1848(2):561‐567. [DOI] [PubMed] [Google Scholar]
- 43. Anroedh S, Hilvo M, Akkerhuis KM, et al. Plasma concentrations of molecular lipid species predict long‐term clinical outcome in coronary artery disease patients. J Lipid Res. 2018;59(9):1729‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tippetts TS, Holland WL, Summers SA. The ceramide ratio: a predictor of cardiometabolic risk. J Lipid Res. 2018;59(9):1549‐1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kester M, Bassler J, Fox TE, Carter CJ, Davidson JA, Parette MR. Preclinical development of a C6‐ceramide NanoLiposome, a novel sphingolipid therapeutic. Biol Chem. 2015;396(6–7):737‐747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The data that support the findings of this study are available in the methods of this article. Further information and requests for reagents are available from the corresponding author, Dr. Todd Fox, upon reasonable request.