

Abstract
Objective
The autoimmune response in rheumatoid arthritis (RA) is marked by the presence of anti–citrullinated protein antibodies (ACPAs). A notable feature of IgG ACPA is the abundant expression of N‐linked glycans in the variable domain. However, the presence of ACPA variable domain glycosylation (VDG) across disease stages, and its response to therapy, are poorly described. To understand its dynamics, we investigated the abundance of IgG ACPA VDG in 1,498 samples from individuals in different clinical stages.
Methods
Using liquid chromatography, we analyzed IgG ACPA VDG profiles in 7 different cohorts from Japan, Canada, The Netherlands, and Sweden. We assessed 106 healthy individuals, 228 individuals with presymptomatic RA, 277 individuals with arthralgia, 307 patients with new‐onset/early RA, and 117 RA patients after prespecified treatment regimens. Additionally, we measured VDG in 234 samples from patients with RA who did or did not achieve long‐term drug‐free remission (DFR) during up to 16 years follow‐up.
Results
IgG ACPA VDG significantly increased (P < 0.0001) toward disease onset and was associated with ACPA levels and epitope spreading prior to diagnosis. A slight increase in VDG was observed in patients with established RA, with a moderate influence of treatment (P = 0.007). In patients in whom DFR was later achieved, IgG ACPA VDG was already reduced at the time of RA onset.
Conclusion
The abundance of IgG ACPA VDG increases toward RA onset and correlates with maturation of the ACPA response. While IgG ACPA VDG levels are fairly stable in established disease, a lower degree of VDG at RA onset correlates with DFR. Although the underlying biologic mechanisms remain elusive, our data support the concept that VDG relates to an expansion of the ACPA response in the pre‐disease phase and contributes to disease development.
INTRODUCTION
Rheumatoid arthritis (RA) is a prevalent, slowly evolving autoimmune disease in which arthralgia is an important pre‐disease manifestation. The autoimmune response that is the most specific for RA is characterized by the presence of anti–citrullinated protein antibodies (ACPAs), which can be present several years before the onset of clinical symptoms. ACPA‐positive patients have a more severe disease course and are less likely to achieve drug‐free remission (DFR) as compared to seronegative patients (1). ACPA responses are known to be dynamic during the transition toward RA, as an increase in ACPA levels combined with a broader epitope recognition profile is associated with the development of clinical symptoms (2). Autoantibody levels are, however, not associated with long‐term treatment response and do not predict DFR (3).
Glycomic analysis has revealed that IgG ACPAs are abundantly glycosylated in their antigen‐binding fragments, expressing complex‐type variable domain glycans that are mainly disialylated and bisected (4). Variable domain glycosylation (VDG) on >90% of the autoantibodies is a notable characteristic of IgG ACPA and distinguishes the molecules from conventional IgG antibodies, which display, next to the conserved presence of glycans in the Fc region, a considerably lower VDG of ~12–14% (4, 5). Glycosylation sites required for the attachment of variable domain glycans are introduced by somatic hypermutation (6).
Although the role and dynamics of IgG ACPA Fc glycans have been studied extensively (7, 8, 9, 10), little is known about the expression levels or potential biologic implications of variable domain glycans on ACPA. As carbohydrates might encode important biologic information and possibly affect cellular functions, it is important to understand VDG dynamics over time in relation to the disease course of RA. Previously, we showed that IgG ACPA VDG can occur several years before RA onset. In a Canadian population, IgG ACPA VDG was predictive of disease development (11, 12). However, how IgG ACPA VDG changes between clinical disease states from healthy, symptom‐free individuals to individuals with arthralgia to patients at RA onset and with established RA has not been elucidated. Additionally, it is unclear whether VDG levels are associated with treatment outcomes, predict DFR and disease flares, or can be modified by treatment.
To understand the characteristics and action of variable domain glycans and thereby their possible contribution to autoreactive B cell responses in RA, we cross‐sectionally investigated the presence and abundance of IgG ACPA VDG in 1,498 samples from an ethnically diverse group of individuals in various stages of disease (Table 1). By analyzing samples from a well‐controlled treatment strategy trial (the Improved [Induction Therapy with Methotrexate and Prednisone in Rheumatoid or Very Early Arthritic Disease] study) that aimed to assess the most effective strategy for inducing remission in early RA (13), we investigated longitudinal changes in VDG in established RA after treatment escalation or treatment tapering. Finally, we longitudinally analyzed IgG ACPA VDG changes in patients from the Leiden Early Arthritis Clinic (EAC) in whom sustained (>1 year) DFR (SDFR) was achieved and those who experienced late disease flares, with an extensive follow‐up of up to 16 years (14).
Table 1.
Characteristics of the study cohorts*
| Cohort (location), diagnosis | Female | Age, mean ± SD years | Arthritis/RA at follow‐up | VDG positive | VDG, median (IQR) % | ACPA positive | ACPA level, median (IQR) AU/ml† | ACPA assay |
|---|---|---|---|---|---|---|---|---|
| Cohort 1 (Nagasaki, Japan), healthy, symptom free (n = 58) | 38 (66) | 67 ± 9.9 | 9 (15.5) | 48 (83.8) | 58.1 (35.6) | 58 (100) | 35.8 | CLEIA (STACIA MEBLux CCP test; MBL) (15) |
| Cohort 2 (Manitoba, Canada) | ||||||||
| Healthy (n = 48) | 32 (66.7) | 37.6 ± 13.5 | 31 (64.6) | 42 (33.3) | 44.9 (69.3) | 37 (77.1) | 54 (135.5) | CCP‐2 kit (Inova Diagnostics) (16) |
| RA onset (n = 25) | 19 (76) | 42 ± 14.7 | – | 21 | 99.9 (46.1) | 22 | 200 (103.3) | CCP‐2 kit (Inova Diagnostics) (16) |
| Cohort 3 (Umeå, Sweden) | ||||||||
| Presymptomatic (n = 228) | 145 (64) | 52.2 ± 9.4 | 228 (100) | 105 (46.1) | 97.4 (53.5) | 168 (73.7) | 126.7 (455.5) | Immunoscan RA anti–CCP‐2 EIA (Euro Diagnostica) (18) |
| 0.5–1.5 years after RA onset (n = 126) | 78 (61) | 59.7 ± 9.3 | – | 116 (92.1) | 94.2 (50.8) | 125 (98.4) | 592.9 (725.3) | Immunoscan RA anti–CCP‐2 EIA (Euro Diagnostica) (18) |
| Cohort 4 (Amsterdam, Reade, The Netherlands), arthralgia (n = 239) | 185 (77) | 48.3 ± 11.6 | 137 (57.3) | 211 (87.9) | 75.3 (49) | 239 (100) | 358 (1,351) | Anti‐CCP ELISA (Axis‐Shield) (19) |
| Cohort 5 (Leiden, The Netherlands; CSA Study) | ||||||||
| Arthralgia (n = 38) | 29 (76.3) | 48.3 ± 12.5 | 26 (68.4) | 27 (71.1) | 70.4 (28.8) | 33 (86.8) | 123 (324) | Anti–CCP‐2 ELISA (Euro Diagnostica) (23) |
| RA onset (n = 26) | 22 (84.6) | 48.1 ± 12.5 | – | 18 (69.2) | 59.1 (49.1) | 21 (80.8) | 25.5 (266.8) | Anti–CCP‐2 ELISA (Euro Diagnostica) (23) |
| Cohort 6 (Leiden, The Netherlands; Improved study) | ||||||||
| RA onset (n = 130) | 88 (67.7) | 51.1 ± 12.5 | – | 117 (90) | 96 (48.2) | 130 (100) | 903.3 (1,101.2) | In‐house anti–CCP‐2 ELISA (3, 22) |
| 4 months after RA onset (n = 117) | 79 (67.5) | 50.6 ± 12.8 | – | 78 (66.7) | 95.9 (45.1) | 117 (100) | 449.9 (806.9) | In‐house anti–CCP‐2 ELISA (3, 22) |
| 8 months after RA onset (n = 112) | 78 (69.6) | 50.7 ± 12.4 | – | 86 (76.8) | 101.7 (50.3) | 112 (100) | 602 (1,061.8) | In‐house anti–CCP‐2 ELISA (3, 22) |
| 12 months after RA onset (n = 117) | 78 (66.7) | 51.0 ± 12.4 | – | 98 (83.8) | 105.2 (48.1) | 117 (100) | 651.7 (962.5) | In‐house anti–CCP‐2 ELISA (3, 22) |
| Cohort 7 (Leiden, The Netherlands; EAC) | ||||||||
| RA onset, DFR not achieved (n = 59) | 42 (71.2) | 49.7 ± 14.5 | – | 59 (100) | 83.8 (46) | 59 (100) | 7,340 (5,984) | In‐house anti–CCP‐2 ELISA (3, 22) |
| RA onset, DFR achieved (n = 36) | 24 (66.7) | 50.8 ± 13.1 | – | 32 (89) | 61.4 (35) | 29 (80.6) | 1,933 (7,296) | In‐house anti–CCP‐2 ELISA (3, 22) |
| Pre‐remission (n = 52) | 37 (71.2) | 54.2 ± 14.8 | – | 37 (71) | 74.05 (30) | 38 (73) | 3,583 (5,302) | In‐house anti–CCP‐2 ELISA (3, 22) |
| DFR (n = 41) | 27 (65.9) | 58.5 ± 13.6 | – | 30 (73.2) | 67.7 (41.5) | 33 (80) | 3,010 (8,975) | In‐house anti–CCP‐2 ELISA (3, 22) |
| SDFR (n = 35) | 27 (77.1) | 54.6 ± 15.1 | – | 22. (62.9) | 73.5 (42) | 29 (82.9) | 2,626 (6,765) | In‐house anti–CCP‐2 ELISA (3, 22) |
| DFR with late flares (n = 11) | 7 (63.6) | 69.4 ± 14.8 | – | 9 (81.8) | 78.3 (26.9) | 10 (91) | 4,210 (10,709) | In‐house anti–CCP‐2 ELISA (3, 22) |
Except where indicated otherwise, values are the number (%). RA = rheumatoid arthritis; VDG = variable domain glycosylation; IQR = interquartile range; CCP = cyclic citrullinated peptide; EIA = enzyme immunoassay; ELISA = enzyme‐linked immunosorbent assay; CSA = Clinically Suspect Arthralgia; EAC = Early Arthritis Cohort; DFR = drug‐free remission; SDFR = sustained DFR.
†Anti–citrullinated protein antibody (ACPA) levels were determined with various assays/standards and thus are not directly comparable with one another.
PATIENTS AND METHODS
Study cohorts
IgG ACPA VDG was analyzed in 1,498 serum samples from individuals in different clinical disease stages including 121 ACPA‐negative RA control samples. Descriptive data on the cohorts are presented in Table 1. Additionally, 247 healthy donor and 150 ACPA‐positive RA control samples, obtained at the Leiden University Medical Center outpatient rheumatology clinic, were assessed to verify the methodology used. Details on patient and public involvement and ethical considerations in the recruitment of individuals and of the study cohorts are available in Supplementary Methods, on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42098.
Cohort 1, healthy, symptom free (Nagasaki, Japan)
Cohort 1 consisted of healthy symptom‐free individuals (n = 58) enrolled in the Nagasaki Island Study (a community‐based prospective cohort study based on resident health examinations) (15) who tested positive for ACPA. The individuals included in the study cohort had no joint symptoms at the time of the most recent resident health examination. These individuals were followed up for a period of up to 3 years. Nine of them (15.5%) developed RA during follow‐up.
Cohort 2, healthy and RA onset (Manitoba, Canada)
Members of cohort 2 were part of the longitudinal research project Early Identification of Rheumatoid Arthritis in First Nations, based at the Arthritis Centre, the University of Manitoba (16). Forty‐eight samples from healthy individuals (first‐degree relatives of patients with RA) were included, as were paired samples obtained from 25 individuals prior to RA onset and in the absence of joint‐ symptoms, and at the time of diagnosis of clinically apparent RA.
Cohort 3, presymptomatic and after RA onset/early RA (Umeå, Sweden)
Cohort 3 comprised 354 samples from the Northern Sweden Health and Disease Study or the Västerbotten Intervention Project. Blood samples were collected and stored in a biorepository (the Northern Sweden Medical Research Biobank). Individuals were considered to have RA if they fulfilled the 1987 American College of Rheumatology classification criteria (17). Two hundred twenty‐eight individuals from the cohort were retrospectively identified as having donated blood before the onset of signs or symptoms of joint disease (defined as presymptomatic), with a median time from blood sampling to onset of signs/symptoms of 4.7 years (interquartile range [IQR] 5.9 years). For 126 individuals (defined as having early RA), blood was obtained 0.5–1.5 years after RA diagnosis (18).
Cohort 4, arthralgia (Amsterdam, Reade, The Netherlands)
Patients in cohort 4 were ACPA‐positive individuals with arthralgia (n = 239) who were seen at rheumatology outpatient clinics in the Amsterdam area (19). These individuals were followed up for a period of up to 10 years, during which 137 (57.3%) developed arthritis.
Cohort 5, arthralgia and RA onset (Leiden, The Netherlands)
Individuals at risk of RA development were recruited for the prospective Clinically Suspect Arthralgia (CSA) cohort at the Leiden University Medical Center outpatient rheumatology clinic and followed up longitudinally (20). Thirty‐eight patients with arthralgia from the CSA were included as cohort 5 in the present study. Paired samples from 26 of these patients were studied, i.e., a sample obtained at the time of arthralgia pre‐RA diagnosis and a sample obtained at the time of diagnosis of clinically apparent RA.
Cohort 6, RA onset and established RA after treatment (Leiden, The Netherlands)
Cohort 6 consisted of patients with RA who were recruited at 12 hospitals in the western area of The Netherlands and were included in the Improved study. The aim of this multicenter randomized controlled trial was to assess the achievement of DFR with treatment alteration every 4 months. Initial treatment was methotrexate (MTX) and prednisone. Prednisone was tapered in patients whose RA was in early remission (defined as a Disease Activity Score [DAS] of <1.6) (21) at 4 months. If disease was still in remission at 8 months, MTX was also tapered. If the DAS was ≥1.6 after prednisone was stopped, it was restarted. Patients in whom early remission was not achieved were randomized to 1 of 2 treatment arms: MTX, prednisone, hydroxychloroquine, and sulfasalazine combination (arm 1) or MTX and adalimumab combination (arm 2) (13). Serum samples obtained at the time of RA onset (n = 130) and at 4 months (n = 117), 8 months (n = 112), and 12 months (n = 117) after diagnosis and prespecified treatment were assessed in the present study.
Cohort 7, RA onset, DFR, SDFR, and late disease flares (Leiden, The Netherlands)
Members of cohort 7 were patients from the Leiden EAC (14). Samples obtained at the time of RA onset from individuals in whom DFR was not later achieved (n = 59) and individuals in whom DFR was later achieved (n = 36) were assessed. Patients in whom DFR was later achieved were followed up longitudinally for up to 16 years; samples obtained at RA onset (n = 36), during the pre‐remission phase (n = 52), DFR (n = 41), SDFR (n = 35), and at the time of late disease flares (n = 11) were included. DFR was defined as the absence of clinical synovitis (swollen joints at physical examination) after discontinuation of disease‐modifying antirheumatic drug (DMARD) treatment (including systemic/intraarticular glucocorticoids). In the 41 patients in whom DFR was achieved, DMARD treatment was stopped after a median of 2.9 years (IQR 1.0–4.9 years). SDFR was defined as the absence of clinical synovitis after cessation of DMARD treatment, that persisted for ≥1 year. SDFR was achieved in the 35 patients after a median of 2.8 years of DMARD treatment (IQR 2.0–5.2 years) and was maintained for a median of 7.1 years (IQR 4.5–11.2 years) after DMARD cessation, demonstrating the sustainability of DMARD‐free remission. Flare was defined as the recurrence of clinical synovitis on joint examination. Among patients in whom SDFR was achieved, data in the medical records were reviewed through September 2021.
Laboratory analyses
IgG ACPA levels in serum samples were analyzed using standard and commercially available anti–cyclic citrullinated peptide (anti‐CCP) assays or in‐house anti‐CCP2 enzyme‐linked immunosorbent assays (ELISAs) as previously described (3, 15, 16, 17, 19, 22, 23). ACPA fine specificity of samples from cohort 6 was determined using in‐house anti–citrullinated vimentin 59–74, anti–citrullinated fibrinogen β36–52 and α27–43, and anti–citrullinated enolase 5–20 ELISAs as previously described (3). ACPA fine specificity of samples from cohort 4 was determined using in‐house IgG anti–citrullinated vimentin 60–75, anti–citrullinated fibrinogen β36–52, α60–74, and α36–50, and anti–citrullinated enolase 5–21 ELISAs.
IgG ACPA capture and VDG analysis using liquid chromatography
Capture of IgG ACPA, total glycan release, and glycan labeling and purification were performed as previously described (11). Briefly, ACPAs were affinity isolated from 25‐μl serum samples using NeutrAvidin Plus resin (Thermo Scientific) coupled with 0.1 μg/μl CCP‐2–biotin followed by IgG capture using FcXL affinity beads (Thermo Scientific). N‐linked glycans were released using 0.5 units of PNGaseF (Roche), subsequently labeled with 2‐anthranilic acid and 2‐picoline borane, and hydrophobic interaction liquid chromatography–solid‐phase extraction purified using GHP membrane filter plates (Pall Life Sciences). Ultra high‐performance liquid chromatography was performed on a Dionex Ultimate 3000 instrument (ThermoFisher Scientific), an FLR fluorescence detector set, and an Acquity BEH Glycan column (Waters). Separation and glycan peak alignment were performed as previously described (11). HappyTools, version 0.0.2, was used for calibration and peak integration (24). The N‐linked glycan abundance in each peak was expressed as the total integrated area under the curve. The cutoff was defined based on phosphate buffered saline control (blank) and blood samples from ACPA‐negative healthy control subjects in the Leiden area, excluding outliers (below 1.5× the 25th percentile or above 1.5× the 75th percentile). The percentage IgG ACPA VDG was calculated based on the formula (sum of the most abundant variable domain glycans/sum of the most abundant Fc glycans) × 100, or ([G2FBS1 + G2FS2 + G2FBS2]/[G0F + G1F + G2F]) × 100, where G0/G1/G2 represents A‐/mono‐/di‐galactosylated, F represents core fucosylated, B represents bisecting N‐acetylglucosamine (GlcNAc), and S1/S2 represents mono‐/disialylated (12). The glycan traits were selected based on previous observations showing their exclusive presence on either the variable domain or the Fc domain of IgG ACPA molecules (12, 25). The sum of the Fc glycans (the amount of N‐linked glycans attached to the conserved Asn297 in the Fc domain of IgG antibodies) remains constant.
Statistical analysis
Continuous data were analyzed using nonparametric methods (Kruskal‐Wallis test for unpaired groups and Mann‐Whitney U test for 2 unpaired groups) and parametric tests (mixed‐effects analysis for matched paired samples accounting for missing values) when appropriate. The mixed‐effects analysis model using restricted maximum likelihood is comparable to repeated‐measures analysis of variance with regard to P values and multiple comparisons tests, but can accommodate missing values. Correlations between IgG ACPA levels (log transformed) and percentages of VDG were assessed with Pearson's correlation coefficient. P values (all 2‐sided) less than 0.05 were considered significant. Logistic and ordinal regression analyses were performed in cohort 4 and cohort 6 to investigate the association of IgG ACPA VDG/IgG ACPA levels with epitope spreading, remission, and early DFR. The unstandardized coefficient (B) represents the mean change in the response given a 1‐unit change in the predictor. The longitudinal and repeated‐measures data from cohort 6 were analyzed by generalized estimating equation (GEE) analysis, as previously described (3). GEE analysis was used to assess VDG changes over time and associations with treatment. The specific covariates and dependent variables are listed in the supplementary tables (https://onlinelibrary.wiley.com/doi/10.1002/art.42098). Statistical calculations were performed using Stata, version 16.1.
RESULTS
IgG ACPA variable domain glycosylation increases toward disease onset and remains stable in established RA
To provide a comprehensive overview of the presence and abundance of IgG ACPA VDG (Figures 1A and B), we analyzed 1,377 ACPA‐positive and 121 ACPA‐negative samples from individuals in different clinical disease stages (Figure 1C and Table 1). Comparable to the results of previous studies (11, 12), we found that variable domain glycans were already present in high percentages (median 56.2%) on IgG ACPA from healthy individuals without symptoms (n = 106) (Figure 1D). Cross‐sectional analysis revealed a significant increase in VDG (median 74.7%) in individuals with clinically identified arthralgia (n = 277) compared to healthy individuals (Figure 1D).
Figure 1.
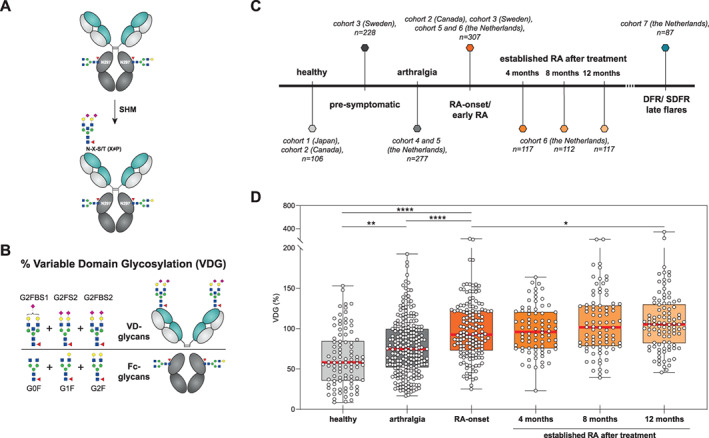
Percentage variable domain glycosylation in IgG anti–citrullinated protein antibodies (ACPAs) from healthy individuals, patients with arthralgia, and patients with rheumatoid arthritis (RA) in different disease stages. A, Depiction of the process by which IgG carries N‐glycans at each N297 residue in the Fc domain and can acquire additional N‐linked glycosylation sites (N‐X‐S/T, X ≠ P) in the variable domain by somatic hypermutation (SHM) (6). B, Depiction of the calculation of IgG ACPA VDG, i.e., (sum of the most abundant variable domain glycans/sum of the most abundant Fc glycans) × 100, or ([G2FBS1 + G2FS2 + G2FBS2]/[G0F + G1F + G2F]) × 100, where G0/G1/G2 represents A‐/mono‐/di‐galactosylated, F represents core fucosylated, B represents bisecting N‐acetylglucosamine (GlcNAc), and S1/S2 represents mono‐/di‐sialylated. The selected glycan traits are exclusively present on either the variable domain or the Fc domain of IgG ACPA. GlcNAc is shown as blue squares, mannose as green circles, galactose as yellow circles, fucose as red triangles, and N‐acetylneuraminic acid as pink diamonds. C, “Timeline” of clinical disease stages, the corresponding analyzed cohorts, and numbers of samples analyzed. D, Percentage IgG ACPA VDG, measured by liquid chromatography, in healthy individuals, patients with arthralgia, patients at the time of RA onset, and patients with established RA 4 months, 8 months, and 12 months after institution of prespecified treatment. Data are presented as box and whisker plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the whiskers represent the minimum to maximum values. Circles represent individual samples. The cross‐sectional data sets from cohorts 1, 2, 4, and 5 were analyzed by Kruskal‐Wallis test with Dunn's post hoc test. The longitudinally obtained data from cohort 6 were analyzed by generalized estimating equation. * = P = 0.037; ** = P = 0.0032; **** = P < 0.0001. DFR = drug‐free remission; SDRF = sustained drug‐free remission.
A further significant increase in VDG of 18% was observed in samples obtained at the time of RA onset (n = 181) (median VDG 92.6% in the combined data sets) (Figure 1D and Table 1). This was, however, not apparent in all individual sample sets, as changes in VDG between the arthralgia phase and RA onset could not be observed in the statistically underpowered longitudinal data set from cohort 5, presumably because the individuals with clinically suspected arthralgia were tested only shortly before the onset of arthralgia (Supplementary Figure 1, on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42098), and an increase in VDG could have occurred earlier. Patients presenting with arthralgia, regardless of whether they did or did not subsequently develop RA, displayed lower VDG than patients tested at the time of RA onset (Supplementary Figure 2E, https://onlinelibrary.wiley.com/doi/10.1002/art.42098). In samples from patients with established RA after prespecified treatment (n = 346), IgG ACPA VDG remained stable, with only a moderate increase after 12 months, to a median of 105.2% (Figure 1D). As previously shown (11), an increase in IgG ACPA VDG toward the time of RA onset was also observed in a Swedish population of ACPA‐positive individuals who later developed RA. The extended data set used here also exhibited a rise in VDG when analyzed per individual in a longitudinal manner (26) (Supplementary Figure 2D); however, there was no significant difference on cross‐sectional analysis (Supplementary Figure 2C).
Overall, the results showed that the presence of variable domain glycans on IgG ACPA was lower in healthy individuals and increased toward RA development. However, in established disease, no further progression of IgG ACPA VDG was observed in this cross‐sectional analysis.
Interconnection between the increase in variable domain glycosylation and maturation of the ACPA immune response
To obtain further insights into IgG ACPA VDG, we investigated the possible association between VDG percentages and the “maturation” of the ACPA response by analyzing IgG ACPA levels and the broadness of the citrullinated epitope recognition profile. Pearson's correlation analysis revealed a strong, highly significant correlation between VDG percentages and IgG ACPA levels among healthy individuals (r = 0.672 and r = 0.728 in cohorts 1 and 2, respectively) and among individuals with arthralgia (r = 0.640) (Figure 2A and Supplementary Figure 3, https://onlinelibrary.wiley.com/doi/10.1002/art.42098). At RA onset and in established RA after prespecified treatment, however, we observed only moderate correlations (r = 0.214, 0.341, 0.362, and 0.215 at RA onset and after 4, 8, and 12 months of treatment, respectively) (Figure 2A and Supplementary Figure 2E, https://onlinelibrary.wiley.com/doi/10.1002/art.42098). Likewise, our data revealed that IgG ACPA with increased VDG showed a significantly broader recognition profile toward multiple citrullinated epitopes (Figures 2B and C). Ordinal regression analyses confirmed these findings in individuals with arthralgia (P < 0.001) (Supplementary Table 1, https://onlinelibrary.wiley.com/doi/10.1002/art.42098) as well as in patients at the time of RA onset (P = 0.004) and over time after treatment (P < 0.001) (Supplementary Table 2, https://onlinelibrary.wiley.com/doi/10.1002/art.42098). Thus, IgG ACPA VDG is associated with IgG ACPA levels and the breadth of the epitope recognition profile, suggesting that these two features of the ACPA response are interconnected.
Figure 2.
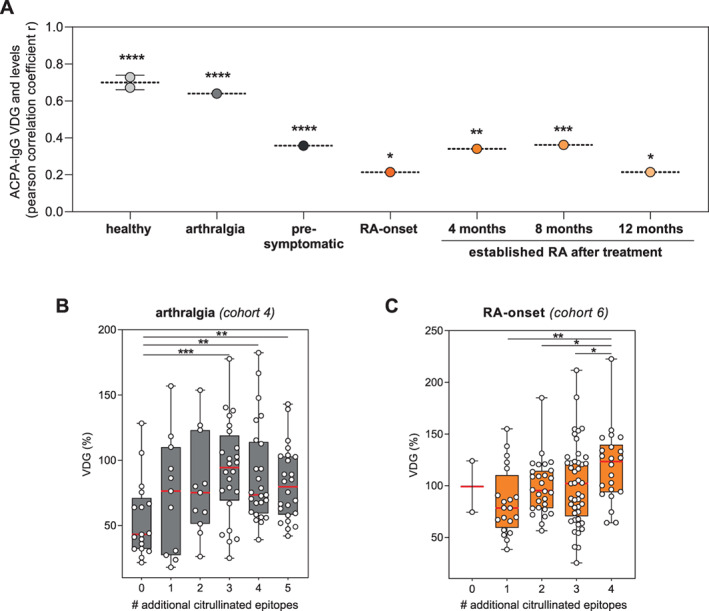
Correlation of IgG ACPA VDG with IgG ACPA levels and epitope spreading (maturation of the ACPA response). A, Pearson's correlation coefficients for the correlation between IgG ACPA VDG and IgG ACPA levels across different disease stages. * = P < 0.05; ** = P < 0.005; *** = P < 0.001; **** = P < 0.0001. For healthy individuals, values from 2 different cohorts are shown. B, VDG percentage on IgG ACPA from individuals with arthralgia, isolated using cyclic citrullinated peptide 2 and tested for binding to 5 additional citrullinated antigens (citrullinated vimentin 60–75, citrullinated fibrinogen β36–52, α60–74, and α36–50, and citrullinated enolase 5–21). ** = P < 0.01; *** = P = 0.0006. C, VDG percentage in IgG ACPA from patients at the time of RA onset, isolated using cyclic citrullinated peptide 2 and tested for recognition of 4 additional citrullinated antigens (citrullinated vimentin 59–74, citrullinated fibrinogen β36–52 and α27–43, and citrullinated enolase 5–20). * = P < 0.05; ** = P = 0.001. In B and C, data are presented as box and whisker plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the whiskers represent the minimum to maximum values. Circles represent individual samples. See Figure 1 for definitions.
Impact of immunosuppression on IgG ACPA variable domain glycosylation
We took advantage of the design of the Improved study (Figure 3A) to investigate whether IgG ACPA VDG predicts early remission in RA or is associated with the intensity of immunosuppression. First, we used the longitudinal data set to identify changes in IgG ACPA VDG over time by analyzing paired samples from patients at RA onset (n = 130) versus at 4 months (n = 117), 8 months (n = 112), and 12 months (n = 117) after disease development. Variable domain glycans appeared to be steadily and abundantly expressed on IgG ACPA after the onset of RA, although minor changes in expression levels were observed over time.
Figure 3.
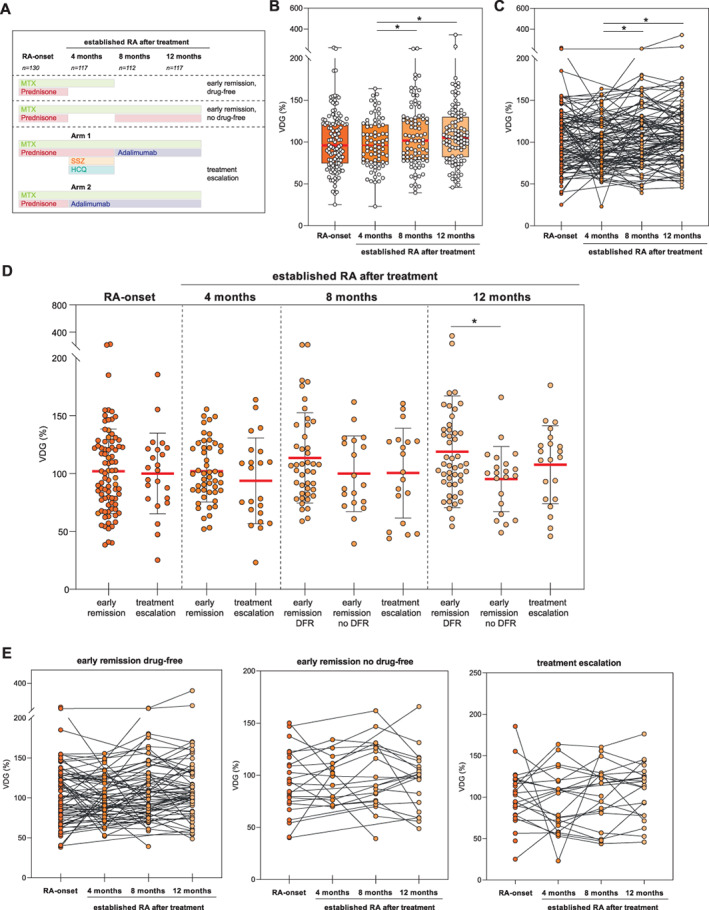
Longitudinal analysis of IgG ACPA VDG at the time of RA onset and in established RA after treatment (cohort 6). A, Treatment protocol. B, Longitudinal data on VDG percentage on IgG ACPA by time since RA onset. Data are presented as box and whisker plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the whiskers represent the minimum to maximum values. Circles represent individual samples. Data were analyzed by mixed‐effects analysis using restricted maximum likelihood and Tukey's test. C, Longitudinal data by time since RA onset, represented as matched pairs. D, VDG percentage in IgG ACPA by treatment group (early remission [drug‐free], early remission [no drug‐free], and treatment escalation) at each assessed time point since RA onset. Data were analyzed by one‐way analysis of variance with Fisher's least significant difference test. Horizontal and vertical bars show the mean ± SD. Circles represent individual samples. E, Longitudinal data by time since RA onset within each treatment group, represented as matched pairs. * = P < 0.05. MTX = methotrexate; SSZ = sulfasalazine; HCQ = hydroxychloroquine (see Figure 1 for other definitions).
A slight but nonsignificant decrease was observed 4 months after disease onset and initiation of treatment with MTX and prednisone (Figures 3B and C and Supplementary Table 3, https://onlinelibrary.wiley.com/doi/10.1002/art.42098). Previous studies have shown a similar decline of IgG ACPA levels after initiation of treatment (3), providing further evidence of a correlation between VDG and IgG ACPA levels. After 4 months, prednisone was tapered such that patients were then treated with MTX only, if early remission (DAS <1.6) had been achieved. If early remission was not achieved, patients were randomized to 1 of 2 treatment escalation arms, i.e., combination treatment with MTX, prednisone, hydroxychloroquine, and sulfasalazine or combination treatment with MTX and adalimumab (13) (Figure 3A). At 8 months, individuals in the early remission group either continued MTX treatment combined with prednisone (no drug‐free group) or, if disease remission persisted, their medication was tapered (drug‐free group). Individuals in the treatment escalation group (arms 1 and 2) continued MTX treatment, in combination with adalimumab. Overall, irrespective of the treatment arm, VDG had increased moderately but significantly at 12 months after RA onset (P = 0.037) (Supplementary Table 3).
When the different treatment groups were compared, marginal but statistically significant effects of immunosuppression on IgG ACPA VDG were observed, with a reduction in VDG 12 months after RA onset (Figures 3D and E and Supplementary Figure 4A, https://onlinelibrary.wiley.com/doi/10.1002/art.42098), though not at 4 months or 8 months. This moderate but significant negative effect of immunosuppression on VDG was confirmed by GEE analysis of changes over time (8 months versus 12 months) (regression coefficient [B] 12.27 [95% confidence interval −7.32, 31.87] in the early remission, drug‐free group versus 6.42 [95% confidence interval −0.35, 13.10] in the early remission, no drug‐free and treatment escalation group; P = 0.007) (Supplementary Table 4, https://onlinelibrary.wiley.com/doi/10.1002/art.42098) and was similar to previously reported findings with regard to IgG ACPA levels (3). Last, we investigated whether VDG percentage at RA onset predicts remission after 4 months and drug‐free remission within the first year. Similar to IgG ACPA levels (3), VDG percentages did not predict early drug‐free remission (Supplementary Table 5, https://onlinelibrary.wiley.com/doi/10.1002/art.42098). Collectively, these results show that IgG ACPA variable domain glycans are expressed at a persistently high level in established RA and show a slight but statistically significant decrease upon immunosuppression.
Decreased VDG during active disease in patients in whom sustained DFR is later achieved
As a next step, we performed cross‐sectional and longitudinal analyses of IgG ACPA VDG in individuals in whom long‐term DFR was achieved or who experienced DFR with late flares. We made use of the unique EAC database including patients who were followed up for a period of up to 16 years after disease onset. Using this database, we were able to identify 41 individuals in whom DFR had been achieved and 35 patients in whom SDFR (>1 year) had been achieved. Longitudinal samples obtained from the same patient at RA onset (n = 36), during active disease (pre‐remission) (n = 52), during DFR (n = 41), during SDFR (n = 35), and when experiencing late disease flares (n = 11) were assessed. Again, the data showed that variable domain glycans are stably expressed in established RA. Intriguingly, however, patients in whom DFR was achieved during follow‐up (n = 36) showed significantly reduced IgG ACPA VDG at the onset of disease compared to age‐ and sex‐matched patients with persistently high disease activity (DAS >3) (n = 59) (median VDG at disease onset 61.4% versus 83.8%) (Figure 4A and Table 1). In contrast, no statistically significant changes were observed when the IgG ACPA VDG percentages were determined over time in the DFR group or any of the other groups analyzed (Figures 4B–D). Thus, these longitudinal data confirm that IgG ACPAs express a constant amount of variable domain glycans after RA onset. The cross‐sectional data also indicate that among individuals in whom long‐term DFR is achieved, fewer glycans are present on IgG ACPA variable domains at the time of RA onset.
Figure 4.
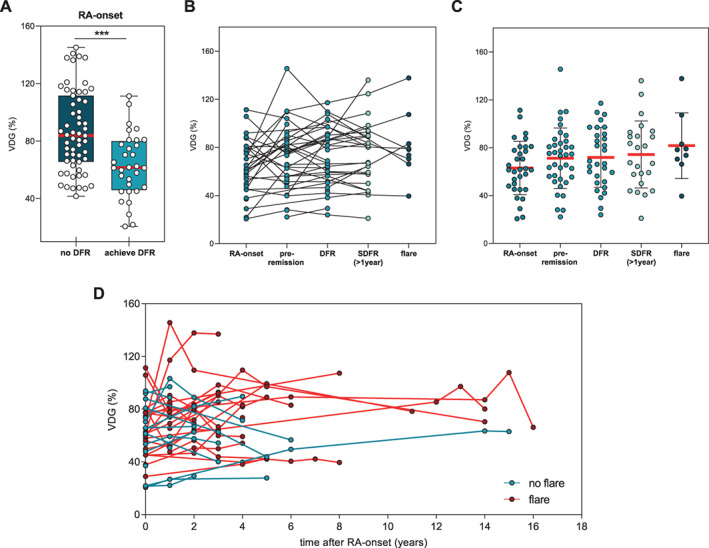
Cross‐sectional and longitudinal analysis of IgG ACPA VDG at the time of RA onset and during DFR (cohort 7). A, VDG percentage on IgG ACPA at the time of RA onset in individuals in whom DFR was not achieved and those in whom DFR was achieved. DFR was defined as the absence of clinical synovitis after discontinuation of disease‐modifying antirheumatic drug treatment. Data are presented as box and whisker plots, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the whiskers represent the minimum to maximum values. *** = P < 0.0001 by Mann‐Whitney U test. B, Data on VDG percentage on IgG ACPA in matched paired samples from patients at the time of RA onset, pre‐remission, during DFR, during SDFR, and during late disease flares. Flare was defined as the recurrence of clinical synovitis on joint examination. C, IgG ACPA VDG data by group (RA onset, pre‐remission, DFR, SDFR, flare). Horizontal and vertical bars show the mean ± SD. Circles represent individual samples. D, IgG ACPA VDG data by assessment time point in longitudinally assessed samples from patients who did and those who did not experience late flares. Circles represent individual samples. See Figure 1 for definitions.
DISCUSSION
A key important characteristic of IgG autoantibodies from patients with RA is the abundant presence of bisected and disialylated glycans in the variable domain. To gain insight into the introduction and occurrence of this unusual antibody feature across different disease stages, we assessed IgG ACPAs in ~1,500 samples from 852 individuals in different clinical disease stages. Moreover, we analyzed the effect of therapy on the degree of VDG on ACPAs. The large sample size increased the power of our study, and we demonstrated that IgG ACPA VDG correlates strongly with the maturation of the ACPA immune response prior to disease onset, while no correlation with age was observed. We found that the abundance of IgG ACPA VDG increased significantly from the time these ACPA‐positive individuals were healthy and symptom‐free (58.1%) toward the pre‐RA phase (arthralgia) (74.7%), with a further increase around the time of disease onset (92.6%). Thus, our data strongly indicate that an increase in IgG ACPA VDG occurs in the asymptomatic phase, with a further increase during progression to arthralgia and ultimately RA diagnosis, although the latter notion requires further detailed research with longitudinal sampling.
In established RA, we noted constant high expression of glycans on the variable domain of IgG ACPA, with a slight, but significant, increase after 12 months (105.2%). This is consistent with our previous observations, estimating >90% VDG on IgG ACPA in RA (4), as well as the finding that >80% of ACPA B cell receptors in RA express N‐linked glycosylation sites in the variable region (27). Our longitudinal data from cohort 6 depict increased VDG levels in individuals in whom treatment was tapered, while patients who received more intensive treatment showed reduced IgG ACPA VDG profiles over time (P = 0.007). This significant impact of immunosuppression was also observed for IgG ACPA levels (22), confirming the correlation between IgG ACPA levels and VDG, which was strongest in the pre‐disease phase. These findings are also in accordance with the notion that variable domain glycans could have a regulatory impact on the ACPA immune response. In this respect, it is intriguing to note that the HLA shared epitope alleles predispose to ACPA harboring VDG rather than to ACPA in general (11), thus linking ACPA VDG with the major genetic risk factor for RA. Indeed, a more in‐depth longitudinal analysis of the correlation between the presence of predisposing HLA–DR4 genes and the presence of VDG revealed a shorter “transit time” to RA in HLA–DR4–positive pre‐disease individuals who still displayed relatively low levels of ACPA variable domain glycans, as compared to HLA–DR4–negative individuals with similar ACPA variable domain glycan levels (26).
Of note, in the longitudinal analysis we observed that individuals in whom long‐term DFR is achieved exhibit lower VDG profiles at disease onset (61.4%) compared to patients in whom long‐term DFR is not achieved (83.8%). The relevance of these findings is unknown, although it is remarkable that long‐term DFR, a relatively rare event in ACPA‐positive RA, was associated with lower VDG on ACPAs.
Importantly, reduced IgG ACPA levels are not the cause of lower VDG, which was controlled by titrating IgG ACPA into healthy serum samples to enable maintenance of a high degree of VDG (Supplementary Figure 5B, on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42098). Thus it is tempting to speculate that variable domain glycans serve as an additional “hit” determining the fate of the autoreactive B cell response and thereby exert an impact on ACPA levels.
Together with previous data showing that N‐linked glycan sites are selectively introduced into ACPA B cell receptor sequences upon somatic hypermutation (27) and that variable domain glycan levels are significantly elevated in ACPA‐positive individuals who subsequently develop RA (12), our results provide evidence that a glycan attached to the variable domain fosters a breach of tolerance of autoreactive B cells. As carbohydrates are known to affect cellular functions, ACPA‐expressing B cells may gain a selection advantage when abundantly expressing glycans in their variable domains. The disialylated, and thus negatively charged, glycans attached to the variable domain, which also have a large steric requirement, might modulate binding to autoantigens or affect B cell receptor signaling of citrullinated antigen–directed B cells. Further, we cannot rule out a possible role of variable domain glycans in effector mechanism, and thereby, autoantibody‐mediated inflammation, similar to findings for Fc glycans. In addition to these areas for further research, it would be interesting to investigate changes in specific variable domain glycan traits in more depth, as altered glycan composition could be associated with defined biologic implications, as also observed for Fc glycans. Recent studies have shown, for example, that not only Fc glycans on total IgG, but also variable domain glycans on IgG ACPA, show a decrease in the bisecting GlcNAc after COVID‐19 (28, 29). Interestingly, variable domain glycans are not only a feature of IgG ACPA in RA, but have also been described in other human autoimmune responses, such as in antineutrophil cytoplasmic antibody–associated vasculitis and Sjögren's syndrome, and have been observed on anti‐hinge and antidrug antibodies (30, 31, 32).
A limitation of our study is that VDG profiles could be detected in only 70% of the samples analyzed, mainly due to a limited amount of serum available for the IgG ACPA capturing and subsequent glycan analysis or to low IgG ACPA levels, as observed in the group of healthy individuals. Especially for rare disease stages, such as for the “DFR with late flares” group, only a limited number of samples were available to us. In addition, ACPAs were captured using the highly sensitive and specific antigen CCP‐2. However, it cannot be excluded that certain ACPA molecules that recognize different citrullinated epitopes and do not interact with CCP‐2 were omitted from the analysis. Importantly though, we did not observe an effect of VDGs on binding affinity to CCP‐2 (data not shown), making selection bias toward higher or lower glycosylated ACPAs unlikely. Another limitation of the study is that conclusions are mainly based on cross‐sectional data derived from samples collected at different sites. Although collection of such data from one site would be highly challenging, the analyses of samples from different sites could be hampered by site‐specific effects. Importantly, however, we also observed an increase in IgG ACPA VDG toward the time of RA onset in the longitudinal data set from cohort 3, including paired samples obtained from individuals when they were presymptomatic and after RA onset, over a period of 15 years, as also previously described (26). Furthermore, our findings of IgG ACPA variable domain glycan levels were concordant across different cross‐sectional cohorts of healthy individuals (58.1% and 44.9%) or individuals with arthralgia (75.3% and 70.4%).
In summary, we have provided a comprehensive overview of the expression of variable domain glycans on IgG ACPA over various clinical disease stages in RA. Although the biologic implications of variable domain glycans attached to antibodies in general and to ACPAs specifically are still largely unexplored, our data show that they are a key characteristic of ACPAs across disease stages in individuals of different ethnicities who develop RA. Our results demonstrate an increase in VDG toward the time of disease onset and, taken together with previous data indicating a selective introduction of these N‐linked glycan sites, suggest that variable domain glycans may serve as a trigger for the maturation of the ACPA immune response. It will therefore be useful to understand the biologic impact of variable domain glycans on the ACPA immune response and its detailed clinical implications.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Ms. Kissel had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Kissel, Hafkenscheid, Tamai, Kawashiri, Kawakami, El‐Gabalawy, van Schaardenburg, Rantapää‐Dahlqvist, Wuhrer, van der Helm‐van Mil, Allaart, van der Woude, Scherer, Toes, Huizinga.
Acquisition of data
Kissel, Hafkenscheid.
Analysis and interpretation of data
Kissel, Wesemael, Wuhrer, van der Helm‐van Mil, van der Woude, Scherer, Toes, Huizinga.
Supporting information
Disclosure Form
Appendix S1 Supplementary materials and methods
Figure S1 ACPA‐IgG capturing and liquid chromatography VDG analysis. (A) Experimental procedure: ACPA were captured using CCP2‐ streptavidin beads, followed by IgG capturing (FcXl beads). N‐linked glycans were released using PNGaseF and 2‐AA labeled for liquid chromatography. G0F, G1F, G2F, G2FBS1, G2FS2 and G2FBS2 glycan peaks are highlighted. A‐/mono‐/di‐galactosylated (G0/G1/G2), fucose attached to the core GlcNAc (F), bisecting GlcNAc (B), mono‐/di‐sialylated (S1/S2). Blue square: N‐acetylglucosamine (GlcNAc), green circle: mannose, yellow circle: galactose, red triangle: fucose, pink diamond: N‐acetylneuraminic acid. (B) UHPLC chromatograms of ACPA‐IgG VDGs from a healthy individual (cohort 1), an individual with arthralgia (cohort 4) and an individual at RA‐ onset and 4, 8 and 12 months after disease development (cohort 6). (C) ACPA‐IgG VDG of individuals diagnosed with RA later in life and sampled prior to symptom‐onset and after RA diagnosis (cohort 3). Data are presented as box and whiskers including all data points. (D) Matched paired samples of pre‐symptomatic individuals and early RA patients (cohort 3). (E) Percentage ACPA‐IgG VDG of individuals with arthralgia that later convert or do not convert to RA and of patients at RA‐onset. Data are presented as box and whiskers including all data points. Kruskal‐Wallis tests were performed. Significant differences are denoted by *(p = 0.0406), ***(p = 0.0001), or ****(p < 0.0001).
Figure S2 ACPA‐IgG VDG percentages correlate with ACPA‐IgG levels. Pearson correlation between ACPA‐IgG VDG percentages and ACPA‐IgG levels (aU/ml). The respective p‐values (two‐tailed) and correlation coefficients are presented.
Figure S3 ACPA‐IgG VDG in individuals with arthralgia from cohort 5 (the Netherlands). (A) Longitudinal analysis of ACPA‐IgG VDG percentages from individuals with clinically suspect arthralgia (CSA), at RA‐ onset and 1 or 2 years after disease development. (B) “Time line” of VDG percentages up to 36 months after 1st visit. Time point of RA‐ onset is depicted in orange.
Figure S4 Longitudinal analysis of ACPA‐IgG VDG at RA‐ onset and in established RA for cohort 6 (the Netherlands) separated for the different treatment arms. (A) ACPA‐IgG VDG comparison of the four treatment groups: early remission, drug‐free; early remission, no drug‐free; arm 1 and arm 2. Treatment specifications are illustrated in figure 3A. Ordinary one‐way ANOVA was performed including the Fisher's LSD test: *p = 0.015. (B) Longitudinal matched paired samples are shown for treatment arm 1 and 2.
Figure S5 ACPA‐IgG VDG over various time‐points and for different ACPA‐IgG titer. (A) “Time‐line” of ACPA‐IgG VDG after RA‐ onset and towards drug‐free remission (DFR) (cohort 7, the Netherlands). ACPA‐IgG VDG mean and SEM of the samples presented in figure 4D is depicted. Individuals that flare later in time are shown in red and individuals that stay in DFR in turquoise. (B) Stable VDG of ACPA‐IgG positive patient serum measured with different titers. ACPA‐IgG titers were lowered by mixing the sera with ACPA negative healthy donor sera. Two individual experiments are presented.
ACKNOWLEDGMENTS
The authors would like to thank personnel of the Department of Biobank Research (Umeå University), the Västerbotten Intervention Programme (Västerbotten, Sweden), the Northern Sweden MONICA study, and the County Council of Västerbotten for providing data and samples. We are grateful to Dr. Jan Wouter Drijfhout (Leiden University Medical Center) for providing the CCP‐2 peptide, Carolien Koeleman (Leiden University Medical Center) for expert assistance with liquid chromatography, and Ellis Niemantsverdriet and Marloes Verstappen (Leiden University Medical Center) for assistance with sample and data collection.
Supported by ReumaNederland (grants 17‐1‐402 and 08‐1‐34), the IMI funded project RTCure (777357), ZonMw TOP (grant 91214031), Target to B! (LSHM18055‐5GF), the Swedish Research Council (VR 2017‐00650), King Gustaf V's 80‐Year Fund, King Gustaf V's and Queen Victoria's Fund, the Swedish Rheumatism Association, and the Canadian Institutes of Health Research MOP grant (77700). Dr. Scherer is recipient of an NWO‐ZonMW clinical fellowship (90714509), an NWO‐ZonMW VENI grant (91617107), an NWO‐ZonMW VIDI grant (09150172010067), and a ZonMW Enabling Technology Hotels grant (435002030) and has received support from the Dutch Arthritis Foundation (15‐2‐402 and 18‐1‐205). Dr. Toes is recipient of a European Research Council (ERC) advanced grant (AdG2019‐884796).
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42098&file=art42098‐sup‐0001‐Disclosureform.pdf.
REFERENCES
- 1. Van der Kooij SM, Goekoop‐Ruiterman YP, de Vries‐Bouwstra JK, Guler‐Yuksel M, Zwinderman AH, Kerstens PJ, et al. Drug‐free remission, functioning and radiographic damage after 4 years of response‐driven treatment in patients with recent‐onset rheumatoid arthritis. Ann Rheum Dis 2009;68:914–21. [DOI] [PubMed] [Google Scholar]
- 2. Willemze A, Trouw LA, Toes RE, Huizinga TW. The influence of ACPA status and characteristics on the course of RA [review]. Nat Rev Rheumatol 2012;8:144–52. [DOI] [PubMed] [Google Scholar]
- 3. de Moel EC, Derksen V, Trouw LA, Bang H, Collee G, Lard LR, et al. In rheumatoid arthritis, changes in autoantibody levels reflect intensity of immunosuppression, not subsequent treatment response. Arthritis Res Ther 2019;21:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hafkenscheid L, Bondt A, Scherer HU, Huizinga TW, Wuhrer M, Toes RE, et al. Structural analysis of variable domain glycosylation of anti‐citrullinated protein antibodies in rheumatoid arthritis reveals the presence of highly sialylated glycans. Mol Cell Proteomics 2017;16:278–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kasermann F, Boerema DJ, Ruegsegger M, Hofmann A, Wymann S, Zuercher AW, et al. Analysis and functional consequences of increased Fab‐sialylation of intravenous immunoglobulin (IVIG) after lectin fractionation. PLoS One 2012;7:e37243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vergroesen RD, Slot LM, Hafkenscheid L, Koning MT, van der Voort EI, Grooff CA, et al. B‐cell receptor sequencing of anti‐citrullinated protein antibody (ACPA) IgG‐expressing B cells indicates a selective advantage for the introduction of N‐glycosylation sites during somatic hypermutation. Ann Rheum Dis 2018;77:956–8. [DOI] [PubMed] [Google Scholar]
- 7. Rombouts Y, Ewing E, van de Stadt LA, Selman MH, Trouw LA, Deelder AM, et al. Anti‐citrullinated protein antibodies acquire a pro‐inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis 2015;74:234–41. [DOI] [PubMed] [Google Scholar]
- 8. Scherer HU, van der Woude D, Ioan‐Facsinay A, el Bannoudi H, Trouw LA, Wang J, et al. Glycan profiling of anti–citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum 2010; 62:1620–9. [DOI] [PubMed] [Google Scholar]
- 9. Bondt A, Hafkenscheid L, Falck D, Kuijper TM, Rombouts Y, Hazes JM, et al. ACPA IgG galactosylation associates with disease activity in pregnant patients with rheumatoid arthritis. Ann Rheum Dis 2018;77:1130–6. [DOI] [PubMed] [Google Scholar]
- 10. Ercan A, Cui J, Chatterton DE, Deane KD, Hazen MM, Brintnell W, et al. Aberrant IgG galactosylation precedes disease onset, correlates with disease activity, and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum 2010;62:2239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kissel T, van Schie KA, Hafkenscheid L, Lundquist A, Kokkonen H, Wuhrer M, et al. On the presence of HLA‐SE alleles and IgG ACPA variable domain glycosylation in the phase preceding the development of rheumatoid arthritis. Ann Rheum Dis 2019;78:1616–20. [DOI] [PubMed] [Google Scholar]
- 12. Hafkenscheid L, de Moel E, Smolik I, Tanner S, Meng X, Jansen BC, et al. N‐linked glycans in the variable domain of IgG anti–citrullinated protein antibodies predict the development of rheumatoid arthritis. Arthritis Rheumatol 2019;71:1626–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heimans L, Wevers‐de Boer KV, Visser K, Goekoop RJ, van Oosterhout M, Harbers JB, et al. A two‐step treatment strategy trial in patients with early arthritis aimed at achieving remission: the IMPROVED study. Ann Rheum Dis 2014;73:1356–61. [DOI] [PubMed] [Google Scholar]
- 14. Van Aken J, van Bilsen JH, Allaart CF, Huizinga TW, Breedveld FC. The Leiden Early Arthritis Clinic. Clin Exp Rheumatol 2003;21 Suppl:S100–5. [PubMed] [Google Scholar]
- 15. Kawashiri SY, Tsuji Y, Tamai M, Nonaka F, Nobusue K, Yamanashi H, et al. Effects of cigarette smoking and human T‐cell leukaemia virus type 1 infection on anti‐citrullinated peptide antibody production in Japanese community‐dwelling adults: the Nagasaki Islands Study. Scand J Rheumatol 2020;22:1–4. [DOI] [PubMed] [Google Scholar]
- 16. Smolik I, Robinson DB, Bernstein CN, El‐Gabalawy HS. First‐degree relatives of patients with rheumatoid arthritis exhibit high prevalence of joint symptoms. J Rheumatol 2013;40:818–24. [DOI] [PubMed] [Google Scholar]
- 17. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988;31:315–24. [DOI] [PubMed] [Google Scholar]
- 18. Rantapää‐Dahlqvist S, de Jong BA, Berglin E, Hallmans G, Wadell G, Stenlund H, et al. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum 2003;48:2741–9. [DOI] [PubMed] [Google Scholar]
- 19. Bos WH, Wolbink GJ, Boers M, Tijhuis GJ, de Vries N, van der Horst‐Bruinsma IE, et al. Arthritis development in patients with arthralgia is strongly associated with anti‐citrullinated protein antibody status: a prospective cohort study. Ann Rheum Dis 2010;69:490–4. [DOI] [PubMed] [Google Scholar]
- 20. Newsum EC, van der Helm‐van Mil AH, Kaptein AA. Views on clinically suspect arthralgia: a focus group study. Clin Rheumatol 2016;35:1347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van der Heijde DM, van 't Hof MA, van Riel PL, Theunisse LM, Lubberts EW, van Leeuwen MA, et al. Judging disease activity in clinical practice in rheumatoid arthritis: first step in the development of a disease activity score. Ann Rheum Dis 1990;49:916–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Moel EC, Derksen V, Stoeken G, Trouw LA, Bang H, Goekoop RJ, et al. Baseline autoantibody profile in rheumatoid arthritis is associated with early treatment response but not long‐term outcomes. Arthritis Res Ther 2018;20:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Steenbergen HW, Mangnus L, Reijnierse M, Huizinga TW, van der Helm‐van Mil AH. Clinical factors, anticitrullinated peptide antibodies and MRI‐detected subclinical inflammation in relation to progression from clinically suspect arthralgia to arthritis. Ann Rheum Dis 2016;75:1824–30. [DOI] [PubMed] [Google Scholar]
- 24. Jansen BC, Hafkenscheid L, Bondt A, Gardner RA, Hendel JL, Wuhrer M, et al. HappyTools: a software for high‐throughput HPLC data processing and quantitation. PLoS One 2018;13:e0200280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rombouts Y, Willemze A, van Beers JJ, Shi J, Kerkman PF, van Toorn L, et al. Extensive glycosylation of IgG ACPA variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann Rheum Dis 2016;75:578–85. [DOI] [PubMed] [Google Scholar]
- 26. Kissel T, van Wesemael TJ, Lundquist A, Kokkonen H, Kawakami A, Tamai M, et al. Genetic predisposition (HLA‐SE) is associated with IgG ACPA variable domain glycosylation in the predisease phase of RA [letter]. Ann Rheum Dis 2021;81:141–3. [DOI] [PubMed] [Google Scholar]
- 27. Vergroesen RD, Slot LM, van Schaik BD, Koning MT, Rispens T, van Kampen AH, et al. N‐glycosylation site analysis of citrullinated antigen‐specific B‐cell receptors indicates alternative selection pathways during autoreactive B‐cell development. Front Immunol 2019;10:2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Derksen V, Kissel T, Lamers‐Karnebeek FB, van der Bijl AE, Venhuizen AC, Huizinga TW, et al. Onset of rheumatoid arthritis after COVID‐19: coincidence or connected? Ann Rheum Dis 2021. DOI: 10.1136/annrheumdis-2021-219859. E‐pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 29. Chakraborty S, Gonzalez J, Edwards K, Mallajosyula V, Buzzanco AS, Sherwood R, et al. Proinflammatory IgG Fc structures in patients with severe COVID‐19. Nat Immunol 2021;22:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vletter EM, Koning MT, Scherer HU, Veelken H, Toes RE. A comparison of immunoglobulin variable region N‐linked glycosylation in healthy donors, autoimmune disease and lymphoma. Front Immunol 2020;11:241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Biermann MH, Griffante G, Podolska MJ, Boeltz S, Sturmer J, Munoz LE, et al. Sweet but dangerous: the role of immunoglobulin G glycosylation in autoimmunity and inflammation. Lupus 2016;25:934–42. [DOI] [PubMed] [Google Scholar]
- 32. Van de Bovenkamp FS, Derksen NI, Ooijevaar‐de Heer P, van Schie KA, Kruithof S, Berkowska MA, et al. Adaptive antibody diversification through N‐linked glycosylation of the immunoglobulin variable region. Proc Natl Acad Sci U S A 2018;115:1901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Appendix S1 Supplementary materials and methods
Figure S1 ACPA‐IgG capturing and liquid chromatography VDG analysis. (A) Experimental procedure: ACPA were captured using CCP2‐ streptavidin beads, followed by IgG capturing (FcXl beads). N‐linked glycans were released using PNGaseF and 2‐AA labeled for liquid chromatography. G0F, G1F, G2F, G2FBS1, G2FS2 and G2FBS2 glycan peaks are highlighted. A‐/mono‐/di‐galactosylated (G0/G1/G2), fucose attached to the core GlcNAc (F), bisecting GlcNAc (B), mono‐/di‐sialylated (S1/S2). Blue square: N‐acetylglucosamine (GlcNAc), green circle: mannose, yellow circle: galactose, red triangle: fucose, pink diamond: N‐acetylneuraminic acid. (B) UHPLC chromatograms of ACPA‐IgG VDGs from a healthy individual (cohort 1), an individual with arthralgia (cohort 4) and an individual at RA‐ onset and 4, 8 and 12 months after disease development (cohort 6). (C) ACPA‐IgG VDG of individuals diagnosed with RA later in life and sampled prior to symptom‐onset and after RA diagnosis (cohort 3). Data are presented as box and whiskers including all data points. (D) Matched paired samples of pre‐symptomatic individuals and early RA patients (cohort 3). (E) Percentage ACPA‐IgG VDG of individuals with arthralgia that later convert or do not convert to RA and of patients at RA‐onset. Data are presented as box and whiskers including all data points. Kruskal‐Wallis tests were performed. Significant differences are denoted by *(p = 0.0406), ***(p = 0.0001), or ****(p < 0.0001).
Figure S2 ACPA‐IgG VDG percentages correlate with ACPA‐IgG levels. Pearson correlation between ACPA‐IgG VDG percentages and ACPA‐IgG levels (aU/ml). The respective p‐values (two‐tailed) and correlation coefficients are presented.
Figure S3 ACPA‐IgG VDG in individuals with arthralgia from cohort 5 (the Netherlands). (A) Longitudinal analysis of ACPA‐IgG VDG percentages from individuals with clinically suspect arthralgia (CSA), at RA‐ onset and 1 or 2 years after disease development. (B) “Time line” of VDG percentages up to 36 months after 1st visit. Time point of RA‐ onset is depicted in orange.
Figure S4 Longitudinal analysis of ACPA‐IgG VDG at RA‐ onset and in established RA for cohort 6 (the Netherlands) separated for the different treatment arms. (A) ACPA‐IgG VDG comparison of the four treatment groups: early remission, drug‐free; early remission, no drug‐free; arm 1 and arm 2. Treatment specifications are illustrated in figure 3A. Ordinary one‐way ANOVA was performed including the Fisher's LSD test: *p = 0.015. (B) Longitudinal matched paired samples are shown for treatment arm 1 and 2.
Figure S5 ACPA‐IgG VDG over various time‐points and for different ACPA‐IgG titer. (A) “Time‐line” of ACPA‐IgG VDG after RA‐ onset and towards drug‐free remission (DFR) (cohort 7, the Netherlands). ACPA‐IgG VDG mean and SEM of the samples presented in figure 4D is depicted. Individuals that flare later in time are shown in red and individuals that stay in DFR in turquoise. (B) Stable VDG of ACPA‐IgG positive patient serum measured with different titers. ACPA‐IgG titers were lowered by mixing the sera with ACPA negative healthy donor sera. Two individual experiments are presented.