

Abstract
Hirayama disease (HD) is a rare, benign, and nonprogressive motor neuron disease (MND) affecting the upper limbs. It usually presents with weakness and amyotrophy in a single upper extremity with an insidious onset between adolescence and the third decade of life. Since its description in 1959, HD has been known under several names and eponyms in Europe and in Asian countries probably due to its heterogeneous clinical features. Thus, the unclear nosological classification makes challenging the differential diagnosis between HD and other neuromuscular conditions, such as MNDs. However, apart from the nosological difficulties and the lack of evidence‐based guideline for diagnosis, the neuroimaging is the mainstay for the diagnosis of HD. Indeed, the specific findings on cervical flexion MRI usually lead to a prompt diagnosis. Here, we reviewed the nosological classifications of HD and its neuroimaging features. Also, we report a case of a 18‐year‐old boy who presented to our Clinic complaining of muscle weakness of the left distal upper limb without other neurological signs. The cervical MRI, in the neutral position, revealed a high T2 signal intensity in the C5‐C7 cervical myelomeres as well as the loss of cervical lordosis, whereas, during neck flexion, it showed the anterior displacement of the posterior dura ad the post‐gadolinium T1‐weighted imaging enhancement of the posterior epidural plexus. These findings are typical for HD allowing the diagnosis as well as the differential diagnosis from other neuromuscular diseases.
Keywords: cervical MRI, flexion MRI, Hirayama, JASSMA, MMA, motor neuron disease
INTRODUCTION
Hirayama disease (HD) is a benign and self‐limiting motor neuron disease (MND) described for the first time in 1959 as the “juvenile muscular atrophy of the unilateral upper extremity” characterized by painless weakness and amyotrophy in the forearm and hand of a single upper limb with an insidious onset between adolescence and the third decade of life, followed by spontaneous arrest within a few years after the clinical onset. 1 Of interest, HD has been described under several names according to its clinical features; however, it differs from other incurable MNDs, such as PLS (ie, primary lateral sclerosis), SMA (ie, spinal muscular atrophy), and ALS (ie, amyotrophic lateral sclerosis), because of its benign and nonprogressive course and pathogenesis. It has been hypothesized that the compression of the spinal cord by dura mater during neck flexion might induce chronic microcirculatory changes in the territory of the anterior spinal artery, which supplies the lower cervical cord, thus causing HD. 2 The nosological difficulties are supported by the heterogeneity of HD in clinical presentation, ranging from monomelic amyotrophy to asymmetric or symmetric bilateral amyotrophy of upper limbs in which the right limb is more affected (right/left ratio 3:1). 3 Hypotrophy usually affects both the hand and forearm muscles sparing of brachioradialis muscle contributing to the typical “oblique amyotrophy” of the ventral forearm. 4 Furthermore, in many patients, the muscle weakness may worse after the exposure to a cold environment (ie, cold paresis); however, cranial nerve dysfunction, sensory disturbances, urinary disturbances, and upper motor neuron signs are usually not seen in HD (with rare exceptions). 5 Here, we report a typical case of an adolescent male with a diagnosis of asymmetric HD supported by typical MRI findings. The purposes of this review are to discuss the nosological classification of HD, clarifying the disease's eponyms according to the clinical features of HD, as well as to emphasize the MRI findings which allow the prompt diagnosis.
Clinical scenario
An 18‐year‐old boy with a noncontributory past medical history presented to our Neuromuscular Clinic complaining of subtle muscle weakness of the distal left upper limb in absence of pain or any other symptoms. This weakness had started 12 months earlier immediately after a gym training (more precisely, patient was lifting loads). The first neurological consultation showed a muscle wasting of the left hand, especially of the interossei muscles; also, nerve conduction studies (NCS) showed an axonal loss in the left ulnar nerve. Moreover, the ultrasound imaging of the left upper limb was not contributory, and the cervical spinal cord MRI only demonstrated the loss of physiological cervical lordosis. Several months later, a spinal cord MRI with gadolinium administration evidenced the reduction of blood flow in the left subclavian artery during the left arm was maintained in the maximum abduction position; the same MRI study, also, showed a high intramedullary T2 signal at C7 and C8 levels, which was more pronounced on the left side when considering the axial T2‐weighted sequences. As a consequence, a vascular disease of the upper limbs was suspected, therefore, the patient underwent thoracic, neck, and upper limb CT scans with iodate contrast administration, although relevant findings have not been found. In the meanwhile, the weakness of the left upper limb worsened with subsequent hypotrophy of the left hypothenar muscles accompanied by severe motor weakness. None of the antiganglioside antibodies (GM1, GM2, GM3, GM4, GD1a, GD1b, GD2, GD3, GT1a, GT1b, GQ1b, and sulfatides) have been found. A new neurological examination revealed a motor weakness in the forearm and hand's muscles, bilaterally, with amyotrophy involving the left hand (Figure 1A). According to the Medical Research Council (MRC), the muscle strength of left arm was 2/5 in the first dorsal interosseous, abductor digiti minimi, and in the finger extensors, whereas it was found to be 4/5 in the other muscles. In the left hand, the hypothenar atrophy was greater compared to thenar muscles, thus giving the appearance of the “reverse split hand syndrome” (Figure 1B). The MRC grading of the right arm was 4/5 and 5/5 for distal and proximal muscles, respectively. Deep tendon reflexes were reduced in both upper limbs, whereas upper motor neuron signs were not found. A new NCS with needle‐electromyography of the upper and lower limbs showed the loss of motor axons in the ulnar nerve, bilaterally (with the left hand being more affected) and the active denervation in the left first dorsal interosseous muscle (Table 1). The most common polyneuropathies were ruled out by serum testing of vitamin B12, folates, and vitamin E as well as by performing the investigation for human immunodeficiency virus. Finally, the cervical spinal cord MRI in both neutral and cervical‐flexion positions demonstrated the anterior shifting of the posterior dura and the post‐gadolinium enhancement of the posterior epidural plexus on T2‐weighted and post‐contrast T1‐weighted images, respectively (Figures 2 and 3). These findings, as well as the clinical course, supported the diagnosis of HD. Further neurophysiological procedures contributed in demonstrating typical findings of HD. Indeed, somatosensory‐evoked potentials (SEPs) from bilateral electrical stimulation of the median nerve at the wrist came back normal, while N13 potential was difficult to elicit during neck flexion; motor‐evoked potentials (MEPs) demonstrated low‐amplitude MEPs from abductor pollicis brevis (ABP) bilaterally. Since the diagnosis of HD, the patient has refused neurosurgical intervention and has undergone conservative treatment: neck immobilization with cervical collar and physiotherapy. Two years after the diagnosis, no clinical progression of the disease has been documented; a recent EMG showed no further signs of denervation.
FIGURE 1.
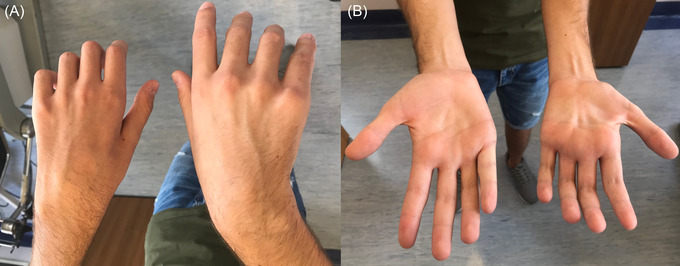
Asymmetric amyotrophy of distal upper limb muscles in patient with Hirayama disease. Moderate to severe muscle wasting of the left hypothenar and interossei muscles and mild atrophy of the right hypothenar muscles (A); in the left hand, the amyotrophy is more severe in the hypothenar muscles compared to thenar with the appearance of the “reverse split hand syndrome” (B)
TABLE 1.
Axonal motor loss in the patient with Hirayama disease
| Motor nerves | Latency (ms) | Amplitude (mV) | Conduction velocity (m/s) |
|---|---|---|---|
| Right median‐APB | |||
| Wrist | 3.4 | 8.3 | |
| Elbow | 7.9 | 7.9 | 55.6 |
| Left median‐APB | |||
| Wrist | 4.25 | 5.6 | |
| Elbow | 8.1 | 6.1 | 59.7 |
| Left ulnar‐ADM | |||
| Wrist | 4 | 0.3 | |
| B. Elbow | 7.35 | 0.5 | 59.7 |
| A. Elbow | 9.9 | 0.3 | 35.3 |
| Axilla | 13.45 | 0.4 | 47.9 |
| Right ulnar‐ADM | |||
| Wrist | 2.8 | 4.7 | |
| B. Elbow | 6.8 | 4.5 | 57.5 |
| A. Elbow | 8.45 | 3.8 | 54.5 |
| Left radial‐EIP | |||
| Forearm | 3.95 | 8.1 | |
| Right common peroneal‐EDB | |||
| Ankle | 3.05 | 8.9 | |
| Fibular head | 10.55 | 8.1 | 40.0 |
| Knee | 11.9 | 8 | 59.3 |
| Left common peroneal‐EDB | |||
| Ankle | 3.9 | 12.8 | |
| Fibular head | 10.75 | 11.7 | 43.8 |
| Knee | 12.55 | 12 | 44.4 |
| Left tibial knee‐AH | |||
| Ankle | 3.7 | 7.1 | |
| Knee | 12 | 7.9 | 43.4 |
| Sensory nerves | Latency (ms) | Amplitude (μV) | Conduction velocity (m/s) |
|---|---|---|---|
| Right median‐Digit II | |||
| 1 | 2.6 | 30.5 | 61.7 |
| 2 | 6.65 | 15.5 | N/A |
| Left ulnar‐Digit V | |||
| Wrist | 2.25 | 19.7 | N/A |
| Before elbow | 5.3 | 19.3 | 75.4 |
| After elbow | 6.8 | 18.2 | 53.3 |
| Axilla | 8.45 | 15.7 | 90.9 |
| Left radial‐thumb | |||
| Forearm | 2.45 | 14.7 | N/A |
| Left sural‐lateral malleolus | 44.8 | ||
| Calf | 2.9 | 14.3 | 61.7 |
Note: Nerve conduction studies show a low‐amplitude left ulnar compound motor action potential and a reduced ratio (<0.6) between the abductor digit minimi and abductor pollicis brevis, bilaterally, supporting the diagnosis.
Abbreviations: ADM, abductor digiti minimi; AH, abductor hallucis; APB, abductor pollicis brevis; EDB, extensor digiti brevis; EIP, extensor indicis proprius; ms, milliseconds; m/s, meters per second; mV, millivolts; N/A, not available; μV, microvolts.
FIGURE 2.
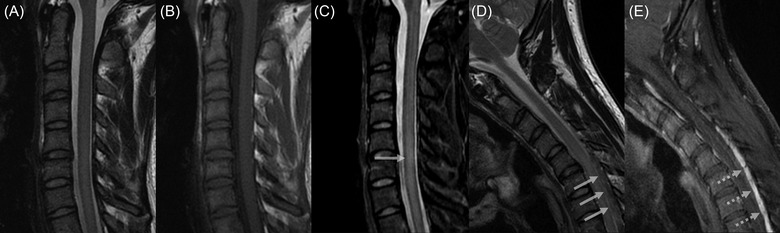
Sagittal T2‐weighted (A) and pre‐contrast T1‐weigted (B) cervical MRI images acquired on neutral position demonstrate the loss of cervical lordosis. Sagittal Short Tau Inversion Recovery image shows mild intramedullary cord hyperintensity (arrow) at the levels of C5‐C7. Flexion T2‐weighted image (D) reveals a 6 mm forward displacement of the posterior cervical dural sac associated with a widening of the posterior laminodural space (arrows). Post‐contrast sagittal T1‐weigted image (E) shows posterior epidural enhancement (dashed arrows) with crescent‐shaped appearance
FIGURE 3.
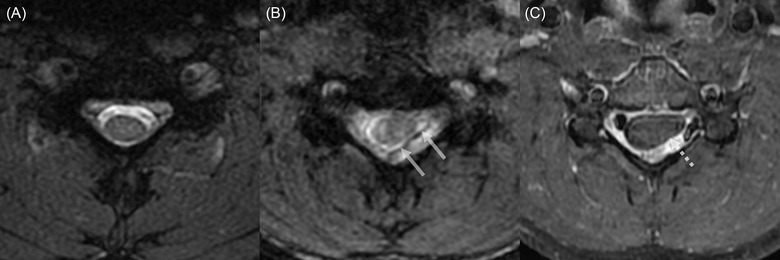
Axial cervical MRI on the neutral (A) and flexion position (B and C). Spoiled T2*‐weighted image at the level of C5 in neutral position shows a normal spinal cord morphology (A). Spoiled T2*‐weighted (B) and post‐contrast T1‐weighted (C) images at the level of C5 reveal the forward displacement of the posterior dural sac (arrows in B) with asymmetrical compression of the spinal cord, more prominent on the left side (dashed arrow in C)
The patient signed the informed consent forms for both diagnostic workup and cervical spinal cord MRI with gadolinium administration. Furthermore, the patient gave the written consent to use its clinical data and images for publication purposes.
Definitions and eponyms
Since the description in 1959 by Hirayama et al., HD has been proposed under several names according to its clinical presentation (Table 2). The term “Brachial MonoMelic Amyotrophy” (MMA) refers to unilateral and distal amyotrophy of a single upper limb, which may spread to contralateral limb in about 20% of cases. 6 However, several atypical cases have been reported, such as the symmetric amyotrophy of the distal upper limbs and the monomelic amyotrophy of the proximal upper limb's muscles. 7 , 8 Preethish‐Kumar et al. coined the term “Distal BiMelic Amyotrophy” (DBMA) to describe a symmetrical amyotrophy of the distal upper extremities with rapid progression to severe motor disability and showing similar MRI features with the previously described entity of MMA/HD (eg, flexion‐induced cervical myelopathy at C8‐T1 myelomers). 9 Also, the flexion‐induced myelopathy may spread from C8‐T1 myelomeres to two segment higher or lower than MMA/HD (eg, C5‐T1 myotomes), leading to a “Proximal BiMelic Amyotrophy” (PBMA) or “Proximo‐Distal biMelic Amyotrophy” (PDMA). 10 However, the absence of sensory, pyramidal, and cerebellar signs, as well as the presence of a cold paresis are common features of HD regardless of the type of muscular atrophy. Hence, we should wonder ourselves whether HD includes MMA or if it may be part of a broader spectrum of nonprogressive and benign MNDs. Furthermore, the marked heterogeneity in the clinical presentation makes the diagnosis of HD challenging, usually requiring experts and trained neurologists. Hence, a nationwide Japanese study has outlined the following diagnostic criteria required for diagnosis of HD: (1) distally dominant muscle weakness and atrophy in the forearm and hand; (2) disease onset between ages 10 years and early 20s; (3) unilateral or unilaterally dominant symptoms and signs; (4) insidious onset followed by arrest of progression; (5) lack of sensory disturbances and tendon reflex abnormalities; and (6) exclusion of other diseases (eg, MNDs, brachial plexopathy, spinal cord tumors, ulnar neuropathy, etc). 11 More recently, further guidelines, based on 24 statements, have been published supporting clinicians in the diagnosis, treatment, and follow‐up of HD. Unfortunately, these recommendations do not represent a standard for diagnosis. 12 In this scenario, as the proposed diagnostic criteria are based mainly on the clinical features, we would highlight the central role of an accurate cervical spinal cord MRI in both neutral and neck‐flexion positions when HD is suspected to achieve an early and correct diagnosis.
TABLE 2.
Nosological classification of the benign and nonprogressive motor neuron diseases
| Eponyms | Clinical presentation | References |
|---|---|---|
| Juvenile muscular atrophy of the unilateral upper extremity | Hirayama et al. in 1959 described the unilateral juvenile amyotrophy of the distal upper‐limb. | 1 |
| Juvenile nonprogressive monomelic amyotrophy (JNPMA) | Hashimoto in 1972 coined the term JNPMA because of the unilateral and nonprogressive amyotrophy of the upper limb. | 38 |
| Monomelic amyotrophy (MMA) | As the amyotrophy may involve the upper or lower limb, Gourie‐Devi et al. in 1984 labeled the term MMA. | 39 |
| Juvenile muscular atrophy of the distal upper extremity (JMADUE) | Biondi et al. in 1989 coined the term JMADUE because of the amyotrophy involved only in the distal upper limb. | 4 , 40 |
| Juvenile asymmetric segmental spinal muscular atrophy (JASSMA) | Pradhan and Gupta in 1997 labeled the term JASSMA because of the bilateral and asymmetric amyotrophy of the upper limbs. | 41 |
| Brachial monomelic amyotrophy (BMMA) | Gourie‐Devi and Nalini in 2003 labeled the term BMMA, which represents a case of HD, distinguishing it from crural MMA. | 10 |
| Distal bimelic amyotrophy | Symmetrical amyotrophy of both upper limbs distal muscles. | 13 |
| Proximal bimelic amyotrophy (PBMA) or proximo‐distal bimelic amyotrophy (PDMA) | PBMA is characterized by the bilateral amyotrophy of the shoulder girdles and arms; PDMA affects bilaterally both proximal and distal muscles of the upper limbs. | 14 |
Further points need some clarifications. Indeed, the use of MEPs and SEPs in the diagnostic workup is still controversial, since some authors reported both normal SEPs and MEPs. 13 However, a significant reduction in amplitude of the N13 cervical response during neck flexion in patients with HD has also been reported. 14 Of interest, the neurophysiological abnormalities of the MEPs have been described in a broad range (14‐100%) of patients with HD. 15 Zheng et al. observed a prolongation of both central motor conduction time and peripheral motor latency in a cohort of 41 patients with HD. 16 According to these studies, we observed an amplitude reduction of the N13 cervical potential during neck‐flexion and the bilateral prolongation of the peripheral motor latency when recording by ABPs, after magnetic stimulation of the cervical roots. These data may support the use of neurophysiology in HD, but their role in the diagnosis is still unclear.
Pathogenesis and clinical presentation
The main pathogenetic hypothesis of HD is that the forward displacement of the lower posterior dural sac during neck flexion might induce the asymmetrical flattening of the anterior lower cervical cord against the vertebral bodies causing subsequently the ischemic injury of the motor neurons located in the spinal anterior horn with the more flattened side corresponding to the more atrophied limb. 17 However, it is still unknown what causes this forward displacement of the dura mater in HD. Some authors theorized that this asymmetric cord compression might be caused by an unequal right‐to‐left distribution of the posterior epidural ligaments, whereas other ones suggested that a disproportion in length between the vertebral column and the dural sac might be behind the HD pathogenesis. 18 , 19 As the imbalance in growth between vertebral column and dural sac could be accentuated during juvenile growth spurt, the “disproportion hypothesis” might explain the juvenile onset and the male preponderance of HD. 20
The role of flexion MRI
MRI has a central role in the diagnostic workup of HD. However, conventional scans are not adequate and usually fail in the diagnosis. Indeed, MRI scans should be acquired with a 1.5‐T or 3.0‐T scanner in both neutral and neck‐flexion positions on the patient lying in the supine position. The optimal scanning protocol includes the following MRI sequences: sagittal and axial T1‐weighted fast/turbo spin‐echo, sagittal and axial T2‐weighted fast/turbo spin‐echo, sagittal and axial T1‐weighted images with fat saturation acquired before and after gadolinium administration with a slice thickness of 3‐4 mm. 21 Flexion MRI should be acquired with a neck‐flexion angle of 35‐40° by using a positioning sponge or a pillow to maintain the flexed position. 22
Table 3 provides the principal MRI findings in HD observed in both neutral and neck‐flexion positions. The main findings in the neutral position are the loss of physiological cervical lordosis, the spinal cord atrophy at C5‐C7 levels, the asymmetric flattening of the spinal cord on axial images as well as the loss of attachment of the posterior dura to the subjacent lamina, and an increased T2 signal intensity of anterior cervical horns without signs of extrinsic compression. 21 , 23 , 24 , 25 , 26 , 27 The loss of the attachment of the posterior dura in the neutral position imaging is reported with a sensitivity and specificity of 93% and 98%, respectively. 23 The cord atrophy and the flattening of the spinal cord in neutral position are associated with a more severe disease and poor recovery after surgical cervical fusion. 26 , 28 , 29 The T2 hyperintensity of anterior cervical spinal horns giving the characteristic “snake eye” appearance is a typical feature of HD, although it has been variably reported with a prevalence of 17‐77% in patients with a confirmed diagnosis of HD. 21 , 27 , 30 , 31 Despite these suggestive MRI signs are frequently observed in HD patients, it remains particularly challenging to identify them on routine nonflexion MRI studies. Indeed, these subtle sings are frequently overlooked, considering the rarity of the disease and if not are specifically searched in the context of HD.
TABLE 3.
Neuroimaging features of Hirayama disease and their prevalence in the present study and in case series 21 , 23 , 24 , 25 , 26 , 27 , 30 , 31
| MRI features | Key sequences | Prevalence |
|---|---|---|
| Neutral position | ||
| Loss of normal cervical lordosis | Sagittal T1WI, T2WI | 50‐100% |
| Cord atrophy at C5‐C7 levels | Sagittal or axial T1WI, T2WI | 59‐100% |
| Asymmetric cervical cord flattering | Axial T1WI, T2WI | 48‐100% |
| Loss of dural attachment | Axial T2WI | 65‐100% |
| Intramedullary cord hyperintensity | Sagittal or axial T2WI | 17‐77% |
| Flexion position | ||
| Displacement of the posterior cervical dural sac | Sagittal T2WI | 100% |
| Compression of the spinal cord | Sagittal or axial T2WI | 100% |
| Epidural flow voids | Sagittal or axial T2WI | 47‐100% |
| Contrast enhancement in epidural space | Sagittal or axial post‐contrast T1WI | 100% |
Abbreviation: WI, weighted imaging.
On the flexion sequences, the loss of dural attachment and the forward displacement of the posterior cervical dural sac which is associated with a widening of the posterior laminodural space, prominent epidural flow voids, and post‐gadolinium enhancement of the posterior epidural plexus represent the pathognomonic imaging features. 25 , 26 , 27 , 32 Among them, the forward displacement of the posterior dural sac is the most relevant sign for the diagnosis of HD on flexion MRI. This displacement is associated with a compression of the spinal cord leading to the asymmetrical reduction of its anteroposterior diameter. 33 However, it is worth considering that the mild displacement of the posterior dural sac may occasionally be observed in normal subjects. Nevertheless, the degree of displacement is significantly greater in HD patients (6‐9 mm) when compared to normal subjects (1‐4 mm); also, in normal conditions, there is a physiological compensation through the intrinsic expansion of the spinal canal in flexion position. 27 , 33 Lai et al. suggested the MRI‐based ratio between the anteroposterior diameter of the maximal forward displacement of the dural sac and the anteroposterior diameter of the spinal canal as a measure distinguishing between HD (increased ratio in flexion position) and normal subjects. 33 This ratio obtained in flexion position has been shown to be directly related to disease length in a subsequent study conducted by Shao et al. evaluating 64 patients with HD. 34 Moreover, the epidural flow voids might be detected on T2‐weighted images as linear hypointense structures in the enlarged epidural space. They are caused by engorgement of the posterior epidural venous plexus during neck flexion, and they are not seen in neutral position. Hou et al. investigated the appearance of MRI features at different neck‐flexion angles in 45 patients with HD. 22 In that study, the anterior displacement of cervical dural sac and the epidural flow voids were significantly more common at 35° compared to 20° (75% vs. 100% and 44% vs. 85%, respectively). 22 Also, the post‐contrast enhancement is observed in the posterior epidural space with a characteristic crescent‐shaped appearance. 25 , 27 , 35 The posterior epidural enhancement typically disappears when the neck returns to neutral position, thus confirming itself to be a congested posterior internal vertebral venous plexus rather than a vascular malformation.
The combination of these MRI features in both neutral and flexion positions provides a high specificity for the diagnosis of HD. 30 , 31 In the study by Gotkine et al., all the patients presenting with the typical MRI features on the cervical flexion MRI had a confirmed HD, while the patients with progressive weakness of the distal upper limb's muscles without these neuroimaging features had the diagnosis of ALS. 30 In a multicentric cohort study published by Lehman et al., the combination of neutral and flexion imaging features had a sensitivity and specificity of 71% and 100% for the diagnosis of HD, respectively. 31
CONCLUSIONS
This review highlights the importance of performing a dynamic flexion and extension cervical MRI in young patients with otherwise unexplained progressive weakness and amyotrophy of the upper limb. In this particular setting, the dynamic flexion and extension MRI is the best diagnostic tool for the diagnosis of HD and it is also useful in the differential diagnosis between HD, incurable, and progressive MNDs, such as distal SMA, ALS, PLS, post‐polio syndrome, and curable conditions, such as cervical myelopathy, syringomyelia, multifocal motor neuropathy with conduction block, and neuralgic amyotrophy. 36 At difference with incurable MNDs and degenerative diseases that are characterized by a poor prognosis, the early diagnosis of HD allows to prompt treatment, thus favoring a good recovery. Conservative treatment is the most common choice providing neck immobilization with collars, physiotherapy, and absolute avoidance of neck flexion positions, whereas surgical treatment is recommended in progressive patients who do not respond to conservative treatment. 37 Hence, HD should be considered as a part of wider spectrum of benign and nonprogressive MNDs ranging from MMA to juvenile asymmetric segmental spinal muscular atrophy (JASSMA), DBMA, PBMA, and PDMA. 14 According to this viewpoint, our case would resemble a JASSMA in which the left upper limb is more affected than the right. Finally, given the heterogeneity of the MMA spectrum disorder, the cervical spinal cord MRI plays a key role to identify the dynamic cervical cord compression leading to the diagnosis of HD.
ACKNOWLEDGEMENTS AND DISCLOSURE
None. The authors declare that there is no conflict of interest.
Open Access Funding provided by Universita degli Studi di Palermo within the CRUI‐CARE Agreement. [Correction added on 10 May 2022, after first online publication: CRUI‐CARE funding statement has been added.]
Iacono S, Di Stefano V, Gagliardo A, et al. Hirayama disease: Nosological classification and neuroimaging clues for diagnosis. J Neuroimaging. 2022;32:596–603. 10.1111/jon.12995
Funding information
None.
REFERENCES
- 1. Hirayama K, Tsubaki T, Toyokura Y, Okinaka S. Juvenile muscular atrophy of unilateral upper extremity. Neurology 1963;13:373‐80. [DOI] [PubMed] [Google Scholar]
- 2. Hirayama K, Tomonaga M, Kitano K, Yamada T, Kojima S, Arai K. Focal cervical poliopathy causing juvenile muscular atrophy of distal upper extremity: a pathological study. J Neurol Neurosurg Psychiatry 1987;50:285‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang YL, Chen CJ. Hirayama disease. Neuroimaging Clin N Am 2011;21:939‐50. [DOI] [PubMed] [Google Scholar]
- 4. Hirayama K. Juvenile muscular atrophy of distal upper extremity (Hirayama disease). Intern Med 2000;39:283‐90. [DOI] [PubMed] [Google Scholar]
- 5. Yoo SD, Kim HS, Yun DH, et al. Monomelic amyotrophy (Hirayama disease) with upper motor neuron signs: a case report. Ann Rehabil Med 2015;39:122‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gourie‐Devi M, Nalini A. Long‐term follow‐up of 44 patients with brachial monomelic amyotrophy. Acta Neurol Scand 2003;107:215‐20. [DOI] [PubMed] [Google Scholar]
- 7. Pradhan S. Bilaterally symmetric form of Hirayama disease. Neurology 2009;72:2083‐9. [DOI] [PubMed] [Google Scholar]
- 8. Yilmaz Ö, Alemdaroǧlu I, Karaduman A, Haliloǧlu G, Topaloǧlu H. Benign monomelic amyotrophy in a 7‐year‐old girl with proximal upper limb involvement: case report. Turk J Pediatr 2011;53:471‐6. [PubMed] [Google Scholar]
- 9. Preethish‐Kumar V, Nalini A, Singh R, et al. Distal bimelic amyotrophy (DBMA): phenotypically distinct but identical on cervical spine MR imaging with brachial monomelic amyotrophy/Hirayama disease. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:338‐44. [DOI] [PubMed] [Google Scholar]
- 10. Preethish‐Kumar V, Polavarapu K, Singh R, et al. Proximal and proximo‐distal bimelic amyotrophy: evidence of cervical flexion induced myelopathy. Amyotroph Lateral Scler Frontotemporal Degener 2016;17:499‐507. [DOI] [PubMed] [Google Scholar]
- 11. Tashiro K, Kikuchi S, Itoyama Y, et al. Nationwide survey of juvenile muscular atrophy of distal upper extremity (Hirayama disease) in Japan. Amyotroph Lateral Scler 2006;7:38‐45. [DOI] [PubMed] [Google Scholar]
- 12. Lyu F, Zheng C, Wang H, et al. Establishment of a clinician‐led guideline on the diagnosis and treatment of Hirayama disease using a modified Delphi technique. Clin Neurophysiol 2020;131:1311‐9. [DOI] [PubMed] [Google Scholar]
- 13. Ammendola A, Gallo A, Iannaccone T, Tedeschi G. Hirayama disease: three cases assessed by F wave, somatosensory and motor evoked potentials and magnetic resonance imaging not supporting flexion myelopathy. Neurol Sci 2008;29:303. [DOI] [PubMed] [Google Scholar]
- 14. Restuccia D, Rubino M, Valeriani M, Mirabella M, Sabatelli M, Tonali P. Cervical cord dysfunction during neck flexion in Hirayama's disease. Neurology 2003;60:1980‐3. [DOI] [PubMed] [Google Scholar]
- 15. Bembenek JP, Kłysz B, Kurkowska‐Jastrzȩbska I. Transcranial magnetic stimulation‐induced motor evoked potentials in Hirayama disease: systematic review of the literature. J Clin Neurophysiol 2020;37:181‐90. [DOI] [PubMed] [Google Scholar]
- 16. Zheng C, Zhu D, Lu F, et al. A double determination of central motor conduction time in the assessment of Hirayama disease. Clin Neurophysiol 2017;128:2369‐74. [DOI] [PubMed] [Google Scholar]
- 17. Hirayama K, Tokumaru Y. Cervical dural sac and spinal cord in juvenile muscular atrophy of distal upper extremity. Neurology 2000;54:1922‐6. [DOI] [PubMed] [Google Scholar]
- 18. Shinomiya K, Dawson J, Spengler DM, Konrad P, Blumenkopf B. An analysis of the posterior epidural ligament role on the cervical spinal cord. Spine (Phila Pa 1976) 1996;21:2081‐8. [DOI] [PubMed] [Google Scholar]
- 19. Kikuchi S, Tashiro K, Kitagawa M, Iwasaki Y, Abe H. A mechanism of juvenile muscular atrophy localized in the hand and forearm (Hirayama's disease)–flexion myelopathy with tight dural canal in flexion. Clin Neurol 1987;27:412‐9. [PubMed] [Google Scholar]
- 20. Toma S, Shiozawa Z. Amyotrophic cervical myelopathy in adolescence. J Neurol Neurosurg Psychiatry 1995;58:56‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khadilkar S, Patel B, Bhutada A, Chaudhari C. Do longer necks predispose to Hirayama disease? A comparison with mimics and controls. J Neurol Sci 2015;359:213‐6. [DOI] [PubMed] [Google Scholar]
- 22. Hou C, Han H, Yang X, et al. How does the neck flexion affect the cervical MRI features of Hirayama disease? Neurol Sci 2012;33:1101‐5. [DOI] [PubMed] [Google Scholar]
- 23. Chen CJ, Hsu HL, Tseng YC, et al. Hirayama flexion myelopathy: neutral‐position MR imaging findings–importance of loss of attachment. Radiology 2004;231:39‐44. [DOI] [PubMed] [Google Scholar]
- 24. Raval M, Kumari R, Dung Dung A, Guglani B, Gupta N, Gupta R. MRI findings in Hirayama disease. Indian J Radiol Imaging 2010;20:245‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hassan KM, Sahni H, Jha A. Clinical and radiological profile of Hirayama disease: a flexion myelopathy due to tight cervical dural canal amenable to collar therapy. Ann Indian Acad Neurol 2012;15:106‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vitale V, Caranci F, Pisciotta C, et al. Hirayama's disease: an Italian single center experience and review of the literature. Quant Imaging Med Surg 2016;6:364‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boruah DK, Prakash A, Gogoi BB, Yadav RR, Dhingani DD, Sarma B. The importance of flexion MRI in Hirayama disease with special reference to laminodural space measurements. AJNR Am J Neuroradiol 2018;39:974‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao MF, Chang HS, Chang KH, et al. Correlations of clinical, neuroimaging, and electrophysiological features in Hirayama disease. Medicine (Baltimore) 2016;95:e4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zou F, Yang S, Lu F, Ma X, Xia X, Jiang J. Factors affecting the surgical outcomes of Hirayama disease: a retrospective analysis of preoperative magnetic resonance imaging features of the cervical spine. World Neurosurg 2019;122:e296‐301. [DOI] [PubMed] [Google Scholar]
- 30. Gotkine M, Abraham A, Drory VE, Argov Z, Gomori JM, Blumen SC. Dynamic MRI testing of the cervical spine has prognostic significance in patients with progressive upper‐limb distal weakness and atrophy. J Neurol Sci 2014;345:168‐71. [DOI] [PubMed] [Google Scholar]
- 31. Lehman VT, Luetmer PH, Sorenson EJ, et al. Cervical spine MR imaging findings of patients with Hirayama disease in North America: a multisite study. AJNR Am J Neuroradiol 2013;34:451‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dejobert M, Geffray A, Delpierre C, Chassande B, Larrieu E, Magni C. Hirayama disease: three cases. Diagn Interv Imaging 2013;94:319‐23. [DOI] [PubMed] [Google Scholar]
- 33. Lai V, Wong YC, Poon WL, Yuen MK, Fu YP, Wong OW. Forward shifting of posterior dural sac during flexion cervical magnetic resonance imaging in Hirayama disease: an initial study on normal subjects compared to patients with Hirayama disease. Eur J Radiol 2011;80:724‐8. [DOI] [PubMed] [Google Scholar]
- 34. Shao M, Yin J, Lu F, Zheng C, Wang H, Jiang J. The quantitative assessment of imaging features for the study of Hirayama disease progression. Biomed Res Int 2015;2015;803148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prior DE, Ghosh PS. Neuroimage: the crescent moon sign of Hirayama disease. Acta Neurol Belg 2021. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 36. Rotondo E, Pellegrino N, Di Battista C, Graziosi A, Di Stefano V, Striano P. Clinico‐diagnostic features of neuralgic amyotrophy in childhood. Neurol Sci 2020;41:1735‐40. [DOI] [PubMed] [Google Scholar]
- 37. Hassan KM, Sahni H. Nosology of juvenile muscular atrophy of distal upper extremity: from monomelic amyotrophy to Hirayama disease — Indian perspective. Biomed Res Int 2013;2013:478516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hashimoto O, Asada M, Ohta M, Kuroiwa Y. Clinical observations of juvenile nonprogressive muscular atrophy localized in hand and forearm. J Neurol 1976;211:105‐10. [DOI] [PubMed] [Google Scholar]
- 39. Gourie‐Devi M, Suresh TG, Shankar SK. Monomelic amyotrophy. Arch Neurol 1984;41:388‐94. [DOI] [PubMed] [Google Scholar]
- 40. Biondi A, Dormont D, Weitzner I, Bouche P, Chaine P, Bories J. MR imaging of the cervical cord in juvenile amyotrophy of distal upper extremity. Am J Neuroradiol 1989;10:263‐8. [PMC free article] [PubMed] [Google Scholar]
- 41. Pradhan S, Gupta RK. Magnetic resonance imaging in juvenile asymmetric segmental spinal muscular atrophy. J Neurol Sci 1997;146:133‐8. [DOI] [PubMed] [Google Scholar]