

Abstract
Background
Mast cells (MC) are powerful inflammatory immune sentinel cells that drive numerous allergic, inflammatory, and pruritic disorders when activated. MC‐targeted therapies are approved in several disorders, yet many patients have limited benefit suggesting the need for approaches that more broadly inhibit MC activity. MCs require the KIT receptor and its ligand stem cell factor (SCF) for differentiation, maturation, and survival. Here we describe CDX‐0159, an anti‐KIT monoclonal antibody that potently suppresses MCs in human healthy volunteers.
Methods
CDX‐0159‐mediated KIT inhibition was tested in vitro using KIT‐expressing immortalized cells and primary human mast cells. CDX‐0159 safety and pharmacokinetics were evaluated in a 13‐week good laboratory practice (GLP)‐compliant cynomolgus macaque study. A single ascending dose (0.3, 1, 3, and 9 mg/kg), double‐blinded placebo‐controlled phase 1a human healthy volunteer study (n = 32) was conducted to evaluate the safety, pharmacokinetics, and pharmacodynamics of CDX‐0159.
Results
CDX‐0159 inhibits SCF‐dependent KIT activation in vitro. Fc modifications in CDX‐0159 led to elimination of effector function and reduced serum clearance. In cynomolgus macaques, multiple high doses were safely administered without a significant impact on hematology, a potential concern for KIT inhibitors. A single dose of CDX‐0159 in healthy human subjects was generally well tolerated and demonstrated long antibody exposure. Importantly, CDX‐0159 led to dose‐dependent, profound suppression of plasma tryptase, a MC‐specific protease associated with tissue MC burden, indicative of systemic MC suppression or ablation.
Conclusion
CDX‐0159 administration leads to systemic mast cell ablation and may represent a safe and novel approach to treat mast cell‐driven disorders.
Keywords: CDX‐0159, KIT, mast cell, monoclonal antibody
This study presents the preclinical characterization, safety, pharmacokinetic and pharmacodynamic activity in a placebo‐controlled phase 1a healthy volunteer study of CDX‐0159, a specific and potent anti‐KIT inhibitory monoclonal antibody. CDX‐0159 inhibits SCF‐dependent KIT and mast cell activation. In a dose‐dependent manner, CDX‐0159 induces suppression of plasma tryptase – a marker of mast cell burden – showing a potential as a therapeutic strategy in mast cell‐driven disorders.Abbreviations: CDX‐0159, anti‐KIT inhibitory monoclonal antibody; FcR, Fc receptor; KIT, KIT proto‐oncogene, receptor tyrosine kinase; MRGPRX2, mas‐related G protein‐coupled receptor‐X2; SCF, stem cell factor

1. INTRODUCTION
During normal homeostasis, mast cells (MCs) can exert protective functions against helminth infections, venoms, and may play a role in wound healing and initiating adaptive responses. 1 , 2 , 3 , 4 However, MCs are better known for their role in driving or contributing to numerous allergic, inflammatory, and autoimmune disorders. 5 , 6 MCs are long‐lived innate immune sentinel cells that reside in tissues across the body, particularly at interfaces with the external environment. MCs initiate and perpetuate immune responses when activated by a multitude of endogenous and exogenous stimuli, including allergen‐specific IgE, autoantibodies, complement, toll‐like receptor (TLR) agonists, alarmins, cytokines, neuropeptides, drugs, and venoms. 5 , 7 , 8 Upon stimulation, MCs immediately release pre‐formed mediators stored in granules (proteases, histamine, serotonin, and cytokines), followed by a second wave of eicosanoids (leukotrienes and prostaglandin D2) and a wide array of inflammatory cytokines and chemokines through de novo synthesis. 9 , 10 , 11 These events lead to a rapid inflammatory response characterized by vasodilation, extravasation, smooth muscle contraction, itch, and recruitment of additional immune cell types, which can manifest in both acute and chronic conditions. 10 , 12
MC activation underlies the etiology of allergic reactions and has been strongly implicated in chronic acute and pruritic conditions, neuroinflammatory disorders, pain, fibrosis, and autoimmune diseases. 5 , 6 , 13 , 14 , 15 , 16 , 17 , 18 , 19 , 20 , 21 , 22 Indeed, therapies that inhibit specific MC triggers, such as anti‐IgE (omalizumab) or mediators (antihistamines) have been approved by health authorities and recommended by guidelines as therapies, 23 , 24 although many patients have limited benefit indicating additional MC triggers or mediators are likely involved. Thus, therapies that lead to comprehensive mast cell suppression may result in broader efficacy in indications where MCs contribute to disease pathophysiology. 6 , 23 , 25
The KIT (c‐KIT/CD117) receptor tyrosine kinase and its only ligand stem cell factor (SCF) are master regulators of MC biology. 26 , 27 , 28 KIT is highly expressed throughout the life of a MC and is also expressed in hematopoietic stem cells, melanocytes, interstitial cells of Cajal, germ cells, and a subset of taste receptor cells. 29 , 30 , 31 , 32 , 33 , 34 MCs arise from multipotent hematopoietic stem cell progenitors, entering circulation as immature progenitors and influx into tissues, where they reach maturity. 18 , 26 KIT phosphorylation by soluble or transmembrane SCF expressed in stromal cells (e.g., fibroblasts, keratinocytes, and endothelial cells) and MCs themselves, regulates their differentiation, tissue migration, adhesion, maturation, survival, and modulates their activation. 28 , 35 , 36 , 37 Similarly, exogenous SCF is required to differentiate, mature, and maintain primary MCs grown in vitro. 38 , 39 Mice deficient in either KIT or SCF lack tissue MCs, 40 , 41 and the KIT tyrosine kinase inhibitor (TKI) imatinib significantly reduces the MC burden (and serum tryptase) after long‐term dosing in patients with asthma or chronic myelogenous leukemia. 42 , 43
KIT‐targeting TKIs have received regulatory approval in indications that are generally driven by activating KIT mutations, and typically exhibit relatively low potency for wild‐type KIT and reactivity to related kinases. 44 , 45 By contrast, monoclonal antibodies can be developed with high affinity and selectivity toward extracellular targets. A KIT‐targeting antibody has been shown to efficiently deplete MCs in mice, though this may be through effector function. 46 , 47 Similarly, we have previously demonstrated that an anti‐KIT mAb reduced skin MCs in dogs. 48 Here, we present the preclinical characterization, safety, pharmacokinetic, and pharmacodynamic activity in a placebo‐controlled phase 1a healthy volunteer study of CDX‐0159, a specific and potent anti‐KIT inhibitory mAb.
2. RESULTS
2.1. CDX‐0159 inhibits SCF‐dependent KIT and mast cell activation
CDX‐0159 is a humanized IgG1/κ monoclonal antibody with a modified Fc domain that was derived from CDX‐0158, a predecessor anti‐KIT antibody with an unmodified IgG1/κ backbone. 48 The antibody was developed from mice immunized with the membrane proximal dimerization domains (Ig 4 and 5) of human KIT (huKIT‐D4D5), which are required for KIT activation by SCF but are not directly involved in ligand binding. 49 CDX‐0159 bound to immobilized purified KIT extracellular domain (huKIT‐ECD) and huKIT‐D4D5 by ELISA with similar potency (Figure 1A). While strong reactivity was observed for purified KIT from human, monkey, cat, and dog, no binding to mouse or rat KIT could be measured (Figure S1), indicative of substantial epitope divergence in rodents. Furthermore, CDX‐0159 bound specifically to KIT but not to other related type‐III receptor tyrosine kinases (Figure S2). CDX‐0159 bound to KIT naturally expressed in the surface of megakaryoblastic leukemia M‐07e cells with an EC50 of 153 ± 22 pM (Figure 1B), and completely blocked binding of fluorescently labeled SCF to M‐07e cells with an IC50 value of 118 ± 6 pM (Figure 1C). Similarly, titration of CDX‐0159 on CHO cells stably transfected to express human KIT (CHO‐KIT) inhibited SCF‐dependent KIT phosphorylation (Figure 1D) with an IC50 of 232 ± 4 pM, or 2–3 orders of magnitude more potently than FDA‐approved KIT‐targeting TKIs imatinib, pexidartinib, and avapritinib (IC50 = 931 ± 78 nM, 87 ± 7 nM, and 175 ± 59 nM, respectively). Consistent with the observed reactivity with monkey KIT, CDX‐0159 inhibited KIT phosphorylation in CHO cells expressing cynomolgus KIT with an IC50 of 230 pM (Figure S3). Similar inhibition of SCF‐dependent signaling through KIT, ERK, and AKT was observed in M‐07e cells (Figure S4), as well as cell proliferation when co‐cultured with SCF (IC50 = 1.11 ± 0.15 nM), in a longer term assay (Figure 1E).
FIGURE 1.
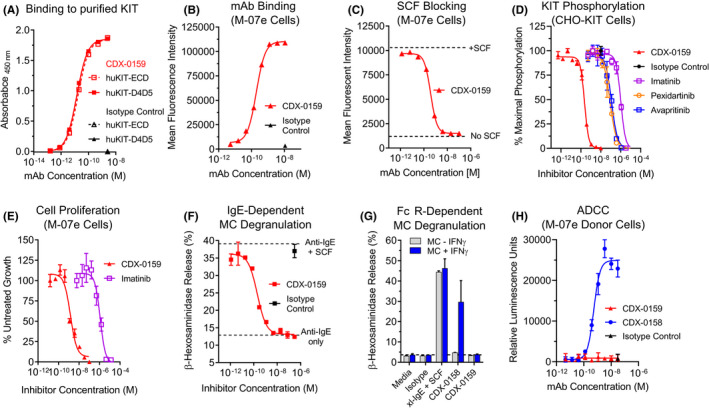
CDX‐0159 Inhibits SCF‐dependent KIT Activation and MC Degranulation. (A) CDX‐0159 binds to purified human KIT extracellular domain (huKIT‐ECD) and a fragment comprised of the membrane proximal dimerization domains Ig4 and Ig5 (huKIT‐D4D5) with indistinguishable potency. Purified KIT proteins were immobilized in ELISA plates followed by titration with CDX‐0159. (B) Binding to KIT‐expressing M‐07e cells was demonstrated by flow cytometry, with an EC50 value of 153 ± 22 pM (C). CDX‐0159 completely blocks binding of 10 nM fluorescently labeled SCF to M‐07e cells with a potency of 118 ± 6 pM. (D) Inhibition of SCF‐dependent KIT tyrosine phosphorylation in CHO cells expressing human KIT is demonstrated for CDX‐0159 and KIT‐targeting TKIs imatinib, pexidartinib, and avapritinib. (E) SCF‐dependent proliferation of M‐07E cells is inhibited by CDX‐0159 more potently than with imatinib. (F) IgE‐dependent MC degranulation as measured by β‐hexosaminidase release is significantly enhanced by addition of 100 ng/mL of SCF. CDX‐0159 fully inhibits SCF‐dependent β‐hexosaminidase release. (G) Fc‐silencing mutations in CDX‐0159 abolish FcγR‐dependent MC activation. In MCs pre‐treated with IFNγ to upregulate FcγRI, CDX‐0158 but not CDX‐0159 induces MC β‐hexosaminidase release. Cross‐linked IgE (xl‐IgE) plus SCF is used as a positive control. (H) CDX‐0159 does not elicit measurable ADCC. Fc‐silencing mutations abolish ADCC observed with CDX‐0158 using a reporter assay using Jurkat cells with an NFAT‐luciferase reporter element under the control of FcγRIII as effector cells, and M‐07e as target cells. All experiments were performed at least 3 independent times. Mean values and S.E.M.s are shown
The activity of CDX‐0159 on primary human MCs differentiated from peripheral blood was also evaluated. Consistent with previously published results SCF addition to human MCs greatly enhanced IgE/FcεR1‐dependent MC degranulation as measured by β‐hexosaminidase release. 50 In this assay, CDX‐0159 titration fully inhibited SCF‐dependent MC degranulation (IC50 = 650 ± 88 pM) (Figure 1F).
2.2. Fc domain mutations abolish FcγR‐dependent MC activation and extend serum half‐life
A significant concern for systemic dosing of KIT‐targeting antibodies is the potential for MC activation and degranulation by KIT‐dependent clustering of MC FcγRs via the antibody Fc domain. 51 Indeed, significant infusion reactions and marked transient elevation of plasma tryptase, a bona fide marker of mast cell degranulation, were observed in patients with gastrointestinal stromal tumors administered CDX‐0158 (NCT02642016) (Table S1, Figure S5). In addition, Fc‐mediated effector functions could result in unwanted effects on other KIT‐bearing cells.
To eliminate the potential for FcγR crosslinking and effector functions, we engineered 3 amino acid substitutions (L234A/L235Q/K322Q) in the Fc region of CDX‐0158 predicted to abolish FcγR binding and expected to similarly impact C1q receptor binding. 52 In addition, we also incorporated a set of modifications (M252Y/S254T/T256E)—herein named YTE—that increase the binding affinity for the neonatal Fc receptor (FcRn) which enhances the pharmacokinetic (PK) properties of the mAb by reducing the rate of in vivo clearance. 53 YTE mutations also reduce binding to FcγRIIIa and reduce ADCC activity. Together, these substitutions in CDX‐0159 completely abolished any measurable binding to all FcγRs (Figure S6). In addition, increased affinity for FcRn was observed. These Fc‐silencing mutations also completely eliminated KIT‐dependent FcγR activation in two different contexts.
The engineered modifications do not affect CDX‐0159 binding or inhibition of the KIT receptor (Figure S7). However, in cultured primary MCs, which express low levels of the high‐affinity FcγRI that can be upregulated by treatment with IFNγ (Figure S8), CDX‐0158 (IgG1 unmutated)‐induced significant degranulation in MCs pre‐treated with IFNγ, comparable in magnitude to that of treatment with cross‐linked IgE with SCF (Figure 1G). By contrast, CDX‐0159 did not elicit any measurable β‐hexosaminidase release under these conditions. Moreover, CDX‐0158‐induced robust antibody‐dependent cell‐mediated cytotoxicity (ADCC) using a FcγRIIIa‐driven NFAT‐luciferase reporter assay and M‐07e as target cells, whereas no ADCC signal could be detected with CDX‐0159 at saturating concentrations (Figure 1H). Taken together, these data demonstrate that the Fc modifications in CDX‐0159 effectively abolish Fc‐dependent effector function and agonist activity leading to MC degranulation.
We also tested the effect of YTE mutations on the pharmacokinetic (PK) properties of CDX‐0159. Consistent with previously published data with antibodies harboring YTE mutations, the pharmacokinetic properties of CDX‐0159 in cynomolgus macaques were indeed enhanced relative to CDX‐0158 as demonstrated by drug exposure levels (24,654 ± 955 day*ug/ml vs. 14,174 ± 866 day*ug/ml) and terminal half‐life (22 ± 5.8 vs. 4.8 ± 3.5 days) (Figure S9).
To support human trials, a GLP‐compliant toxicology study was performed with CDX‐0159 in cynomolgus monkeys with dose levels of 1, 10, and 75 mg/kg administered every two weeks for 13 weeks. The highest dose level of CDX‐0159 provided significant drug exposure maintaining circulating CDX‐0159 concentrations of at least 1950‐fold over the IC50 for in vitro MC KIT inhibition, throughout the dosing and 8‐week recovery period (Figure 2A). CDX‐0159 was well tolerated with no significant changes in body weight, coagulation, clinical chemistry, ophthalmology, electrocardiography, respiratory, or neurological function during the treatment and recovery period. Observations attributed to CDX‐0159‐included minimal increases in the myeloid to erythroid ratio in bone marrow smears in the animals dosed at 10 and 75 mg/kg at the end of dosing (day 85), with evidence of recovery by day 141 despite continued drug exposure in the recovery period (Figure 2B). In addition, recoverable minimal/mild decreases in red blood cell mass parameters were observed (Figure 2C). Hair color changes were noted in some high‐dose‐treated animals, consistent with inhibition of KIT activity in follicular melanocytes. 54
FIGURE 2.
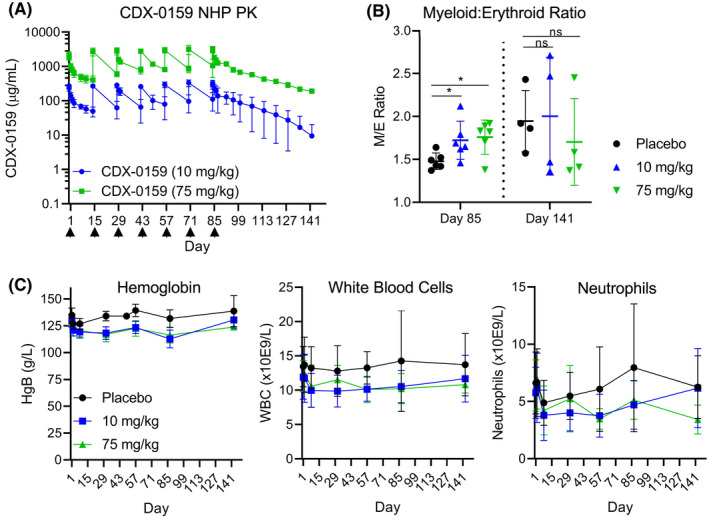
Repeat dosing of CDX‐0159 in non‐human primates does not induce significant myelosuppression. (A) CDX‐0159 administration every two weeks results in high drug levels and exposure at 10 and 75 mg/kg throughout the dosing period 13weeks and post‐treatment recovery (8 weeks). Arrows denote dosing. (B) In bone marrow smears, CDX‐0159‐induced marginal increases in myeloid/erythroid ratios at the end of treatment which were not seen at the end of the recovery period. Cohort means (n = 6 for on‐treatment and n = 4 for recovery animals) and S.E.M. values are shown. p‐values; ns: not significant; *: p < 0.05. (C) Repeat CDX‐0159 administration induces mild rapid decreases in hemoglobin values (left) without further decline despite high and prolonged drug exposure. A similar pattern is observed in total leukocyte count (center) and neutrophils (right), although the total counts exhibit greater intra‐subject variability. Cohort means (n = 10 for on‐treatment and n = 4 for recovery animals) and S.E.M. values are shown. In all cases, no meaningful differences in hematology values between genders were observed
2.3. CDX‐0159 administration to healthy human subjects was generally well tolerated
The safety, pharmacokinetics, and pharmacodynamic activity of CDX‐0159 in human healthy subjects were evaluated in a randomized, double‐blind, placebo‐controlled, Phase 1 single, ascending dose study. A single dose of CDX‐0159 at 0.3, 1, 3, and 9 mg/kg, or placebo was intravenously administered to healthy subjects (Table 1). A CDX‐0159 intravenous infusion was well tolerated at all doses. The most common adverse event (AE)s observed in thirteen out of twenty‐four subjects who received CDX‐0159 were mild, self‐limited infusion reactions that were consistently described as areas of local itching associated with erythema, and induration/hives, which started during the infusions (Table 2). None of the infusion reactions were associated with clinically significant changes in vital signs or difficulty breathing and they all spontaneously resolved within a few hours of completing the infusion without any medications, and no recurrence of symptoms after the initial reactions resolved.
TABLE 1.
CDX‐0159 healthy volunteer patient demographics
| CDX−0159 | Placebo (n = 8) | |||||
|---|---|---|---|---|---|---|
| 0.3 mg/kg (n = 6) | 1 mg/kg (n = 6) | 3 mg/kg (n = 6) | 9 mg/kg (n = 6) | Total (n = 24) | ||
| Age (median, range, years) | 29.5 (24–55) | 27.5 (26–44) | 39.5 (23–55) | 25.5 (20–53) | 29.0 (20–55) | 33.5 (24–51) |
| Gender | ||||||
| Female, N (%) | 2 (33%) | 4 (67%) | 3 (50%) | 3 (50%) | 12 (50%) | 4 (50%) |
| Male, N (%) | 4 (67%) | 2 (33%) | 3 (50%) | 3 (50%) | 12 (50%) | 4 (50%) |
| Race | ||||||
| White, N (%) | 2 (33%) | 3 (50%) | 6 (100%) | 1 (17%) | 12 (50%) | 1 (12%) |
| Black/African American, N (%) | 4 (67%) | 3 (50%) | 0 (0%) | 5 (83%) | 12 (50%) | 7 (88%) |
| Ethnicity | ||||||
| Not Hispanic or Latino | 6 (100%) | 6 (100%) | 6 (100%) | 6 (100%) | 24 (100%) | 8 (100%) |
| Weight (median, range, kg) | 79.0 (60.5–89.9) | 76.8 (55.1–95.4) | 74.3 (57.4–94.1) | 68.7 (59.7–85.8) | 76.0 (55.1 – 95.4) | 71.6 (62.0–84.7) |
| Baseline Tryptase Levels (median, range, ng/ml) | 3.9 (2–21.2) | 2.3 (1.8–3.8) | 3.8 (1.7–8.4) | 3.4 (1.9–4.7) | 3.4 (1.7–21.2) | 3.1 (1.9–6.7) |
TABLE 2.
Treatment‐emergent adverse events occurring in 3 or more subjects
| CDX−0159 | Placebo (n = 8) | |||||
|---|---|---|---|---|---|---|
| 0.3 mg/kg (n = 6) | 1 mg/kg (n = 6) | 3 mg/kg (n = 6) | 9 mg/kg (n = 6) | Total (n = 24) | ||
| Any Event | 2 (33%) | 5 (83%) | 6 (100%) | 6 (100%) | 19 (79%) | 5 (63%) |
| Infusion‐related reaction | 2 (33%) | 5 (83%) | 5 (83%) | 1 (17%) | 13 (54%) | 0 (0%) |
| WBC count decreased | 0 (0%) | 0 (0%) | 0 (0%) | 4 (67%) | 4 (17%) | 1 (13%) |
| Neutrophil count decreased | 0 (0%) | 0 (0%) | 0 (0%) | 4 (67%) | 4 (17%) | 1 (13%) |
| Sensation of Foreign Body in the Throat | 0 (0%) | 0 (0%) | 0 (0%) | 3 (50%) | 3 (13%) | 0 (0%) |
Consistent with observations from the non‐human primate toxicology studies, CDX‐0159 treatment resulted in modest changes in hematologic parameters (Figure 3). Mild decreases in hemoglobin levels generally within the normal range were observed, which rapidly stabilized despite the presence of high plasma drug levels (Figure 3A). The relatively low incidence and magnitude of decreases in hemoglobin support the conclusion that CDX‐0159 had minimal effects on erythropoiesis despite high exposure. In addition, all CDX‐0159 treatment groups showed variable, asymptomatic decreases relative to placebo on circulating neutrophils, which were also reflected in the total leukocyte count and did not further decline during the course of the study (Figure 3B,C). Individual subjects in all of the treatment groups including placebo crossed threshold values per Common Terminology Criteria for Adverse Events (CTCAE) grades with many of the low values observed in subjects with low baseline values.
FIGURE 3.
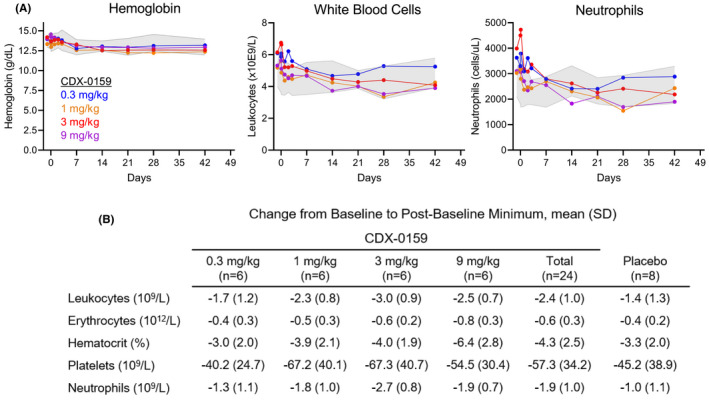
CDX‐0159 induces mild transient decreases in hemoglobin and neutrophil parameters in healthy human subjects. Mean levels of hemoglobin (A) leukocytes (B) and neutrophils (C) after a single dose of CDX‐0159 in healthy subjects are shown. Gray shaded area represents the 95% confidence interval values from placebo‐treated subjects. (D) Maximal mean post‐baseline values for several hematology parameters are shown
CDX‐0159 serum levels were dose proportional and indicative of a PK profile with high exposure and long half‐life at higher doses, consistent with the expected effects of the YTE substitutions (Figure 4A and Table S2). Mean terminal phase half‐life increased from 15 days at 0.3 mg/kg to 32 days at 9 mg/kg, which exceeds the average expected half‐life of 10–21 days for human IgG, 55 and that of CDX‐0158 (6 days at 9 mg/kg) (Table S2).
FIGURE 4.
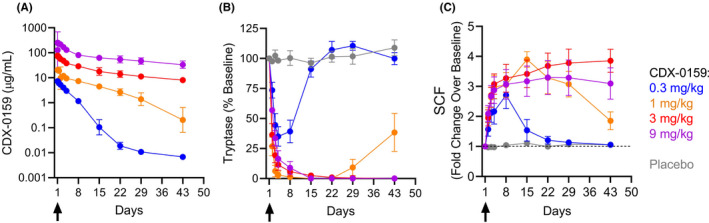
CDX‐0159 induces systemic MC depletion at saturating doses. (A) The PK analysis showed a dose‐dependent increase in levels of CDX‐0159 as expected. Results indicate a PK profile and volume of distribution consistent with those of a monoclonal antibody through this dosing range with evidence of target mediated clearance below approximately 5 μg/ml. Geometric means and 95% confidence intervals are shown. (B) Dose‐dependent reductions in plasma tryptase are observed after a single i.v. dose of CDX‐0159. At the 1 mg/kg dose or above, tryptase suppression below the level of assay detection (1 ng/ml) is observed in all subjects for at least 3 weeks at 1 mg/kg or 6 weeks for 3 and 9 mg/kg. No notable tryptase modulation was observed in placebo‐treated patients. Tryptase values below the lower limit of quantitation were assigned a value of 0 ng/ml. Dose cohort means and S.E.Ms are reported. (C) Dose‐dependent increases in SCF levels were observed, mirroring the pattern of tryptase suppression. No significant modulation of SCF was observed in placebo‐treated subjects. Subjects with baseline levels below the LLoQ (100 pg/ml) were set to 100 pg/ml for normalization. Dose cohort means and S.E.Ms are reported
2.4. CDX‐0159 induces dose‐dependent suppression of plasma tryptase and increases in SCF
Mature tissue MCs store proteolytic enzymes such as tryptase in granules, ready for deployment when they are activated by a variety of stimuli. MCs also release small amounts of tryptase in the steady state resulting in circulating tryptase levels that correlate to the number of MCs in tissues. 56 , 57 Therefore, we measured plasma tryptase levels as an indicator of the potential impact of CDX‐0159 on tissue MC numbers (Figure 4B). Baseline tryptase levels (4.0 ± 3.8 ng/ml) were within the normal expected range, with the exception of one subject who had elevated (21.2 ng/ml) levels at baseline. Tryptase reduction was manifest in all subjects dosed with CDX‐0159 at 24 h after the infusion. At the lowest dose of 0.3 mg/kg, plasma tryptase was reduced by approximately 50%, including the subject with high baseline tryptase. At higher doses, tryptase levels were reduced below the lower limit of assay quantitation (LLoQ) (1 ng/ml) in all CDX‐0159 treated subjects for at least 3 weeks at 1 mg/kg, 6–14 weeks at 3 mg/kg and over 18 weeks at 9 mg/kg (Figure 4B and Figure S10). No significant change in tryptase was observed in the placebo group. These data are consistent with a sustained systemic impact of CDX‐0159 on MC number.
In a subset of subjects who experienced infusion reactions plasma tryptase levels from samples collected during or shortly after the infusion were slightly elevated (3.58 ± 0.45 ng/ml) relative to pre‐dose values (3.55 ± 0.50 ng/ml) consistent with a low level of MC activation in those subjects (Figure S5) and the observed mild infusion reactions. However, the magnitude of the tryptase elevations in CDX‐0159‐treated subjects was lower than that observed in patients with gastrointestinal stromal tumors administered CDX‐0158 (Figure S5).
CDX‐0159 allosterically blocks binding of SCF to KIT, which may result in an elevation of SCF plasma levels from accumulating unbound SCF. We therefore measured plasma SCF as another pharmacodynamic biomarker of KIT engagement and inhibition by CDX‐0159 (Figure 4C). We observed dose‐proportional increases in SCF levels that mirrored the effects of CDX‐0159 on plasma tryptase. Together with the effect of the antibody on tryptase levels, these data provide additional evidence of systemic KIT saturation with CDX‐0159 at doses of 1 mg/kg and above.
3. DISCUSSION
MCs are powerful tissue‐resident inflammatory cells that respond to a wide array of endogenous and environmental stimuli and are the key effector cells in many allergic, pruritic, and inflammatory disorders. MC‐targeting strategies have generally focused on neutralizing known triggers (IgE) or individual mediators (histamine and leukotrienes), with inherently limited efficacy. By targeting the KIT receptor tyrosine kinase, which is central to MC differentiation and survival, broader and systemic MC suppression may be achieved in disorders where MCs play an important role, independent of the causative trigger, or dominant mediators.
The data presented herein demonstrate that the anti‐KIT mAb CDX‐0159 is a potent inhibitor of KIT signaling and MC activation induced by SCF. Moreover, in healthy subjects, CDX‐0159 induces rapid, profound, and durable tissue MC suppression as reflected by reductions in plasma tryptase levels. The duration of tryptase suppression was clearly dose‐dependent, and levels remain below the level of quantitation for at least 3, 6, and 18 weeks after a single dose of 1, 3, and 9 mg/kg, respectively. The observed tryptase reductions follow similar kinetics to dose‐dependent increase in circulating SCF, indicative of systemic KIT saturation at higher doses. These effects on circulating tryptase are unprecedented, as small molecule KIT inhibitors have only partial effects on tryptase levels that require months to achieve. 43 We attribute this dramatic effect to the much higher and more specific KIT antagonist activity of CDX‐0159 on MCs.
Importantly, the administration of CDX‐0159 was not accompanied by any severe infusion reactions. The related antibody that targeted KIT with an unmodified IgG1 Fc domain (CDX‐0158) resulted in significant (Grade 2 and several Grade 3) infusion reactions (Table S1), despite pre‐medications, which were clearly related to MC activation as demonstrated by rapid increases in circulating tryptase levels (Figure S5). Similarly, the severe infusion reactions reported for another anti‐KIT mAb with unmodified IgG1 Fc domain were demonstrated to be the result of FcγR crosslinking and MC activation. 51 The Fc‐silencing mutations engineered in CDX‐0159 clearly had a profound effect in eliminating Fc‐mediated MC activation and limiting the magnitude of infusion reactions. In this study, only half of the CDX‐0159‐dosed patients experienced any infusion reactions, which were mild and resolved rapidly without any medication or without decreasing the infusion rate.
The importance of KIT/SCF signaling in hematopoiesis is well documented and an important safety consideration for KIT antagonists. Overall, the effects of CDX‐0159 on circulating hematology parameters were clinically asymptomatic and mostly maintained within normal range, consistent with observations from our multi‐dose toxicology study with observations through 21 weeks. Surprisingly, the most prominent effect in human volunteers was mild decreases in neutrophil levels, which appeared soon after dosing and did not decrease further over time, and while saturating drug levels were still present, suggesting maximal effect on neutrophil decreases were observed as inferred from PK and pharmacodynamic (PD) measurements. The mechanistic reason for the mild effect in neutrophils is unclear and could reflect interference with certain aspects of KIT‐dependent neutrophil differentiation or survival, or indirect modulation of neutrophil tissue distribution as a result of MC suppression. Interestingly, decreases in neutrophil count have also been reported with other approved mAbs that affect MC activity such as the anti‐IgE omalizumab and the anti‐IL4Rα mAb dupilumab. 23 , 58 , 59 In addition to its role in hematopoiesis, KIT signaling is important in other processes and has been described to have a role in hair pigmentation and spermatogenesis. 31 , 32 , 60 , 61 Importantly, the data from preclinical models suggest these effects are fully reversible. We observed patches of hair lightening in our 21 weeks, high‐dose monkey study and this finding could be expected in humans as this has been observed with other KIT inhibitors. 54 , 62 In our healthy volunteer study, there were no reports of hair color changes. The effect of CDX‐0159 on fertility parameters will be studied in relevant preclinical models.
Despite being a single‐dose study, the high exposure achieved with CDX‐0159 resulted in long‐lasting pharmacodynamic effects. For example, the terminal half‐life of CDX‐0159 at a single 3 mg/kg dose was 23 days and was accompanied with tryptase suppression for >6 weeks. The extended half‐life is attributed to modifications engineered into the Fc domain, which increase binding to the FcRn and reduce clearance of the antibody. The half‐life for CDX‐0159 in this study was approximately five‐fold longer than what was observed for CDX‐0158 in a phase 1 clinical trial (Table S2).
The unprecedented and dramatic decrease in circulating tryptase observed in this study is expected to reflect a concomitant decrease in tissue MC numbers. In a nonclinical study with healthy dogs, CDX‐0158 depleted the majority of skin MCs within 2 weeks, with evidence of apoptosis in the remaining MCs, although this may be at least partially driven by effector function. 48 Furthermore, studies with imatinib and avapritinib have demonstrated partial reductions in both tryptase and mast cells in several studies when given at high doses for an extended time. 42 , 43 , 63 Emerging data from an ongoing study with CDX‐0159 in chronic inducible urticaria (NCT04548869) confirm that CDX‐0159 administration leads to significant depletion of skin MCs, consistent with the reduction in circulating tryptase shown in this manuscript.
The long‐term consequence of mast cell ablation or suppression remains to be determined. MCs are thought to play a predominant role in Th2 inflammatory responses, although they can also release Th1 cytokines and pro‐fibrotic cytokines such as TGF‐β or fibroblast growth factors. Mice lacking MCs show no overt phenotype but take longer to clear certain parasitic infections, consistent with MC involvement in Th2 immunity. 1 , 64 In addition, murine MCs have been implicated in combating bacterial infections, and in wound healing, although their exact contribution to these processes is still not fully understood. 65 , 66 Chronic dosing with imatinib, which partially depletes MCs, or omalizumab, which eliminates an important regulator of MCs, has not shown any toxicities that can be directly ascribed to MC impairment. Importantly, other mAbs that inhibit the activity of key Th2 cytokines such as IL‐4, IL‐13, IL‐5, or eliminate eosinophils have demonstrated favorable long‐term safety profiles to date. 15 , 67 , 68 , 69 , 70
MCs have been directly implicated in the pathogenesis of numerous inflammatory disorders and are the key effector cell in chronic urticarias. Mast cell activation by known triggers (inducible urticaria) or unidentifiable triggers (spontaneous urticaria) leads to the formation of itchy wheals and/or angioedema, and a significantly impaired quality of life. 71 , 72 While inhibition of specific triggers, such as IgE or mediators, such as histamine leads to limited response rates, it is expected that depletion or metabolic suppression of MC may have broader impact in chronic urticarias and other MC‐driven disorders. As such, the activity and safety of CDX‐0159 are currently being explored in both chronic inducible urticaria and chronic spontaneous urticaria (NCT04548869 and NCT04538794).
The present study demonstrates for the first time that potent and specific KIT inhibition with a monoclonal antibody can achieve profound suppression of tissue MCs without significantly impacting hematopoiesis. This surprising dichotomy may reflect KIT redundancy with other pathways in the stem cell niche or may be due to a yet unappreciated mechanistic aspect of CDX‐0159. For instance, it has been shown that hematopoietic stem cells (HSCs) and MCs have different thresholds for SCF/KIT signaling, as evidenced by SCF variants with partial agonism that can fully activate HSCs but not MCs. 73 Importantly, the approach described herein circumvents limitations inherent to KIT‐targeting TKIs, which typically suffer from relatively lower potency and poor tolerability due to reactivity with multiple other kinases, hindering their use in chronic inflammatory disorders.
In summary, the preclinical and clinical safety and pharmacodynamic data presented in this manuscript show that CDX‐0159 is well tolerated and impacts tissue mast cells systemically, indicating it may have broad utility in diseases with MC involvement. Importantly, this potent MC‐targeted agent will be an invaluable tool to elucidate the true role of MCs in numerous disorders, which has previously been difficult to discern.
Conflict of Interest
DA, MBM, LC, JG, AC, LAV, PAM, LJT, TRH, TK, DY, EC, and MHC are full‐time employees of Celldex Therapeutics. SBS is a full‐time employee of Boehringer‐Ingelheim. MM reports grants and/or personal fees from Allakos, Amgen, Aralez, ArgenX, AstraZeneca, Celldex, Centogene, CSL Behring, FAES, Genentech, GIInnovation, Innate Pharma, Kyowa Kirin, Leo Pharma, Lilly, Menarini, Moxie, Novartis, Roche, Sanofi/Regeneron, Third HarmonicBio, UCB, and Uriach. MK is a full‐time employee of Altasciences, which has received research grant/funding (institution) from Actelion Pharmaceuticals, Acurx Pharmaceuticals, Bioxcel Therapeutics, Grifols, Jazz Pharmaceuticals, Novus Therapeutics, Pfizer, DynPort Vaccine Company, Novo Nordisk, FDA/NIH, and ViroDefense. MK holds a leadership role at Altasciences. JG and RG are inventors in patent applications No: 63/140,642 and 63/140,621.
AUTHOR CONTRIBUTIONS
DA, RG, MBM, JG, LAV, LJT, TRH, and TK contributed to nonclinical experimental design, analysis, and interpretation. DA, MM, RG, LJT, TRH, TK, DY, EC, MK, and MHC contributed to the design, execution, analysis, and interpretation of clinical data. MK was the Principal Investigator for the conduct of the study. SBS, MBM, LC, AC, LAV, and PAM contributed to experimental execution and data analysis. DA, MM, RG, LJT, TRH, TK, DY, EC, MK, and MHC drafted the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
We thank patients, volunteers, and investigators for participating in the CDX‐0158 and CDX‐0159 clinical trials. We would like to thank Celldex employees for critical review of the manuscript. We wish to acknowledge the important contributions of Elsa Paradise, Lynn Aneiro, Charlene Micklus, Theresa Belotti, Eric Forsberg, Ami Fields, Montu Patel, and Nick Bahns to this work for their clinical and nonclinical operational and technical support. The authors would like to acknowledge the contributions of André Dumais (Charles River Laboratories).
Alvarado D, Maurer M, Gedrich R, et al. Anti‐KIT monoclonal antibody CDX‐0159 induces profound and durable mast cell suppression in a healthy volunteer study. Allergy. 2022;77:2393–2403. doi: 10.1111/all.15262
Funding information
This work was sponsored by Celldex Therapeutics.
REFERENCES
- 1. Varricchi G, Rossi FW, Galdiero MR, et al. Physiological roles of mast cells: collegium internationale allergologicum update 2019. Int Arch Allergy Immunol. 2019;179:247‐261. [DOI] [PubMed] [Google Scholar]
- 2. Dudeck A, Köberle M, Goldmann O, et al. Mast cells as protectors of health. Journal of Allergy and Clinical Immunology. 2019;144:S4‐S18. [DOI] [PubMed] [Google Scholar]
- 3. Dahlin JS, Maurer M, Metcalfe DD, Pejler G, Sagi‐Eisenberg R, Nilsson G. The ingenious mast cell: contemporary insights into mast cell behavior and function. Allergy. 2021;77(1):83‐99. [DOI] [PubMed] [Google Scholar]
- 4. Maurer M, Köberle M, Metz M, Biedermann T. Mast cells: Promoters of health and modulators of disease. Journal of Allergy and Clinical Immunology. 2019;144:S1‐S3. [DOI] [PubMed] [Google Scholar]
- 5. Jönsson F, Mast DM. Mast cells and company. Front Immun. 2012;3. 10.3389/fimmu.2012.00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Church MK, Kolkhir P, Metz M, Maurer M. The role and relevance of mast cells in urticaria. Immunol Rev. 2018;282:232‐247. [DOI] [PubMed] [Google Scholar]
- 7. Caslin HL, Kiwanuka KN, Haque TT, et al. Controlling mast cell activation and homeostasis: work influenced by bill paul that continues today. Front Immunol. 2018;9:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Theoharides TC, Alysandratos K‐D, Angelidou A, et al. Mast cells and inflammation. Biochimica Et Biophysica Acta (BBA) ‐ Molecular Basis of Disease. 2012;1822(1):21‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gaudenzio N, Sibilano R, Marichal T, et al. Different activation signals induce distinct mast cell degranulation strategies. J Clin Invest. 2016;126:3981‐3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elieh Ali Komi D, Wöhrl S, Bielory L. Mast cell biology at molecular level: a comprehensive review. Clinic Rev Allerg Immunol. 2020;58:342‐365. [DOI] [PubMed] [Google Scholar]
- 11. Halova I, Rönnberg E, Draberova L, Vliagoftis H, Nilsson GP, Draber P. Changing the threshold‐Signals and mechanisms of mast cell priming. Immunol Rev. 2018;282:73‐86. [DOI] [PubMed] [Google Scholar]
- 12. Paivandy A, Pejler G. Novel strategies to target mast cells in disease. J Innate Immun. 2021;13:131‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tordesillas L, Berin MC, Sampson HA. Immunology of food allergy. Immunity. 2017;47:32‐50. [DOI] [PubMed] [Google Scholar]
- 14. Tomasiak‐Łozowska MM, Klimek M, Lis A, Moniuszko M, Bodzenta‐Łukaszyk A. Markers of anaphylaxis – a systematic review. Advances in Medical Sciences. 2018;63:265‐277. [DOI] [PubMed] [Google Scholar]
- 15. Maurer M, Khan DA, Elieh Ali Komi D, Kaplan AP. Biologics for the use in chronic spontaneous urticaria: when and which. J Allergy Clin Immunol In Pract. 2021;9:1067‐1078. [DOI] [PubMed] [Google Scholar]
- 16. Xu Y, Chen G. Mast cell and autoimmune diseases. Mediators Inflamm. 2015;2015:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Siiskonen H, Harvima I. Mast cells and sensory nerves contribute to neurogenic inflammation and pruritus in chronic skin inflammation. Front Cell Neurosci. 2019;13:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Méndez‐Enríquez E, Hallgren J. Mast cells and their progenitors in allergic asthma. Front Immunol. 2019;10:821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bradding P, Arthur G. Mast cells in asthma ‐ state of the art. Clin Exp Allergy. 2016;46:194‐263. [DOI] [PubMed] [Google Scholar]
- 20. Wang F, Yang T‐LB, Kim BS. The return of the mast cell: new roles in neuroimmune itch biology. J Invest Dermatol. 2020;140:945‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Héron A, Dubayle D. A focus on mast cells and pain. J Neuroimmunol. 2013;264:1‐7. [DOI] [PubMed] [Google Scholar]
- 22. Strattan E, Hildebrandt GC. Mast cell involvement in fibrosis in chronic graft‐versus‐host disease. IJMS. 2021;22:2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maurer M, Rosén K, Hsieh H‐J, et al. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. N Engl J Med. 2013;368:924‐935. [DOI] [PubMed] [Google Scholar]
- 24. Zuberbier T, Abdul Latiff AH, Abuzakouk M, et al. The International EAACI/GA2LEN/EuroGuiDerm/APAAACI guideline for the definition, classification, diagnosis and management of urticaria. Allergy. 2022;77(3):734‐766. doi: 10.1111/all.15090 [DOI] [PubMed] [Google Scholar]
- 25. Kolkhir P, Elieh‐Ali‐Komi D, Metz M, Siebenhaar F, Maurer M. Understanding human mast cells: lesson from therapies for allergic and non‐allergic diseases. Nat Rev Immunol. 2021;5. 10.1038/s41577-021-00622-y [DOI] [PubMed] [Google Scholar]
- 26. Valent P, Akin C, Hartmann K, et al. Mast cells as a unique hematopoietic lineage and cell system: from Paul Ehrlich’s visions to precision medicine concepts. Theranostics. 2020;10:10743‐10768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Draber P, Halova I, Polakovicova I, Kawakami T. Signal transduction and chemotaxis in mast cells. Eur J Pharmacol. 2016;778:11‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lennartsson J, Rönnstrand L. Stem cell factor receptor/c‐kit: from basic science to clinical implications. Physiol Rev. 2012;92:1619‐1649. [DOI] [PubMed] [Google Scholar]
- 29. Broudy VC. Stem cell factor and hematopoiesis. Blood. 1997;90:1345‐1364. [PubMed] [Google Scholar]
- 30. Lammie A, Drobnjak M, Gerald W, Saad A, Cote R, Cordon‐Cardo C. Expression of c‐kit and kit ligand proteins in normal human tissues. J Histochem Cytochem. 1994;42:1417‐1425. [DOI] [PubMed] [Google Scholar]
- 31. Cardoso HJ, Figueira MI, Correia S, Vaz CV, Socorro S. The SCF/c‐KIT system in the male: Survival strategies in fertility and cancer: SCF/c‐KIT System in Male Fertility and Cancer. Mol Reprod Dev. 2014;81:1064‐1079. [DOI] [PubMed] [Google Scholar]
- 32. Botchkareva NV, Khlgatian M, Jack Longley B, Botchkarev VA, Gilchrest BA. SCF/c‐kit signaling is required for cyclic regeneration of the hair pigmentation unit. FASEB J. 2001;15:645‐658. [DOI] [PubMed] [Google Scholar]
- 33. Ashman LK. The biology of stem cell factor and its receptor C‐kit. Int J Biochem Cell Biol. 1999;31:1037‐1051. [DOI] [PubMed] [Google Scholar]
- 34. Choo E, Dando R. The c‐kit receptor tyrosine kinase marks sweet or umami sensing T1R3 positive adult taste cells in mice. Chem Percept. 2021;14:41‐46. [Google Scholar]
- 35. Miyazawa K, Williams D, Gotoh A, Nishimaki J, Broxmeyer H, Toyama K. Membrane‐bound Steel factor induces more persistent tyrosine kinase activation and longer life span of c‐kit gene‐encoded protein than its soluble form. Blood. 1995;85:641‐649. [PubMed] [Google Scholar]
- 36. Rasky A, Habiel DM, Morris S, et al. Inhibition of the stem cell factor 248 isoform attenuates the development of pulmonary remodeling disease. Am J Physiol Lung Cell Mol Physiol. 2020;318:L200‐L211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wershil BK. The rat c‐kit ligand, stem cell factor, induces c‐kit receptor‐ dependent mouse mast cell activation in vivo. Evidence that signaling through the c‐kit receptor can induce expression of cellular function. J Exp Med. 1992;175:245‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rådinger M, Jensen BM, Kuehn HS, Kirshenbaum A, Generation GAM. Isolation, and Maintenance of Human Mast Cells and Mast Cell Lines Derived from Peripheral Blood or Cord Blood. In: Coligan JE, Bierer BE, Margulies DH, Shevach EM, Strober W, editors. Current Protocols in Immunology. John Wiley & Sons, Inc. 2010. im0737s90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Elst J, Sabato V, van der Poorten MLM , et al. Peripheral blood cultured mast cells: phenotypic and functional outcomes of different culture protocols. J Immunolo Methods 2021;492:113003. [DOI] [PubMed] [Google Scholar]
- 40. Zsebo KM, Williams DA, Geissler EN, et al. Stem cell factor is encoded at the SI locus of the mouse and is the ligand for the c‐kit tyrosine kinase receptor. Cell. 1990;63:213‐224. [DOI] [PubMed] [Google Scholar]
- 41. Grimbaldeston MA, Chen C‐C, Piliponsky AM, Tsai M, Tam S‐Y, Galli SJ. Mast cell‐deficient W‐sash c‐kit mutant KitW‐sh/W‐sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. 2005;167:835‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cerny‐Reiterer S, Rabenhorst A, Stefanzl G, et al. Long‐term treatment with imatinib results in profound mast cell deficiency in Ph+ chronic myeloid leukemia. Oncotarget. 2015;6(5):3071‐3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cahill KN, Katz HR, Cui J, et al. KIT inhibition by imatinib in patients with severe refractory asthma. N Engl J Med. 2017;376:1911‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Abbaspour Babaei M, Kamalidehghan B, Saleem M, Huri HZ, Ahmadipour F. Receptor tyrosine kinase (c‐Kit) inhibitors: a potential therapeutic target in cancer cells. Drug Des Devel Ther. 2016;10:2443‐2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046‐1051. [DOI] [PubMed] [Google Scholar]
- 46. Brandt EB, Strait RT, Hershko D, et al. Mast cells are required for experimental oral allergen–induced diarrhea. J Clin Invest. 2003;112:1666‐1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chhabra A, Ring AM, Weiskopf K, Schnorr PJ, Gordon S, Le AC, et al. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci Transl Med. 2016;8:351ra105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. London CA, Gardner HL, Rippy S, et al. KTN0158, a humanized anti‐KIT monoclonal antibody, demonstrates biologic activity against both normal and malignant canine mast cells. Clin Cancer Res. 2017;23:2565‐2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yuzawa S, Opatowsky Y, Zhang Z, Mandiyan V, Lax I, Schlessinger J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell. 2007;130:323‐334. [DOI] [PubMed] [Google Scholar]
- 50. Gilfillan AM, Beaven MA. Regulation of mast cell responses in health and disease. Crit Rev Immunol. 2011;31:475‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. L’Italien L, Orozco O, Abrams T, et al. Mechanistic insights of an immunological adverse event induced by an anti‐KIT antibody drug conjugate and mitigation strategies. Clin Cancer Res. 2018;24:3465‐3474. [DOI] [PubMed] [Google Scholar]
- 52. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5. 10.3389/fimmu.2014.00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006;281:23514‐23524. [DOI] [PubMed] [Google Scholar]
- 54. Moss KG, Toner GC, Cherrington JM, Mendel DB, Laird AD. Hair depigmentation is a biological readout for pharmacological inhibition of KIT in mice and humans. J Pharmacol Exp Ther. 2003;307:476‐480. [DOI] [PubMed] [Google Scholar]
- 55. Booth BJ, Ramakrishnan B, Narayan K, et al. Extending human IgG half‐life using structure‐guided design. mAbs. 2018;1–13. 10.1080/19420862.2018.1490119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Caughey G. Tryptase genetics and anaphylaxis. J Allergy Clin Immunol. 2006;117:1411‐1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Caughey GH. Mast cell proteases as pharmacological targets. Eur J Pharmacol. 2016;778:44‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Acer E, Kaya Erdogan H, Yüksel Çanakçı N, Saracoglu ZN. The effect of omalizumab on hematological and inflammatory parameters in patients with chronic spontaneous urticaria. Cutan Ocul Toxicol. 2019;38:5‐8. [DOI] [PubMed] [Google Scholar]
- 59. Çildağ S, Şentürk T. The effect of omalizumab treatment on IgE and other immunoglobulin levels in patients with chronic spontaneous urticaria and its association with treatment response. Pdia. 2018;35:516‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Alexeev V, Yoon K. Distinctive role of the cKit receptor tyrosine kinase signaling in mammalian melanocytes. J Invest Dermatol. 2006;126:1102‐1110. [DOI] [PubMed] [Google Scholar]
- 61. Yoshinaga K, Nishikawa S, Ogawa M, et al. Role of c‐kit in mouse spermatogenesis: identification of spermatogonia as a specific site of c‐kit expression and function. Development. 1991;113(2):689‐699. [DOI] [PubMed] [Google Scholar]
- 62. Ricci F, De Simone C, Del Regno L, Peris K. Drug‐induced hair colour changes. Eur J Dermatol. 2016;26:531‐536. [DOI] [PubMed] [Google Scholar]
- 63. Gilreath J, Tchertanov L, Deininger M. Novel approaches to treating advanced systemic mastocytosis. CPAA. 2019;11:77‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Herbert D, Douglas B, Zullo K. Group 2 innate lymphoid cells (ILC2): type 2 immunity and helminth immunity. IJMS. 2019;20:2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zimmermann C, Troeltzsch D, Giménez‐Rivera VA, et al. Mast cells are critical for controlling the bacterial burden and the healing of infected wounds. Proc Natl Acad Sci USA. 2019;116:20500‐20504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Weller K, Foitzik K, Paus R, et al. Mast cells are required for normal healing of skin wounds in mice. FASEB J. 2006;20:2366‐2368. [DOI] [PubMed] [Google Scholar]
- 67. Guttman‐Yassky E, Bissonnette R, Ungar B, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143:155‐172. [DOI] [PubMed] [Google Scholar]
- 68. Bagnasco D, Caminati M, Ferrando M, et al. Anti‐IL‐5 and IL‐5Ra: efficacy and safety of new therapeutic strategies in severe uncontrolled asthma. Biomed Res Int. 2018;2018:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ortega HG, Brusselle GG, Humbert M, Yancey SW. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198‐1207. [DOI] [PubMed] [Google Scholar]
- 70. Xiao X, Lin L, Zhu C, et al. Efficacy and safety of nemolizumab for treatment of adult atopic dermatitis: a meta‐analysis of randomized clinical trials. J Investig Allergol Clin Immunol. 2021;31:190‐192. [DOI] [PubMed] [Google Scholar]
- 71. Zuberbier T, Urticaria MM. Current opinions about etiology, diagnosis and therapy. Acta Derm Venereol. 2007;87(3):196‐205. [DOI] [PubMed] [Google Scholar]
- 72. Magerl M, Altrichter S, Borzova E, et al. The definition, diagnostic testing, and management of chronic inducible urticarias ‐ The EAACI/GA 2 LEN/EDF/UNEV consensus recommendations 2016 update and revision. Allergy. 2016;71:780‐802. [DOI] [PubMed] [Google Scholar]
- 73. Ho CCM, Chhabra A, Starkl P, et al. Decoupling the functional pleiotropy of stem cell factor by tuning c‐Kit signaling. Cell. 2017;168:1041‐1052.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material