

Abstract
Background
Parkinsonian features have been described in patients harboring variants in nuclear genes encoding for proteins involved in mitochondrial DNA maintenance, such as TWNK.
Objectives
The aim was to screen for TWNK variants in an Italian cohort of Parkinson's disease (PD) patients and to assess the occurrence of parkinsonism in patients presenting with TWNK‐related autosomal dominant progressive external ophthalmoplegia (TWNK‐adPEO).
Methods
Genomic DNA of 263 consecutively collected PD patients who underwent diagnostic genetic testing was analyzed with a targeted custom gene panel including TWNK, as well as genes causative of monogenic PD. Genetic and clinical data of 18 TWNK‐adPEO patients with parkinsonism were retrospectively analyzed.
Results
Six of 263 PD patients (2%), presenting either with isolated PD (n = 4) or in combination with bilateral ptosis (n = 2), carried TWNK likely pathogenic variants. Among 18 TWNK‐adPEO patients, 5 (28%) had parkinsonism.
Conclusions
We show candidate TWNK variants occurring in PD without PEO. This finding will require further confirmatory studies. © 2022 Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson Movement Disorder Society.
Keywords: TWNK, twinkle, Parkinson's disease, parkinsonism, mitochondrial DNA
Pathogenesis of Parkinson's disease (PD) has long been associated with mitochondrial dysfunction. 1 Dopaminergic neurons of the substantia nigra pars compacta seem to be particularly vulnerable to mitochondrial damage. 2 Although sequencing of mitochondrial DNA (mtDNA) failed to reveal pathogenic mutations associated with PD, population‐specific common variants defining mtDNA haplogroups have been implicated as possible risk factors. 3 In addition, age‐related accumulation of somatic mtDNA deletions in the substantia nigra has been reported to occur more significantly in PD patients than in age‐matched controls.4, 5 Moreover, the regulation of mtDNA copy number seems to be affected in PD, leading to a relative mtDNA depletion.6, 7
Pathogenic variants in nuclear genes encoding for proteins primarily involved in mtDNA maintenance, such as POLG, TWNK, MPV17, OPA1, DGUOK, or SLC25A46,8, 9, 10, 11, 12, 13 have been described in patients with mitochondrial syndromes featuring parkinsonian signs as part of their complex phenotypic manifestation. 14 Among these genes, TWNK encodes for the mitochondrial twinkle helicase, which is essential for mtDNA replication. 15 Pathogenic variants in TWNK have been associated with different phenotypes, ranging from autosomal dominant progressive external ophthalmoplegia 16 (adPEO) to rare autosomal recessive syndromes, such as mtDNA depletion syndrome, Perrault syndrome, infantile‐onset spinocerebellar ataxia, mitochondrial recessive ataxia syndrome, and sensory ataxia neuropathy dysarthria and ophthalmoplegia.17, 18, 19, 20, 21 Thus far, 10 TWNK‐adPEO patients also presenting with parkinsonism have been reported in the literature.9, 22, 23, 24, 25, 26, 27
We here explore the frequency of TWNK variants in an Italian cohort of PD patients and describe the associated clinical and neuroradiological phenotypes (TWNK‐PD). In addition, we reassess clinical and genetic data from a cohort of adPEO‐carrying TWNK pathogenic variants and manifesting parkinsonism (TWNK‐adPEO‐P) and compare the clinical and genetic findings of TWNK‐PD and TWNK‐adPEO‐P. Finally, we evaluate the defects of mtDNA maintenance in either blood or muscle biopsies (when available) associated with detected TWNK variants.
Patients and Methods
We included in the study 263 patients with a diagnosis of PD made by a movement disorder specialist and referred for genetic testing for diagnosis at the IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico (Milan, Italy) or at IRCCS Mondino Foundation (Pavia, Italy). Data were consecutively collected from 2017 to 2021. Patients with adPEO were evaluated at the Mitochondrial Diseases Center of the IRCCS Institute of Neurological Sciences (Bologna, Italy). Of 302 adPEO, data from 18 carrying TWNK variants were retrospectively analyzed. The Ethics Committees of the IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico, Mondino Foundation, and Institute of Neurological Sciences (Comitato Etico Interaziendale Bologna‐Imola, CE‐BI 13036) approved the study, and all patients provided written informed consent to study participation.
Genetic Analysis
PD patients underwent either next‐generation sequencing of a targeted panel of 19 genes associated to PD and parkinsonism (Table S1), which included POLG, TWNK, OPA1, and SLC25A46 (Haloplex Technology, Agilent, Santa Clara, CA, United States), or whole exome sequencing (WES) with subsequent analysis of the same virtual gene panel (Twist Core Exome Kit, Twist Biosciences, San Francisco, CA, United States). Exon rearrangements were assessed through Multiplex Ligation Probe Amplification (MS‐MLPA) using Salsa MLPA Probemix P051‐D2 or P052 Parkinson (MRC‐Holland, Amsterdam, Netherlands) following manufacturer's instructions. All TWNK‐adPEO‐P patients had been diagnosed through WES and subsequent analysis of the same virtual gene panel used for the PD cohort. Further information is available in Appendix S1.
Results
Genetic Results
Monoallelic variants of TWNK were detected in 6 of 263 (2%) patients with PD (162 men, 62%; 85 with positive family history for PD, 32%; mean age at onset 51.35 ± 13.03 years) (Table 1; Fig. 1; Table S3 in Appendix S1). The TWNK variants identified in PD were searched in in‐house exomes of Italian non‐PD subjects (n = 2529) and in two databases, Network for Italian Genomes (n = 1492) and NIG—Exomes from Italy (n = 1686). None of the variants were present in any databases, except for the p.G540R that was found in the heterozygous state in a pediatric patient with neurodevelopmental syndrome (one of 5707, 0.0001752). None of the TWNK‐PD and TWNK‐adPEO‐P patients carried additional pathogenic variants or genomic rearrangements in other autosomal dominant or autosomal recessive PD‐related genes. The variant identified in patient 4 was detected also in the mother who had also developed PD. To obtain support for the functional relevance of the TWNK variants identified in PD patients, we performed molecular modeling. Our in silico analysis predicted that all the identified variants could have negative effects on twinkle activity (see Appendix S1, Fig. S3).
TABLE 1.
Genetic data of TWNK variants reported in this article
| Patient | Phenotype | Base change | AA change | Zygosity | dbSNP (rs number) | gnomAD* v2.1.1 | Italian controls** | ACMG criteria*** | Classification |
|---|---|---|---|---|---|---|---|---|---|
| TWNK‐PD | |||||||||
| Patient 1 | EOPD |
c.500 T > C |
p.L167P | Het | – | – | – |
PM1m, PM2s PP2su, PP3m |
LP |
| Patient 2 | EOPD |
c.1112 G > A |
p.R371Q | Het |
rs 143309797 |
13/129′034 (0.0001007) |
– | PM1s, PM2su, PP2su, PP3m | LP |
| Patient 3 | EOPD |
c.1381 G > A |
p.E461K | Het |
rs 776518524 |
4/113′770 (0.00003516) |
– |
PM1s, PM2su PP2su, PP3m |
LP |
| Patient 4 |
PD with ptosis |
c.1618 G > A |
p.G540R | Het |
rs 568256888 |
3/113′770 (0.00002637) |
1/5707 (0.0001752) |
PM1m, PM2s PP2su, PP3m PP1su |
P |
| Patient 5 |
PD with ptosis |
c.1966 A > C |
p.K656Q | Het | – | – | – |
PM2su, PP2su PP3su |
VUS‐LP |
| Patient 6 | PD |
c.2010 G > C |
p.Q670H | Het |
rs 778236767 |
– | – | PM2su, PP2su, PP3su | VUS‐LP |
| TWNK‐adPEO‐P | |||||||||
| Patient 7 | adPEO‐P |
c.907 C > T |
p.R303W | Het |
rs 1159929268 |
1/113′466 (0.000008813) |
– | PM1m, PM5m, PP5m, PM2su PP2su, PP3su | LP |
| Patient 8 | adPEO‐P | ||||||||
| Patient 9 | adPEO‐P |
c.1001 G > A |
p.R334Q | Het |
rs 28937887 |
– | – |
PM1m, PP5s PM5m, PM2su PP2su, PP3su, BP6su |
P |
| Patient 10 | adPEO‐P | ||||||||
| Patient 11 | adPEO‐P |
c.1609 T > C |
p.Y537H | Het |
rs 144001072 |
33/113′770 (0.0002941) |
‐ |
PM1m, PM2su PP2su, BP4su |
VUS‐LP |
AA, amino acids; ACMG, American College of Medical Genetics; EOPD early‐onset Parkinson's disease; Het, heterozygous (monoallelic); LP, likely pathogenic; P, pathogenic; VUS, variant of unknown significance; VUS‐LP, VUS with likely pathogenic effect; adPEO, autosomal dominant progressive external ophthalmoplegia.
** In‐house exomes of patients with neurological diseases (non‐PD, n = 2529), Network for Italian Genomes (n = 1492), and NIG—Exomes from Italy (n = 1686).
*** ACMG criteria: m, moderate; s, strong; su, supporting.
*Minor allele frequency in European (non‐Finnish) population. All variants were absent in European (Finnish) population.
FIG 1.
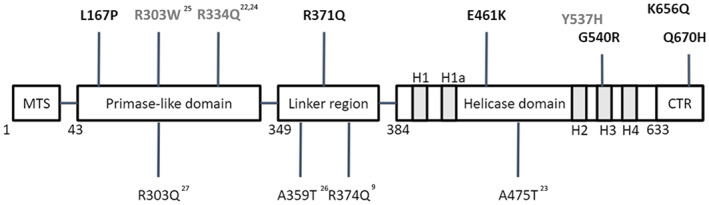
Distribution of TWNK variants in patients with parkinsonism. TWNK variants here reported (bold) are in the upper part of the figure (black = TWNK‐PD patients; gray = TWNK‐adPEO‐P). Variants related to previously reported TWNK‐adPEO‐P patients are in the lower part (see references 9, 22, 23, 24, 25, 26, 27). MTS, mitochondrial targeting sequence (1–42); primase‐like domain (43–348); linker region (349–383); helicase domain (384–632), H1 (409–422), H1a (439–446), H2 (512–517), H3 (535–558), and H4 (569–587); CTR, C‐terminal region (633–684). adPEO, autosomal dominant progressive external ophthalmoplegia. [Color figure can be viewed at wileyonlinelibrary.com]
Clinical Features
Clinical features are summarized in Table S3, Appendix S1. Two TWNK‐PD patients also manifested ptosis without clinical and video‐oculographic limitations of gaze. Interestingly, the mother of patient 4 was affected by ptosis and developed PD at age 60 years (see Appendix S1). Of 18 TWNK‐adPEO patients, 5 (28%) also presented with parkinsonism. A more exhaustive clinical description of all TWNK‐PD and TWNK‐adPEO‐P cases reported here is available in Appendix S1. Mean age at onset of parkinsonism was significantly younger in TWNK‐PD than in TWNK‐adPEO‐P patients (52.7 vs. 73.6 years, P = 0.0018). Two TWNK‐adPEO‐P patients were treated with levodopa (L‐dopa) without a clear benefit. Other neurological features like postural tremor, head tremor, and apraxia appeared to be more frequent in TWNK‐adPEO‐P patients but without reaching statistical significance.
mtDNA Deletions and Copy Number
Quantitative analysis of mtDNA copy number between TWNK‐PD patients and age‐ and sex‐matched controls showed no differences (Appendix S1 in Fig. S1). In skeletal muscle biopsies, mtDNA amount and 7sDNA were comparable among groups, whereas quantifiable mtDNA deletions were detected only in a subgroup of TWNK‐adPEO patients (Appendix S1, Fig. S2). COX negative fibers in muscle biopsies of patients 7 and 10 were 2.3% and 0.9%, respectively.
Discussion
Our study focused on the role of TWNK in patients from two cohorts: (1) a PD cohort of patients who consecutively underwent genetic testing, including TWNK; and (2) a retrospective adPEO cohort known to carry a heterozygous TWNK pathogenic variant and presenting parkinsonism. Interestingly, carriers of a TWNK variant were not rare among our cohort of PD patients (6 of 263, 2%). The screening in non‐PD subjects indicates that these variants are not common in the Italian population. Most relevantly, TWNK variants were also found in PD patients lacking ptosis, a hallmark of twinkle ‐related myopathy, suggesting that the diagnostic screening of this gene should be considered also in PD patients without other signs suggestive of a mitochondrial disease.
Before this study, parkinsonism cases associated with TWNK variants without ptosis, PEO, or any other sign of myopathy have not been reported9, 22, 23, 24, 25, 26, 27 (Appendix S1, Table S1). Our findings suggest that TWNK variants could be related to a clinical picture indistinguishable from idiopathic PD with good response to L‐dopa and development of L‐dopa complications (motor fluctuations and dyskinesias), only occasionally featuring ptosis.
Parkinsonism was relatively frequent in the TWNK‐adPEO group (5 of 18, 28%). In line with previous reports, the phenotype of TWNK‐adPEO‐P was characterized by a complex association of neurological and nonneurological signs suggestive of an underlying mitochondrial syndrome (Appendix S1, Tables S3 and S4). Compared to TWNK‐PD subjects, TWNK‐adPEO‐P patients showed a later onset of parkinsonian features (on average in the seventh decade of life) and, when assessed, a poorer response to L‐dopa due to the development of adverse effects. In these patients, the cause of parkinsonian features was accompanied by the frequent co‐occurrence of cardiovascular risk factors or magnetic resonance imaging evidence of vascular encephalopathy. In fact, I123 ioflupane scintigraphy (DaTSCAN) was repeatedly negative in a TWNK‐adPEO‐P patient, challenging the hypothesis of nigrostriatal degeneration as causative of parkinsonian features. These observations suggest that the atypical parkinsonian syndrome of TWNK‐adPEO seems a distinct phenotype compared to that of TWNK‐PD. A possible confounding factor is the different age of the two cohorts, having the PD group an earlier age of onset. TWNK‐PD patients did not show any muscular involvement or complained of muscular symptoms; thus, electromyography (EMG) and muscle biopsy were not performed. Of note, mtDNA copy number abnormalities were not observed in this group (Appendix S1, Fig. S1). On the contrary, among TWNK‐adPEO‐P patients, 3 of 5 patients showed clinical and/or EMG signs of myopathic involvement (Appendix S1, Table S3), and 2 patients had a muscle biopsy typical of mitochondrial myopathy (Appendix S1, Table S3). However, quantification of mtDNA deletions, as well as of mtDNA copy number or DNA 7S performed on muscle biopsy, failed to reveal differences with controls (Appendix S1, Fig. S2), probably due to the rare COX negative fibers in their muscle biopsy, confirming that myopathy in these patients is frequently very mild or even subclinical. Among TWNK‐PD patients, 4 carried a pathogenic or likely pathogenic variant, whereas the remaining 2 carried variants formally classified as variants of unknown significance with likely pathogenic effect according to the American College of Medical Genetics guidelines (see Supplementary Methods in Appendix S1; Table 1). The co‐segregation of the p.G540R variant with PD in the family of patient 4 further supports its likely pathogenic role. However, more evidence is needed to prove the role of these variants in the pathogenesis of PD, especially those identified in “pure” PD. Indeed, the lack of family history and functional demonstration of the impact on mtDNA raises caution in assigning a definitive association with PD. The distribution of the variants across the twinkle protein showed the involvement of all functional domains, without any clustering (Fig. 1). Although TWNK variants are more frequent in the N‐terminal domain and linker region of the protein, we failed to demonstrate any evidence of clear genotype–phenotype correlations.
PD is a complex multifactorial disease, in which several predisposing genetic factors could interplay to promote the development of the disease. To our knowledge, the presence of other genetic contributors to the development of parkinsonian features in patients with mitochondrial syndromes, such as variants in PD‐related genes, has not been ruled out to date. In our series of TWNK‐PD and TWNK‐adPEO‐P patients, the co‐occurrence of possibly causative variants in classical PD genes has been excluded, strengthening the likely pathogenic role of TWNK in contributing to PD. However, the development of parkinsonism in patients carrying variants in TWNK, as well as in other mtDNA maintenance genes, remains somehow enigmatic. Not all carriers eventually develop parkinsonism, denoting an incomplete penetrance of this trait. This incomplete knowledge has relevant implications on genetic counseling related to PD risk in families carrying TWNK variants.
In summary, we described the presence of TWNK variants in patients with PD or parkinsonism, with or without signs of myopathy. Our findings strengthen the relation between the TWNK gene and PD. Screening in larger PD populations, segregation analysis in additional familial cases, and functional studies are needed to confirm these interesting new observations.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical analysis: A. Design, B. Execution, C. Review and critique; (3) Manuscript: A. Writing of the first draft, B. Review and critique.
M.P.: 1A, 1B, 1C, 2A, 2B, 2C, 3A
G.F.: 1A, 1C
E.M.: 1A, 1B, 1C, 2A, 2B, 2C
L.C.:1C, 2B
R.M.: 1C
C.M.: 1C
M.L.V.: 1C
R.L.: 1C
I.P.: 1C
D.O.: 1C
M.V.: 1C
D.R.: 1C, 2B, 2C
F.D.B.: 2B
A.C.: 1C
B.M.: 1D, 3A, 3B
M.F.: 1D, 3A, 3B
G.P.C.: 3B
A.A.: 2C, 3B
B.G.: 3B
E.M.V.: 1A, 1B, 2C, 3B
V.C.: 1A, 1B, 2A, 2C, 3A, 3B
A.D.F.: 1A, 1B, 2A, 2B, 2C, 2A, 3B
Full financial disclosures for the previous 12 months
A.D.F. reports advisory board fees from Sanofi and speaking honoraria from Sanofi and Zambon. V.C. reports consultant and advisory board fees from GenSight Biologics, Pretzel Therapeutics, Stealth Biotherapeutics, and Chiesi Farmaceutici and speaker honoraria from Chiesi Farmaceutici, First Class, and Medscape. M.F. reports consultant and advisory board fees from Pretzel Therapeutics. None of the other authors reports any conflicts of interest.
Supporting information
APPENDIX S1. Supporting Information
Acknowledgments
V.C. acknowledges the support of the Italian region Emilia‐Romagna funding (ER‐MITO project—Programma di ricerca Regione‐Università 2010‐2012, PRUa1RI‐2012‐008). A.D.F. and V.C. acknowledge the Italian Ministry of Health (Ricerca Corrente funding). M.F. acknowledges the support from the ALF agreement (ALFGBG‐965954). We are grateful to the patients and families for participating in this study. We are thankful to the “Network for Italian Genomes” group for sharing genetic data from sequences of Italian subjects.
Alessio Di Fonzo, Valerio Carelli, and Enza Maria Valente share senior authorship.
Relevant conflicts of interest/financial disclosures: A.D.F. reports advisory board fees from Sanofi and speaking honoraria from Sanofi and Zambon. V.C. reports consultant and advisory board fees from GenSight Biologics, Pretzel Therapeutics, Stealth Biotherapeutics, and Chiesi Farmaceutici and speaker honoraria from Chiesi Farmaceutici, First Class, and Medscape. None of the other authors reports any conflicts of interest.
Funding agencies: Italian Ministry of Health Ricerca Corrente 2020–2021 (PARKNET project) to A.D.F., E.M.V., and V.C.; Italian region Emilia‐Romagna funding (ER‐MITO project—Programma di ricerca Regione‐Universitàa 2010–2012, PRUa1RI‐2012‐008) to V.C.
Data Availability Statement
Raw data were generated at IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico (Milan, Italy), IRCCS Mondino Foundation (Pavia, Italy), and IRCCS Institute of Neurological Sciences (Bologna, Italy). Derived data supporting the findings of this study are available from the corresponding author on request.
References
- 1. Rani L, Mondal AC. Emerging concepts of mitochondrial dysfunction in Parkinson's disease progression: pathogenic and therapeutic implications. Mitochondrion 2020;50:25–34. 10.1016/j.mito.2019.09.010 [DOI] [PubMed] [Google Scholar]
- 2. Brichta L, Greengard P. Molecular determinants of selective dopaminergic vulnerability in Parkinson's disease: an update. Front Neuroanat 2014;8:1–16. 10.3389/fnana.2014.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Giannoccaro MP, La MC, Rizzo G, Carelli V. Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson's disease. Mov Disord 2017;32(3):346–363. 10.1002/mds.26966 [DOI] [PubMed] [Google Scholar]
- 4. Gu G, Reyes PF, Golden GT, et al. Mitochondrial DNA deletions/rearrangements in Parkinson disease and related neurodegenerative disorders. J Neuropathol Exp Neurol 2002;61(7):634–639. 10.1093/jnen/61.7.634 [DOI] [PubMed] [Google Scholar]
- 5. Bender A, Krishnan KJ, Morris CM, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 2006;38(5):515–517. 10.1038/ng1769 [DOI] [PubMed] [Google Scholar]
- 6. Dölle C, Flønes I, Nido GS, et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun 2016;7:13548. 10.1038/ncomms13548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grünewald A, Rygiel KA, Hepplewhite PD, Morris CM, Picard M, Turnbull DM. Mitochondrial DNA depletion in respiratory chain‐deficient Parkinson disease neurons. Ann Neurol 2016;79(3):366–378. 10.1002/ana.24571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase γ mutations: clinical and molecular genetic study. Lancet 2004;364(9437):875–882. https://www.embase.com/search/results?subaction=viewrecord&from=export&id=L39221065%0A 10.1016/S0140-6736(04)16983-3 [DOI] [PubMed] [Google Scholar]
- 9. Baloh RH, Salavaggione E, Milbrandt J, Pestronk A. Familial parkinsonism and Ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch Neurol 2007;64(7):998–1000. [DOI] [PubMed] [Google Scholar]
- 10. Garone C, Rubio JC, Calvo SE, et al. MPV17 mutations causing adult‐onset multisystemic disorder with multiple mitochondrial DNA deletions. Arch Neurol 2012;69(12):1648–1651. 10.1001/archneurol.2012.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carelli V, Musumeci O, Caporali L, et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann Neurol 2015;78(1):21–38. 10.1002/ana.24410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caporali L, Bello L, Tagliavini F, et al. DGUOK recessive mutations in patients with CPEO, mitochondrial myopathy, parkinsonism and mtDNA deletions. Brain 2018;141(1):e3. 10.1093/brain/awx301 [DOI] [PubMed] [Google Scholar]
- 13. Bitetto G, Malaguti MC, Ceravolo R, et al. SLC25A46 mutations in patients with Parkinson's disease and optic atrophy. Park Relat Disord 2020;74(March):1–5. 10.1016/j.parkreldis.2020.03.018 [DOI] [PubMed] [Google Scholar]
- 14. Manini A, Abati E, Pietro CG, Corti S, Ronchi D. Mitochondrial DNA homeostasis impairment and dopaminergic dysfunction: a trembling balance. Ageing Res Rev 2022;76:101578. 10.1016/j.arr.2022.101578 [DOI] [PubMed] [Google Scholar]
- 15. Peter B, Falkenberg M. Twinkle and other human mitochondrial DNA helicases: structure, function and disease. Genes (Basel) 2020;11(4):408. 10.3390/genes11040408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spelbrink JN, Li F, Tiranti V, et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding twinkle, a phage T7 gene 4‐like protein localized in mitochondria. Nat Genet 2001;28:223–231. [DOI] [PubMed] [Google Scholar]
- 17. Remtulla S, Emilie Nguyen CT, Prasad C, Campbell C. Twinkle‐associated mitochondrial DNA depletion. Pediatr Neurol 2019;90:61–65. 10.1016/j.pediatrneurol.2018.08.007 [DOI] [PubMed] [Google Scholar]
- 18. Morino H, Pierce SB, Matsuda Y, et al. Mutations in twinkle primase‐helicase cause Perrault syndrome with neurologic features. Neurology 2014;83(22):2054–2061. 10.1212/WNL.0000000000001036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nikali K, Suomalainen A, Saharinen J, et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins twinkle and Twinky. Hum Mol Genet 2005;14(20):2981–2990. 10.1093/hmg/ddi328 [DOI] [PubMed] [Google Scholar]
- 20. Hakonen AH, Goffart S, Marjavaara S, et al. Infantile‐onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet 2008;17(23):3822–3835. 10.1093/hmg/ddn280 [DOI] [PubMed] [Google Scholar]
- 21. Hudson G, Deschauer M, Busse K, Zierz S, Chinnery PF. Sensory ataxic neuropathy due to a novel C10Orf2 mutation with probable germline mosaicism. Neurology 2005;64(2):371–373. 10.1212/01.WNL.0000149767.51152.83 [DOI] [PubMed] [Google Scholar]
- 22. Van Goethem G, Lofgren A, Dermaut B, Ceuterick C, Martin J, Van Broeckhoven C. Digenic progressive external Ophthalmoplegia in a sporadic patient: recessive mutations in POLG and C10orf2 / twinkle. Hum Mutat 2003;22:175–176. 10.1002/humu.10246 [DOI] [PubMed] [Google Scholar]
- 23. Liu Z, Ding Y, Du A, Zhang B, Zhao G, Ding M. A novel twinkle (PEO1) gene mutation in a Chinese family with adPEO. Mol Vis 2008;14:1995–2001. [PMC free article] [PubMed] [Google Scholar]
- 24. Vandenberghe W, Van Laere K, Debruyne F, Van Broeckhoven C, Van Goethem G. Neurodegenerative parkinsonism and progressive external Ophthalmoplegia with a twinkle mutation. Mov Disord 2009;24(2):308–309. 10.1002/mds.22275 [DOI] [PubMed] [Google Scholar]
- 25. Brandon BR, Diederich NJ, Soni M, et al. Autosomal dominant mutations in POLG and C10orf2: association with late onset chronic progressive external ophthalmoplegia and parkinsonism in two patients. J Neurol 2013;260:1931–1933. 10.1007/s00415-013-6975-2 [DOI] [PubMed] [Google Scholar]
- 26. Kiferle L, Orsucci D, Mancuso M, et al. twinklemutation in an Italian family with external progressive ophthalmoplegia and parkinsonism: a case report and an update on the state of art. Neurosci Lett 2013;556:1–4. 10.1016/j.neulet.2013.09.034 [DOI] [PubMed] [Google Scholar]
- 27. Breen DP, Munoz DG, Lang AE. twinkle‐associated familial parkinsonism with Lewy pathology cause or predisposition? Neurology 2020;95:644–647. 10.1212/WNL.0000000000010674 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. Supporting Information
Data Availability Statement
Raw data were generated at IRCCS Foundation Ca' Granda Ospedale Maggiore Policlinico (Milan, Italy), IRCCS Mondino Foundation (Pavia, Italy), and IRCCS Institute of Neurological Sciences (Bologna, Italy). Derived data supporting the findings of this study are available from the corresponding author on request.