

SUMMARY
Translating ribosome affinity purification (TRAP) utilizes transgenic plants expressing a ribosomal protein fused to a tag for affinity co‐purification of ribosomes and the mRNAs that they are translating. This population of actively translated mRNAs (translatome) can be interrogated by quantitative PCR or RNA sequencing. Condition‐ or cell‐specific promoters can be utilized to isolate the translatome of specific cell types, at different growth stages and/or in response to environmental variables. While advantageous for revealing differential expression, this approach may not provide sufficient sensitivity when activity of the condition/cell‐specific promoter is weak, when ribosome turnover is low in the cells of interest, or when the targeted cells are ephemeral. In these situations, expressing tagged ribosomes under the control of these specific promoters may not yield sufficient polysomes for downstream analysis. Here, we describe a new TRAP system that employs two transgenes: One is constitutively expressed and encodes a ribosomal protein fused to one fragment of a split green fluorescent protein (GFP); the second is controlled by a stimulus‐specific promoter and encodes the second GFP fragment fused to an affinity purification tag. In cells where both transgenes are active, the purification tag is attached to ribosomes by bi‐molecular folding and assembly of the split GFP fragments. This approach provides increased sensitivity and better temporal resolution because it labels pre‐existing ribosomes and does not depend on rapid ribosome turnover. We describe the optimization and key parameters of this system, and then apply it to a plant–pathogen interaction in which spatial and temporal resolution are difficult to achieve with current technologies.
Keywords: translation, ribosome, Arabidopsis, Hyaloperonospora arabidopsidis, technical advance
Significance Statement
Translating ribosome affinity purification (TRAP) has been modified to allow with increased sensitivity the isolation of RNA from sets of cells in which the activity of condition/cellspecific promoters is weak, ribosome turnover is low, or cells whose nature is ephemeral. Based on the use of a split linker constituted of the GFP driven by a pathogen‐inducible promoter, this new TRAP system enabled efficient isolation of translated RNA from pathogen‐infected leaf cells.
INTRODUCTION
Translating ribosome affinity purification (TRAP) was pioneered in plants (Kage et al., 2020; Mustroph, Zanetti, et al., 2009; Zanetti et al., 2005) and mice (Heiman et al., 2008) as a technique to reproducibly isolate actively translated mRNAs from genetically defined populations of cells. TRAP utilizes transgenic organisms expressing a tagged ribosomal protein that is incorporated into ribosomes by the cell. Cell constituents are fractionated under conditions that preserve associations between ribosomes and the mRNAs being translated (i.e., polysomes). Tagged polysomes are then purified by immunoprecipitation (IP) using an antibody against the tag. RNA extracted from the IP polyribosomes contains the translatome, i.e., the collection of mRNAs that are associated with and being translated by ribosomes (sometimes referred to as ribosome‐nascent chain complex‐bound mRNAs). Conventional transcriptomics (RNA sequencing [RNAseq], microarrays) typically reports the abundance of all polyadenylated transcripts. Thus, traditional transcriptomics cannot capture the control that an organism exerts on translation (Adams, 2008). Contrastingly, the translatome correlates more closely to the proteome (Wang et al., 2013) and can therefore provide a more accurate assessment of physiological status. In addition, conventional transcriptomic analyses are typically performed using mRNA purified from homogenized organs comprised of various differentiated cell types, making it impossible to distinguish the transcriptomes of various cell types that comprise the organ. On the other hand, TRAP can be customized to reveal the translatome in specific cell types by expressing the tagged ribosomal protein under the control of a cell type‐specific promoter. Similarly, responses to environmental stimuli could be studied by expressing the ribosomal protein using a stimulus‐dependent promoter. In these approaches, the organ is harvested and the tagged polysomes are purified from the mixed population of tagged and untagged polysomes (Mustroph, Juntawong, & Bailey‐Serres, 2009) (Figure 1a). This approach enriches for polysomes from the cell type of interest because the tagged ribosomal protein is expressed only in the specific cell type(s) or conditions in which the promoter is active. mRNA is then isolated from the immunopurified polysomes and subjected to RNAseq or other analyses, yielding cell type‐ or stimulus‐specific translatomes.
Figure 1.
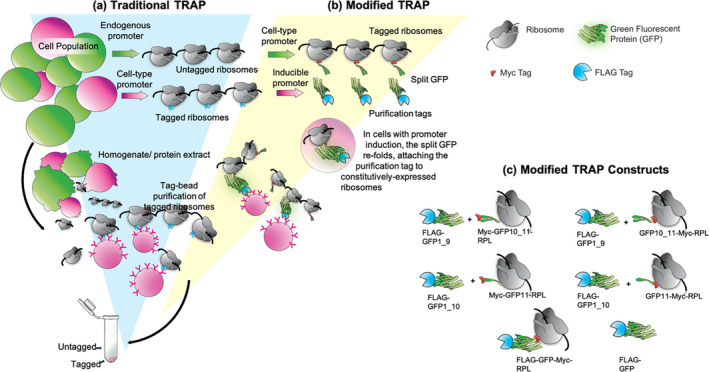
Schematic of traditional and modified translating ribosome affinity purification (TRAP).
(a) In traditional TRAP, a promoter drives expression of a ribosomal protein fused to an epitope tag. The tag allows for affinity purification of polyribosomes and associated RNAs. Placing the tagged ribosomal protein gene under the control of a conditionally active promoter (red) can provide enrichment of ribosomes from specific cells of interest (red) in a complex mixture (green).
(b) The split green fluorescent protein (GFP) TRAP system employs two transgenes: One gene is driven by a condition‐specific promoter that drives the expression of the purification tag and part of a split GFP linker protein (red). The second gene is driven by a constitutive promoter and encodes the remaining portion of the split GFP, fused to an epitope tag and the ribosomal protein for incorporation into a translating ribosome (green). Assembly of the split GFP attaches the purification tag to pre‐existing ribosomes, thereby increasing the proportion of tagged ribosomes in cells of interest, compared to the traditional one‐gene system that requires replacement of endogenous, untagged ribosomes with those that incorporate the tagged protein.
(c) Diagrams representing the constructs used for optimization via transient expression in Nicotiana benthamiana. In these experiments, different splits of GFP and different positions of a constitutive Myc tag were compared to control constructs (FLAG‐GFP‐Myc‐RPL and FLAG‐GFP). [Colour figure can be viewed at wileyonlinelibrary.com]
Applications of TRAP to many organisms, including plants, have shown that translatome analysis can reveal information that is not present in the transcriptome. For example, studies of the translatome in Arabidopsis seedlings exposed to a period of hypoxia identified a specific group of mRNAs that allows for acclimation to hypoxia (Mustroph, Zanetti, et al., 2009). These mRNAs were enriched in several specific cell types and encode proteins aiding in stress tolerance, development, and metabolism (Mustroph, Zanetti, et al., 2009). TRAP analysis of plant–bacteria interactions demonstrated that translational control is a significant component of immune response regulation and delineated a link between metabolism and plant immune responses via specific changes in translation (Meteignier et al., 2017; Xu et al., 2017; Yoo et al., 2020). A recent TRAP‐based study using Arabidopsis showed that a jasmonic acid mutant displayed no difference with the wild type at the level of the transcriptome, but identified a large difference in association of mRNA with ribosomes, providing an explanation for the greatly decreased proteome and strong phenotypes researchers had previously noted under the same conditions (Kimberlin et al., 2021). These studies suggest that additional comparisons of transcriptome and translatome induced by biotic or abiotic stresses will reveal new information that would not be evident with transcriptomic data alone.
Because ribosome biogenesis in plants is not completely understood and ribosomes appear to be relatively stable with a turnover rate of approximately 6–8 days (Salih, Duncan, Li, Trösch, & Millar, 2020), it may be disadvantageous to directly express the ribosomal protein‐purification tag fusion under the control of stimulus‐specific promoters. Low ribosome turnover would retard the presence of a sufficient share of tagged active ribosomes with newly expressed tagged‐ribosomal proteins, hampering the purification of required amounts of mRNA by TRAP for subsequent analyses. This shortcoming limits the utility of this technique for short‐term treatments. In addition, application of TRAP could be problematic for cells in which ribosomal protein synthesis is repressed, as appears to be the case in cells under stress (An et al., 2020; Salih, Duncan, Li, OLeary, et al., 2020). To overcome these limitations, we designed an alternative to conventional TRAP in which pre‐existing modified ribosomes could be tagged soon after application of the stimulus. We optimized this system, validated its efficiency, and applied it to the interaction between Arabidopsis and a filamentous oomycete pathogen.
RESULTS
Design principles of the new TRAP system
Our need for a modified TRAP system arose from the desire to study plant–pathogen interactions between Arabidopsis and its oomycete pathogen Hyaloperonospora arabidopsidis (Hpa) (Herlihy et al., 2019). Specifically, we wanted to profile plant cells that were in direct contact with pathogen feeding structures called haustoria (Bozkurt & Kamoun, 2020). These ‘haustoriated’ cells are difficult to analyze because they are present at relatively low numbers even in heavily infected leaves and cannot be purified from non‐haustoriated leaf cells. Moreover, transcriptome data indicate that Hpa infection has a suppressive effect on ribosome biogenesis (Wang et al., 2011). We reasoned that the potential limitations posed by ribosomal protein turnover and cell specificity could be addressed by a split linker protein system encoded by two transgenes (Figure 1b): One constitutively expressed transgene would encode the ribosomal protein fused to one half of the split linker protein. The second gene would be expressed under the control of a stimulus‐specific promoter (e.g., a pathogen‐inducible promoter) and encode the other half of the split linker, fused to a purification tag for TRAP. The split linker would assemble in cells where both transgenes are expressed, effectively fusing the ribosomal protein to the purification tag, thereby enabling the efficient tagging of pre‐existing ribosomes from the cells of interest and enrichment from a whole‐leaf extract by TRAP.
Selection of linker systems
The success of the strategy described above is predicated upon stable and strong association of the ribosomal protein with the inducible purification tag via the split linker protein. Therefore, it was important to select a modular linker system from which two protein fragments can be expressed from separate genes and bind to one another with high affinity and specificity in plant cells. The ideal linker should also be small to prevent interference with ribosomal function (e.g., less than 50 kDa) and non‐toxic for plant cells, and the association must withstand affinity purification steps in chaotropic buffers. Several systems meeting these criteria were available: split ubiquitin, streptavidin–streptag‐II, iDimerize, PDZ domain–ligand peptides, and split green fluorescent protein (GFP) (Chalfie, 1995; Korndörfer & Skerra, 2002; Lee & Zheng, 2010). Due to issues with potential disruption of cellular functions by ubiquitin, the need for an additional ligand for iDimerize, the size burden of StrepTactin, and a lack of specificity of PDZ domain peptides, we chose the split GFP as a linker. GFP was also an attractive choice due to its use in previous TRAP studies, which validated it as devoid of effects on ribosome biogenesis, function, or purification (Ron et al., 2014).
GFP has been used in many organisms as a reporter due to its intrinsic fluorescence (Chalfie, 1995). The protein is composed of 11 β‐sheets that form a barrel around an internal chromophore. Importantly, fragments of GFP can be expressed from two different genes as a split protein that will spontaneously assemble when those fragments are in close proximity, forming a fully active chromophore (Blakeley et al., 2012; Ito et al., 2013). ‘Superfolder’ GFP variants are composed of fragments that associate with high affinity, thus comprising a stable linker that would not disassociate during TRAP (Cabantous & Waldo, 2006). Moreover, GFP's intrinsic fluorescence would provide a visual confirmation that both genes are expressed and that the split GFP halves have assembled in the desired cell type. Therefore, we selected the ‘superfolder’ GFP described in (Pedelacq et al., 2006) that refolds robustly from a denatured state, emits fluorescence proportional to protein abundance, and has been validated for efficient and specific assembly from split configurations. This GFP was used previously for a tripartite split protein that was further mutagenized to improve reconstitution and folding efficiency (Cabantous et al., 2013).
In addition to the linker, careful consideration was given to tags and spacers (short amino acid sequences that provide flexibility between the different parts of our system) for the system. Two tags were required: one for affinity purification, positioned under the control of the inducible promoter, and one for assessing the abundance and integrity of the protein regulated by the constitutive promoter. We selected the FLAG tag (DYKDDDDK) for purification because it had been used in conjunction with anti‐FLAG beads for purification and elution in previous TRAP protocols (Mustroph, Juntawong, & Bailey‐Serres, 2009; Zanetti et al., 2005). We selected a Myc tag (EQKLISEEDL) for detection of the ribosomal protein because this tag works reliably in plant studies (Walter et al., 2004). We also designed spacers in between GFP, tags, and RPL (Figure S1). These sequences are intended to allow for interactions and refolding of the linker with minimal impact on ribosome function or localization.
Validation of split GFP as an efficient linker
Our first experiments were designed to (i) compare the efficiency of split GFP reconstitution when expressed as different split fragments and with different positions of tags/spacers; (ii) define the timeframe in which assembly of split GFP occurs; and (iii) observe whether the resultant protein complex localizes to the cytosol. These experiments employed transient expression via agroinfiltration in Nicotiana benthamiana (Vaghchhipawala et al., 2011), followed by confocal microscopy for detection of GFP activity and Western blotting to detect the FLAG and Myc tags.
Eight plasmid constructs were created in which every gene was placed under the control of the constitutive cauliflower mosaic virus 35S promoter (CaMV35S, referred to here as 35Sp) (Kay et al., 1987) (Figure 1c). The Arabidopsis ribosomal gene RPL18b was selected because it has been used successfully in previous TRAP protocols (Zanetti et al., 2005). Two different splits of the GFP open reading frame (Cabantous et al., 2013) were tested: (i) a fragment encoding β‐sheets 1 through 9 (hereafter called GFP1_9) for refolding with a second fragment encoding β‐sheets 10 and 11 (GFP10_11) and (ii) a fragment encoding β‐sheets 1 through 10 (GFP1_10) along with the corresponding 11th β‐sheet (GFP11) (Figure 1c). Two assemblies for each GFP fragment were created, positioning the Myc epitope either at the N‐terminus of the GFP fragment‐RPL fusion protein or between the GFP fragment and the RPL (Figure 1c). For the second gene of the system, the FLAG tag was fused to the N‐terminus of GFP1_9 and GFP1_10 to position the tag for IP. Two additional assemblies, FLAG‐GFP‐Myc‐RPL and FLAG‐GFP, were constructed for use as positive controls.
Each pair of split GFP plasmids were co‐infiltrated into leaves of N. benthamiana, and the timing, intensity, and subcellular localization of GFP activity in epidermal cells were assayed by microscopy. At 3 days post‐infiltration (dpi), fluorescence was detectable in all samples (Figure 2). Fluorescence increased in all samples over time. The GFP1_9/10_11 combination fluoresced more strongly than the GFP1_10/11 combination. The position of the Myc tag seemed to affect fluorescence from the GFP1_10/11 assemblies: Very little fluorescence was emitted from the samples expressing GFP1_10 + GFP11‐Myc‐RPL compared to the samples expressing GFP1_10 + Myc‐GFP11‐RPL18b. The placement of the Myc tag in this configuration could interfere with GFP refolding, ribosome function, or ribosomal protein interactions, leading to reduced fluorescence. Both splits exhibited nucleo‐cytoplasmic localization similar to the non‐split FLAG‐GFP‐Myc‐RPL18b control, as expected for a ribosome tagged with GFP (Figure 2). These results indicate that the split GFP assembles quickly and fluoresces brightly without affecting RPL18b localization. The combination of FLAG‐GFP1_9 + Myc‐GFP10_11 appeared to be the best configuration for the system, because it fluoresced more strongly than the other combinations.
Figure 2.
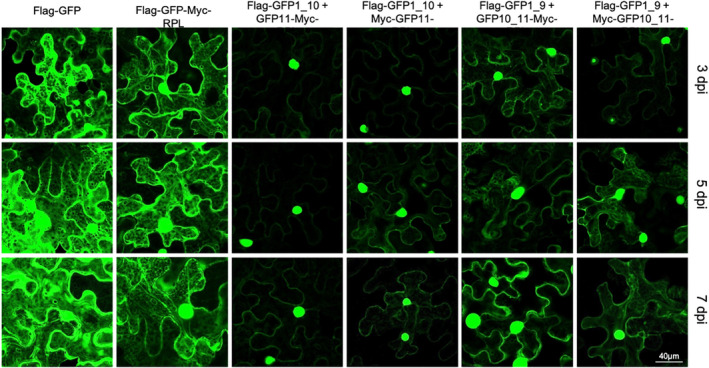
Comparison of fluorescence from green fluorescent protein (GFP) protein splits and different tag positions. Confocal microscopy was used to assay GFP activity and subcellular localization in Nicotiana benthamiana leaves transiently transformed through infiltration of Agrobacterium containing the genes of interest in T‐DNA vectors. Images were recorded as z‐stacks of multiple planes. dpi, days post‐infiltration with Agrobacterium; labels correspond to the constructs that are illustrated in Figure 1(c). [Colour figure can be viewed at wileyonlinelibrary.com]
Validation of assemblies for two‐gene TRAP: protein expression and purification
Our next set of experiments tested (i) whether the association between split fragments of GFP was strong enough to allow for recovery of the tagged ribosomes through isolation of polysomes via sucrose cushion ultracentrifugation and IP and (ii) whether ribosome yield was affected by the location of the GFP split or the position of the Myc tag. Infiltrated N. benthamiana leaves were harvested at 3 and 7 dpi, and three different samples from each time point were collected to track protein abundance at different steps in the IP procedure. The first sample was collected from clarified leaf extract (CLE), comprised of the soluble fraction from leaf tissue homogenized in detergent buffer and clarified via filtration and centrifugation (Figure 3a). The second sample was collected from the resuspension of the pellet recovered after ultracentrifugation through a sucrose cushion (sucrose ultracentrifugation pellet [SUP]); only dense macromolecular complexes such as ribosomes and polyribosomes can penetrate the sucrose cushion (Figure 3b). The third sample was collected from molecules IP from the resuspended SUP with beads coated with anti‐FLAG antibodies (Figure 3c). These samples were assayed via Western blotting (Figure 4). An anti‐FLAG antibody was used to detect the FLAG‐GFP1_9 and FLAG‐GFP1_10 fragments, as well as the full‐length FLAG‐GFP‐Myc‐RPL and FLAG‐GFP controls. The anti‐Myc antibody was used to detect the GFP10_11‐RPL and GFP11‐RPL fragments, with each variation of the Myc tag position.
Figure 3.
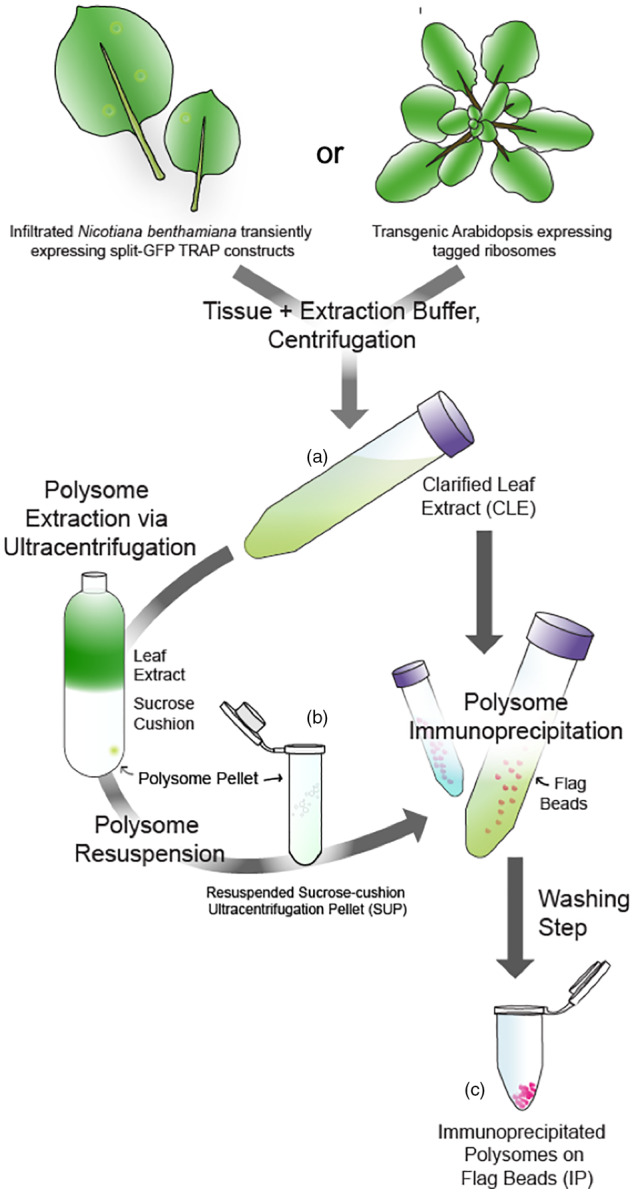
Schematic of the translating ribosome affinity purification (TRAP) workflow.
(a) Tissue from transiently transformed Nicotiana benthamiana or stably transformed Arabidopsis was collected, flash‐frozen, and homogenized with extraction buffer. Clarification of the homogenate via centrifugation yielded the clarified leaf extract (CLE), containing all soluble proteins.
(b) Polysome concentration was achieved by ultracentrifugation of CLE over a sucrose cushion followed by resuspension, yielding the sucrose cushion ultracentrifugation pellet (SUP) containing all protein complexes with sufficient density to penetrate the cushion. Note that this step was omitted from the experiments with transgenic Arabidopsis. (c) Immunopurification of tagged polyribosomes from either CLE (Arabidopsis) or SUP (N. benthamiana) employed FLAG beads to yield immunoprecipitated (IP) polysomes. [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 4.
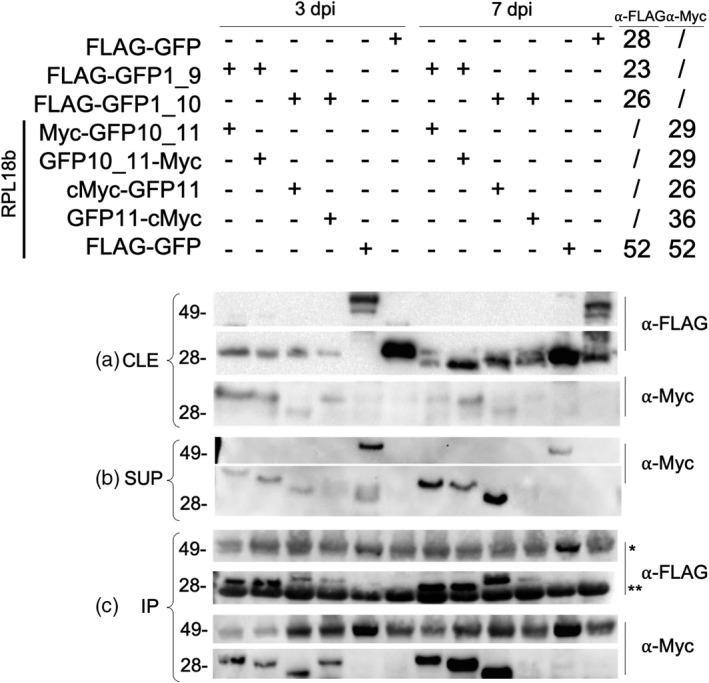
Western blot analysis of translating ribosome affinity purification (TRAP) proteins from transient expression in Nicotiana benthamiana.
(a) Samples from clarified leaf extract (CLE) at 3 and 7 dpi were probed with anti‐FLAG and anti‐Myc to detect GFP1_9 or 1_10 fragments (containing FLAG tags) or GFP10_11‐RPL or GFP11‐RPL fragments (containing Myc tags).
(b) Sucrose ultracentrifugation pellet (SUP) samples probed with anti‐Myc. No proteins were detected on this blot with anti‐FLAG.
(c) IP samples probed with anti‐FLAG and anti‐Myc. The chains of the anti‐FLAG antibody used for purification are indicated by ‘*’ and ‘**’, corresponding respectively to the heavy (approximately 50 kDa) and light (approximately 25 kDa) chains.
We expected to detect bands from every transiently expressed protein at both time points from the CLE samples when probing with anti‐FLAG and anti‐Myc, because the CLE contains all proteins expressed in the leaf. Accordingly, proteins from every transgene were evident in samples from both 3 and 7 dpi (Figure 4a). As a control, we collected and assayed tissue infiltrated with only FLAG‐GFP1_9 or only FLAG‐GFP1_10. These proteins were detected with anti‐FLAG in CLE samples but not in the SUP or IP samples, as expected (not shown).
Only proteins from macromolecular complexes dense enough to penetrate the sucrose cushion should be detected in samples taken from the SUP or the IP. Transiently expressed proteins should be detected in these fractions only if they were incorporated into ribosomes at the time of tissue collection. Accordingly, the full‐length FLAG‐GFP‐Myc‐RPL fusion protein control was detected with anti‐Myc in the SUP (Figure 4b) and IP (Figure 4c), while the FLAG‐GFP protein was not detected in either sample. Each split GFP fusion protein should be detectable in SUP and IP only if it assembled with its cognate split, it was incorporated into ribosomes, and the association between the GFP fragments was sufficiently strong to withstand the sucrose centrifugation and subsequent IP with anti‐FLAG. Accordingly, all Myc‐tagged GFP fragments were detectable with anti‐Myc in the SUP (Figure 4b) and IP (Figure 4c). Surprisingly, no FLAG‐tagged proteins were detectable in the SUP (not shown), perhaps due to the detergent‐heavy nature of the sample preventing the monoclonal anti‐FLAG antibody to bind the FLAG tag. However, the FLAG‐tagged GFP fragments were detected with anti‐FLAG in the IP from the SUP, demonstrating that these proteins were indeed present in the SUP (Figure 4c). In both the IP and SUP, the anti‐Myc blots showed low abundance of the assemblies with GFP11‐Myc‐RPL (Figure 4b,c), possibly due to low expression of the proteins, consistent with the low fluorescence detected from this split (Figure 2).
These results demonstrate that while all tagged proteins were present in the CLE, only tagged proteins designed to incorporate into or link with ribosomes were present in the SUP and IP samples, implying their incorporation into ribosomes. As expected, free GFP was excluded from SUP and IP samples, due to its inability to penetrate the sucrose cushion. These data indicate that transiently expressed fusion proteins from our split linker TRAP system can assemble, are incorporated into ribosomes, and can be immunoprecipitated.
Validation of assemblies for TRAP‐RNAseq: RNA extraction and analysis
Having established that tagged ribosomal proteins could be from homogenized leaf tissue, we tested whether RNA could be isolated from these samples with quality and yield sufficient for downstream analyses. RNA from aliquots of the fractions used in Western blotting from the CLE, SUP, and IP was extracted and quantitated by spectrophotometry. RNA quality was assessed by both agarose gel electrophoresis and chip‐based capillary electrophoreses. A greater amount of RNA was extracted from SUP samples compared to IP samples (Table S1; 2–7.9 μg per sample compared to 0.1–1.4 μg per sample). No RNA was detected from the IP of the free GFP samples, consistent with no detection of protein in Western blotting (Figure 4c). The following RNA electrophoresis profiles were expected for CLE and SUP samples: 25S, 18S cytosolic rRNAs, and the 23S and 16S plastidial rRNAs, with the SUP samples enriched in the cytosolic rRNAs compared to the CLE samples. The IP samples were expected to contain only 25S and 18S cytosolic rRNAs, with little or no plastidial rRNAs because the corresponding ribosomes are not tagged. All samples displayed the expected patterns (Figure 5a). IP samples from GFP1_9 + Myc‐GFP10_11‐RPL, GFP1_9 + GFP10_11‐Myc‐RPL, and the GFP‐Myc‐RPL positive control exhibited higher RNA yields compared to GFP1_10 + GFP11‐RPL splits, consistent with results from GFP imaging (Figure 2) and Western blotting (Figure 4). RNA samples from the higher‐yielding splits were selected for an additional validation step on an Agilent Bioanalyzer using the 7 dpi SUP and the 3 and 7 dpi IP samples (Figure 5b). All IP samples scored an RNA integrity number of 8 or above, indicating very good quality for sequencing. Each sample had a total RNA yield of 1 μg per sample or greater (Figure 5b), surpassing the minimum amount needed for most sequencing applications.
Figure 5.
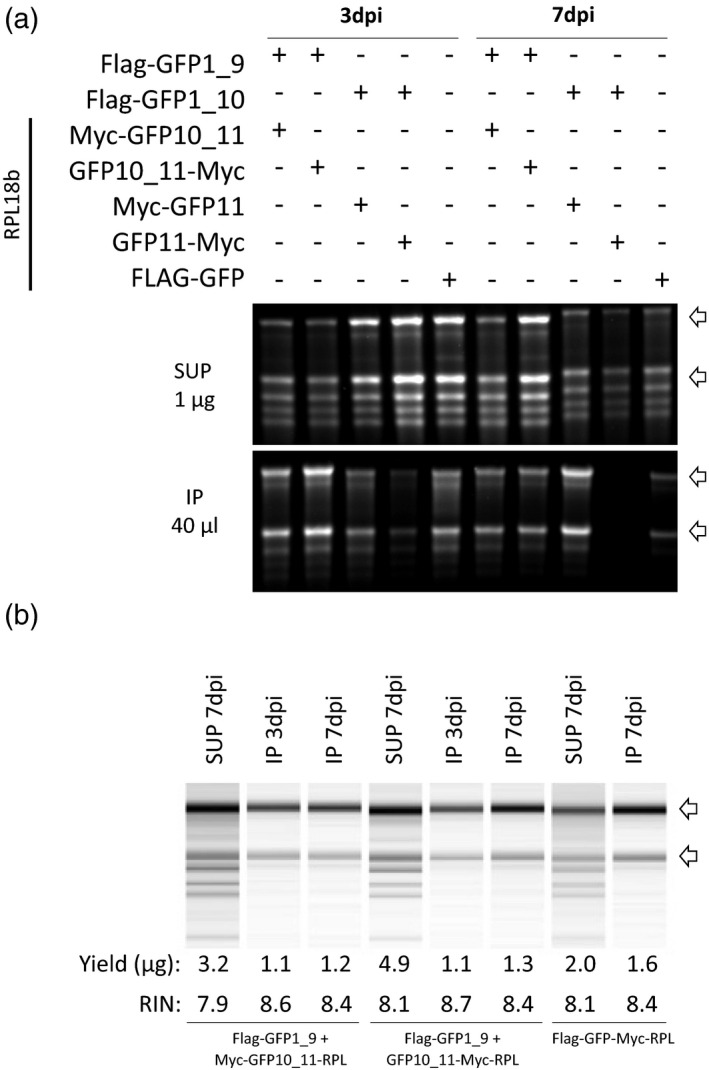
RNA analysis of the sucrose ultracentrifugation pellet (SUP) and immunoprecipitated (IP) RNA samples. (a) RNA gel electrophoresis of SUP samples at 3 and 7 dpi. One microgram of RNA from each sample was dried, resuspended with RNA loading buffer, and run on the gel. (b) Capillary electrophoresis analysis of IP samples at 3 and 7 dpi. The total amounts of RNA extracted were dried, resuspended, and loaded. Banding patterns, yield, and RNA integrity number (RIN) values are shown for each sample.
The data from fluorescence, Western blotting, and RNA analyses indicated that GFP1_9 + GFP10_11 assembly pairs performed better than GFP1_10 + GFP11 assembly pairs, possibly resulting from issues with GFP refolding or protein accumulation. The position of the Myc tag had little influence on the RNA quality and yield, although FLAG‐GFP1_9 + Myc‐GFP10_11‐RPL led to slightly higher fluorescence than the FLAG‐GFP1_9 + GFP10_11‐Myc‐RPL combination (Figure 2). We therefore selected FLAG‐GFP1_9 + Myc‐GFP10_11‐RPL for application of the split GFP TRAP technology to Arabidopsis.
Selection and validation of Arabidopsis promoters
The split GFP TRAP system was tested to study the interactions between Arabidopsis and the oomycete pathogen Hpa, in order to determine the translatome from plant cells that contain pathogen feeding structures called haustoria, present primarily in mesophyll cells (Herlihy et al., 2019). Therefore, we searched the literature for promoters that would provide the desired cell specificity for the two gene fusions: a constitutive, mesophyll‐specific promoter for the expression of Myc‐GFP10_11‐RPL and an Hpa‐responsive promoter to drive the expression of FLAG‐GFP1_9 specifically in haustoriated cells, to specifically tag ribosomes in haustoriated cells. Two promoters fulfilled these criteria: the PLASTOCYANIN 2 (PETE2, AT1G20340) promoter, as a 1790‐bp DNA sequence that was previously shown to be predominantly active in the mesophyll (Vorst et al., 1993), and a 2.5‐kb fragment of the promoter for Arabidopsis Downy Mildew Resistant 6 (DMR6, ATG5G24530), which encodes a defense‐associated protein required for susceptibility to Hpa and was previously reported to be active almost exclusively in haustoriated cells (Van Damme et al., 2008).
To validate the localization of activity from these promoters under our experimental conditions, the PETEp and DMR6p promoter fragments, cloned as described in previous publications, were fused to the Escherichia coli β‐glucuronidase (GUS) gene or the GFP gene and introduced into Arabidopsis. As expected, 3‐week‐old PETEp‐GUS transgenic plants exhibited uniform GUS activity in the mesophyll, with an absence of activity in all major veins (Figure 6a). DMR6p‐GUS transgenic plants exhibited GUS activity from 3 dpi with Hpa onwards in cotyledons of seedlings (Figure 6b). However, GUS staining was not restricted to haustoriated cells in seedlings. Contrastingly, in infected true leaves of 3‐week‐old plants, GUS and GFP activities were almost exclusively confined to haustoriated cells (Figure 6c,d; Figure S2). We consequently analyzed true leaves from adult plants rather than cotyledons from seedlings.
Figure 6.
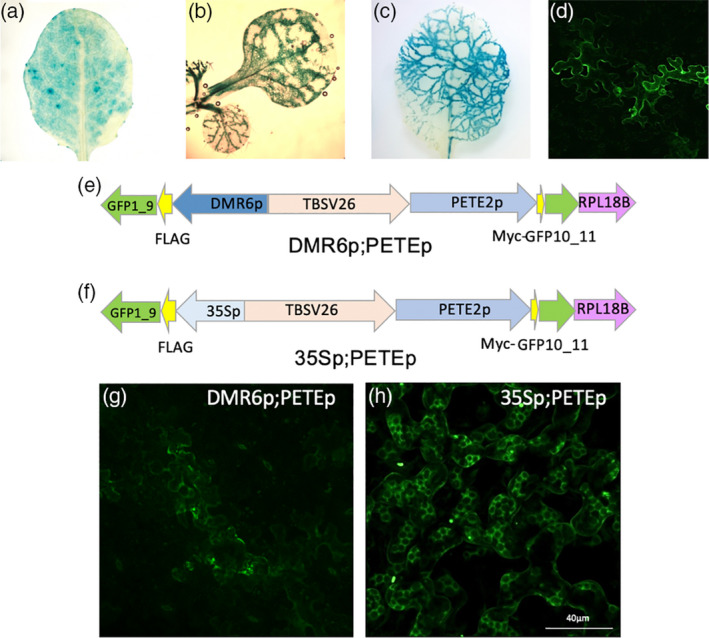
Characterization of PETE and DMR6 promoter activity.
(a–d) Histochemical staining for β‐glucuronidase (GUS) activity in soil‐grown transgenic Arabidopsis (at variable ages) expressing (a) PETEp‐GUS in a true leaf from a 3‐week‐old plant, (b) DMR6p‐GUS in Hpa‐infected cotyledons from 16‐day‐old seedlings at 6 dpi, (c) DMR6p‐GUS in Hpa‐infected true leaves from a 3‐week‐old plant at 6 dpi, or (d) DMR6p‐GFP in Hpa‐infected true leaves from a 3‐week‐old plant at 6 dpi.
(e, f) Plasmid maps for (e) DMR6p;PETEp and (f) 35Sp;PETEp.
(g, h) Confocal microscopy images of GFP fluorescence from 3‐week‐old transgenic Arabidopsis expressing (g) DMR6p;PETEp (at 6 dpi after infection with Hpa) and (h) 35Sp;PETEp (uninfected). [Colour figure can be viewed at wileyonlinelibrary.com]
Trials of the split GFP TRAP system with Arabidopsis infected by Hpa
For the use of TRAP of haustoriated cells in Arabidopsis, a single plasmid was constructed with two assemblies: PETEp‐Myc‐GFP10_11‐RPL and DMR6p‐FLAG‐GFP1_9, separated by the TBSV26 insulator to prevent promoter interference (Hily et al., 2009) (Figure 6e). This plasmid was named ‘DMR6p;PETEp’. A second plasmid, named ‘35Sp;PETEp’, was constructed to collect ribosomes from all mesophyll cells, regardless of pathogen presence or absence (Figure 6f). This plasmid was identical to DMR6p;PETEp except that the DMR6 promoter was replaced by 35Sp. In infected DMR6p;PETEp plants, GFP activity was detected in haustoriated cells and followed hyphal patterns within the leaf (Figure S2), similar to those reported from DMR6p‐GUS plants (Figure 6c), demonstrating that the DMR6 promoter drives the expression of FLAG‐GFP1_9 in the presence of the pathogen and that GFP1_9 refolds with PETEp‐driven Myc‐GFP10_11‐RPL (Figure 6g). No fluorescence was detected in uninfected DMR6p;PETEp lines (not shown). As expected, the 35Sp;PETEp plants showed constitutive fluorescence in all mesophyll cells (Figure 6h).
To further confirm that our chosen promoters were active when anticipated, reverse transcription quantitative real‐time PCR (RT‐qPCR) was performed with tissue collected at 0, 4, and 6 dpi from infected and mock replicates with the DMR6p;PETEp and 35Sp;PETEp lines (Figure 7). In both lines, DMR6 had very low expression at 0 dpi (in both mock and infected conditions) and expression did not change at any other time point for mock conditions. For replicates where plants were infected with Hpa, DMR6 mRNA content increased over the course of infection, consistent with previously published data (Van Damme et al., 2008). The expression of GFP1_9 was also analyzed in the same conditions (Figure 7). In samples from 35Sp;PETEp lines, transcript levels of GFP1_9 (driven by 35Sp) were unaffected by pathogen infection. In samples from DMR6p;PETEp lines, GFP1_9 mRNA content followed that of DMR6, demonstrating that the DMR6 promoter (driving expression of GFP1_9) is pathogen‐responsive. Combined with the fluorescence seen only in haustoriated cells/hyphal patterns, we inferred that the DMR6 promoter drives expression of GFP1_9 only in haustoriated cells and therefore the tagged ribosomes purified from the DMR6p;PETEp lines come from haustoriated cells.
Figure 7.
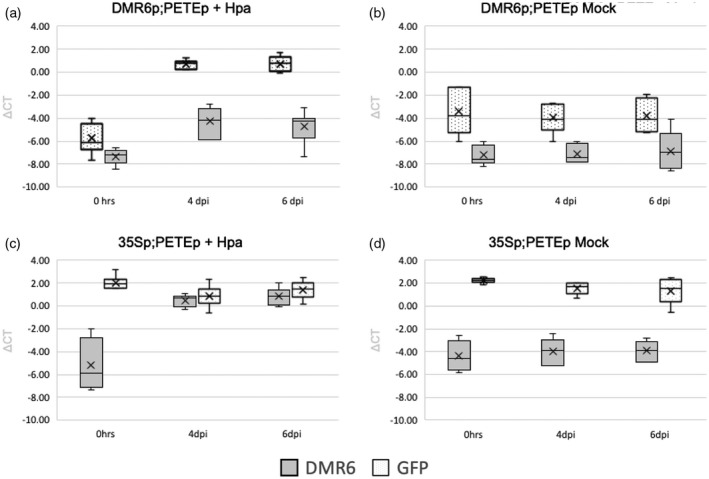
DMR6p and 35Sp activity over the course of infection. RT‐qPCR data are shown from 0, 4, and 6 dpi in infected and mock samples. Actin 2 was used for normalization. (a) DMR6p;PETEp line, infected with Hpa. Data from seven replicates. (b) DMR6p;PETEp line, mock control. Data from four replicates. (c) 35Sp;PETEp line, infected with Hpa. Data from six replicates. (d) 35Sp;PETEp line, mock control. Data from six replicates.
Tissues from Hpa‐infected and mock‐infected DMR6p;PETEp and 35Sp;PETEp lines were collected for TRAP. The experiment was repeated 15 times so that the most similar replicates could be chosen for analysis, thereby reducing the noise from biological variability in the Arabidopsis–Hpa interaction. Quality control assays were run on each of the 15 replicates, and four or five replicates for each condition were selected for TRAP and RNA extraction (Figure S3). Two RNA samples, respectively comprising the transcriptome and translatome, were taken from each replicate: one from the CLE and one from the IP. The TRAP RNA extraction procedure differed slightly from the protocol used for samples from infiltrated N. benthamiana: We did not include the sucrose cushion step (as it was implemented previously to test the strength of association between the linker fragments), and the CLE samples were incubated twice with FLAG beads to increase RNA yields. We observed a higher yield and the typical ribosomal banding pattern from the total RNA sample, and a lower yield and the expected ribosomal banding pattern from the TRAP RNA sample (9–15 versus 1–10.9 μg per sample, respectively; Figure 8; Figure S4, Table S2). RNA yields from the DMR6p;PETEp lines ranged from 1.1 to 1.3 μg per sample. This relatively low yield was anticipated due to the limited number of haustoriated cells in the leaves. No TRAP RNA was recovered from uninfected DMR6p;PETEp, which was expected because the DMR6 promoter is inactive under those conditions. The quality and quantity of RNA from all other samples were sufficient for RNAseq. Analyses of all samples and the corresponding RNAseq data representing the translatome of haustoriated cells compared to the mesophyll as a whole will be described in a forthcoming manuscript.
Figure 8.
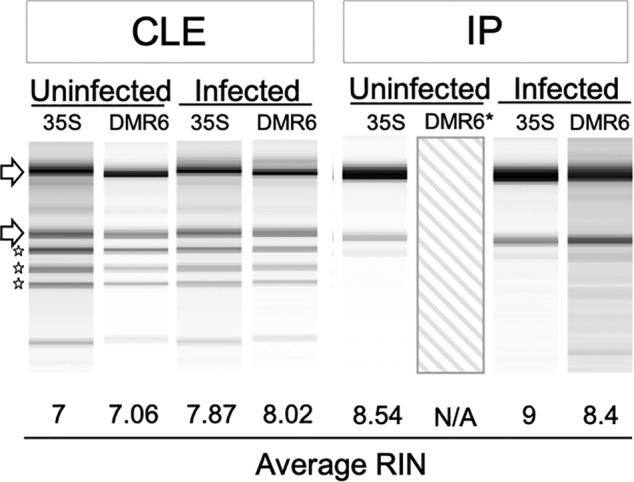
Capillary electrophoresis of RNA from transgenic Arabidopsis. Banding patterns and RNA integrity number (RIN) values for RNA isolated from clarified leaf extract (CLE) or immunoprecipitated (IP) samples from infected or uninfected plants. ‘35S’ denotes the 35Sp;PETEp line, while ‘DMR6’ denotes the DMR6p;PETEp line. Average RIN values were calculated from five samples, and a representative image was chosen for each condition. *No image or RIN is present for uninfected IP samples from the ‘DMR6’ line because no tagged ribosomes were recovered from the immunopurification of this sample.
CONCLUSIONS
We have designed a new TRAP system applicable to any cell‐specific or stimulus‐specific context in plants, based on a split GFP linker. We applied this system to Arabidopsis–Hpa interactions, using promoters that enable specific enrichment of ribosomes from haustoriated cells from a heterogeneous, whole‐leaf sample of haustoriated and non‐haustoriated cells. To demonstrate the viability of this design, we optimized and validated several important features: (i) Split superfolder GFP can refold efficiently in our system, providing an approach for adding purification tags to pre‐existing ribosomes for efficient recovery via IP. GFP florescence was also validated as a visual marker for expression and assembly of the fragments of the split GFP. (ii) The best split of the GFP gene was between β‐sheets 9 and 10, and the best configuration of linkers and tags was FLAG‐GFP1_9 + GFP10_11‐Myc‐RPL. (iii) Association of the split GFP fragments is strong enough to withstand centrifugation through a sucrose cushion, demonstrating that the GFP10_11‐Myc‐RPL fusion protein will not dissociate from the FLAG‐GFP1_9 protein during TRAP. (iv) The amount and quality of the RNA extracted from IP ribosomes are sufficient for qPCR or RNAseq. We expect that these findings should be broadly applicable to any application of the split GFP system for TRAP in plants and potentially other organisms.
With respect to our goal of obtaining the translatome of haustoriated cells, we established the following: (i) The DMR6 promoter is activated specifically in haustoriated plant cells only in true leaves, not in cotyledons. (ii) The DMR6p;PETEp and 35Sp;PETEp assemblies performed well in transgenic Arabidopsis infected with Hpa: IP of ribosomes from the CLE allowed for purification/concentration of RNA in amounts and quality sufficient for RNAseq. Our subsequent analyses of the translatomes from haustoriated cells, to be published elsewhere, confirmed that this system provides new insights into plant–oomycete interactions, and we expect that this approach could be applied productively to many different plant–pathogen interactions, provided that a specific pathogen‐inducible promoter is available for the interaction under study.
Existing TRAP technology provides a powerful tool for researchers to interrogate the translatome. Previous applications of TRAP have employed a single transgene, driven by a strong, constitutive promoter, or less commonly, by a promoter that is active in specific cell types or in response to specific conditions to produce a tagged ribosomal protein that enables TRAP (Urquidi Camacho et al., 2020). Our two‐gene system enables cell‐type or condition‐specific attachment of a tag to an existing pool of ribosomes through refolding of a split GFP linker. This feature will increase the sensitivity of TRAP in situations where a tagged ribosomal protein from a single gene is unlikely to be incorporated into ribosomes with sufficient efficiency for robust yields (e.g., when ribosome protein turnover is slow or suppressed or when the biological material is to be analyzed shortly after induction). Moreover, the two‐gene system can provide an additional level of specificity through customization of two promoters instead of just one. Altogether, we anticipate that this system will be applicable to diverse biological contexts in which TRAP is applied to profile a specific target cell population from a complex assemblage.
EXPERIMENTAL PROCEDURES
Arabidopsis thaliana and N. benthamiana growth and transformation
Arabidopsis thaliana and N. benthamiana were grown under 120 μE m−2 sec−1, at 22°C, under a 16 h light/8 h dark photoperiod on soil (Sunshine Mix #1; Sungro Horticulture, Agawam, MA, USA) and were watered from below with 300 mg L−1 Miracle‐Gro Fertilizer (24‐8‐16 NKP; Scotts, Marysville, OH, USA). Arabidopsis ecotype Columbia 0 (Col‐0, CS70000) was used for all experiments. Stably transformed Arabidopsis lines were generated by floral dipping using Agrobacterium tumefaciens GV3101 (pMP90) (Clough Steven & Bent Andrew, 2008). For transient expression in N. benthamiana, leaves of 5‐week‐old plants were infiltrated with a suspension of A. tumefaciens GV3101 (pMP90) carrying the constructs or p19 (Batoko et al., 2000; Yu & Pilot, 2014) with the following modifications: The bacteria were grown overnight in LB medium supplemented with appropriate antibiotics, washed twice in solution containing 10 mm MgCl2 and 100 μm acetosyringone, and were diluted to a final OD600 of 0.05 in the same solution.
Hpa propagation and infection
Hpa isolate Noco2 was used for all experiments, as described in (McDowell et al., 2011; Reignault et al., 1996). Sporangiospore suspensions of 5 × 104 spores ml−1 were prepared in water from sporulating plants and sprayed on seedlings or 3‐week‐old Arabidopsis plants. Inoculated plants were covered overnight, and then uncovered and kept under short‐day (8 h, 22°C/16 h, 20°C day/night) conditions. Control plants were sprayed with water alone. Infected plant material for TRAP was harvested into liquid nitrogen at 6 dpi, prior to sporulation.
GUS and trypan blue staining
GUS staining was performed with fresh leaf tissue. Leaves were fixed under vacuum in sodium phosphate buffer (50 mm, pH 7.2) containing 0.5% Triton and 1.5% formaldehyde for 45 min. Fixed leaves were washed three times for 5 min in phosphate buffer containing 0.5% Triton, 500 μm potassium ferrocyanide, and 500 μm potassium ferricyanide and infiltrated with wash buffer as above containing 1 mm X‐Gluc (GoldBio, St Louis, MO, USA). Samples were incubated at 37°C in the dark until staining became visible and were cleared with ethanol for imaging. Trypan blue staining of Hpa hyphae, haustoria, and spores was performed as previously described (McDowell et al., 2011) with the following modification: Leaves were incubated for 90 sec at 90°C, incubated at room temperature for 5 min, and de‐stained with chloral hydrate.
Fluorescence microscopy
Prior to imaging, leaves were infiltrated with water via syringe or vacuum and placed on slides with the abaxial side facing the slide cover. Fluorescent imaging was performed on a Zeiss LSM 880 AxioObserver confocal laser scanning microscope with Airyscan. Images were collected using a 25× lens and processed with Zen Black software. GFP was excited at 488 nm and detected from 505 to 540 nm.
Western blotting
Aliquots of extracts taken at different stages of the TRAP procedure were prepared with 1 m DTT and 4× NuPAGE LDS sample buffer, denatured at 90°C for 10 min, and analyzed by SDS‐PAGE (4–12% polyacrylamide NuPAGE MES gels; Life Technologies, Carlsbad, CA, USA). Proteins were transferred to a nitrocellulose membrane (GE Healthcare, Chicago, IL, USA) by wet electroblotting. The membrane was treated with Ponceau red stain and blocked in OneBlock (Genesee Scientific, San Diego, CA, USA). Proteins were detected using anti‐FLAG (monoclonal anti‐FLAG M2, 1:1000; Sigma, St Louis, MO, USA) or anti‐Myc (Clone A‐14, 1:1000; Santa Cruz) primary antibodies and horseradish peroxidase (HRP)‐conjugated anti‐mouse secondary antibody (1:10 000; ThermoFisher Scientific Waltham, MA, USA) in OneBlock buffer. HRP activity was detected using an ECL‐Plus Western blotting detection system (GE Healthcare), with luminescence captured by a ChemiDoc CCD camera imaging system (Bio‐Rad, Hercules, CA, USA).
Multiple fragment cloning using in‐fusion
Primers were designed for each fragment with 10–20‐bp overlaps corresponding to the fragments that would be adjacent to the amplified fragment in each corresponding clone. Fragments were amplified from gBlocks Gene Fragments (Integrated DNA Technologies, Coralville, IA, USA), Col‐0 genomic DNA, or existing fragments by PCR with KOD Hotstart DNA polymerase (Toyobo, New York, NY, USA). Amplified fragments were gel‐purified and used for In‐Fusion Multiple Fragment Cloning (Clontech, Mountain View, CA, USA) according to the manufacturer's directions. Clones were verified by sequencing. Information and sequences of all DNA constructs and assemblies produced in this work are available upon request.
Translating ribosome affinity purification
TRAP was performed according to Mustroph, Juntawong, and Bailey‐Serres (2009) with the following modifications: For N. benthamiana samples, 8 ml of extraction buffer was added to 1.5 g of tissue ground in liquid N2. The resulting leaf extract was clarified twice via centrifugation, with a miracloth filtering step between centrifugations. CLE (6 ml) was loaded on top of a 1.7 m sucrose cushion containing detergents (DOC, PTE, and Detergent Mix) in the same concentrations as in the extraction buffer. Following centrifugation, the pellet was resuspended in 500 μl of extraction buffer by agitation on a spinning wheel at 4°C overnight, followed by removal of insoluble debris by centrifugation for 1 min at 8200 g at 4°C. The resuspended polysomes were added to extraction buffer to a final volume of 5 ml with the addition of 100 μl washed FLAG beads (Sigma). Polysomes were incubated with the beads for 2 h with gentle rocking at 4°C. The beads were washed as follows: one wash with 5 ml extraction buffer and four washes with 5 ml wash buffer with 5‐min incubation periods between washes. For Arabidopsis samples, CLE was added directly to previously washed FLAG beads, following the same incubation and wash protocol as above. To account for a lower number of tagged ribosomes, CLE samples from the Arabidopsis DMR6p;PETEp line were saved and used for a second consecutive incubation step with an additional 100 μl of washed FLAG beads.
RNA extraction and quantification
RNA was extracted from Arabidopsis tissue, CLE, resuspended polysomes, and FLAG beads using either 1 ml of TRI REAGENT (Sigma) or the ISOLATE II RNA Plant kit (Bioline; Meridian Life Science, Memphis, TN, USA). RNA integrity was confirmed by either agarose gel electrophoresis or capillary electrophoresis in an Agilent 2100 Bioanalyzer according to the manufacturer's instructions. cDNA was synthesized from 1.5 μg of RNA using the SuperScript IV Reverse Transcriptase kit (ThermoFisher Scientific) according to the manufacturer's instructions in a 10‐μl reaction volume. qPCR was performed with 5 μl of the product (diluted 50 times in water), 5 μl of primer mix (1 μm each), and 10 μl of 2× PowerUp SYBR Green Master Mix (Applied Biosystems, Waltham, MA, USA) on an Applied Biosystems 7500 Real‐Time PCR System (2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 sec at 95°C, 15 sec at 55°C, and 1 min at 72°C). mRNA levels were calculated by the 2−ΔΔCt method, using Arabidopsis Actin 2 (AT3G18780) as the reference gene.
Infected plant nucleic acid isolation and pathogen quantification via qPCR
Arabidopsis tissue infected with Hpa was harvested at 6 dpi and ground to a fine powder in liquid nitrogen. DNA was extracted using the BioSprint 96 DNA Plant kit (Qiagen, Hilden, Germany) according to the manufacturer's specifications. The protocol from (Anderson & McDowell, 2015) was followed with modifications for the use of TaqMan polymerase, reagents, and probes instead of SYBR Green reagents (Applied Biosystems).
CONFLICT OF INTEREST
The authors have no conflicts of interest.
Supporting information
Figure S1. Illustration of amino acid spacers placed between components of TRAP assemblies.
Figure S2. DMR6p:GUS activity in 3‐week‐old Arabidopsis leaves infected with Hpa.
Figure S3. Pathogen biomass assays for replicate selection.
Figure S4. Capillary electrophoresis analysis of RNA from transgenic Arabidopsis.
Table S1. Total yield (μg) of RNA extracted from SUP and IP Nicotiana benthamiana samples.
Table S2. RNA yield (μg) and quality from Arabidopsis TRAP conditions.
Contributor Information
Kasia Dinkeloo, Email: kdinkeloo@utexas.edu.
John M. McDowell, Email: johnmcd@vt.edu.
Guillaume Pilot, Email: gpilot@vt.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author, GP, upon reasonable request.
REFERENCES
- Adams, J.U. (2008) Transcriptome: connecting the genome to gene function. Nature Education, 1, 195. [Google Scholar]
- An, H. , Ordureau, A. , Körner, M. , Paulo, J.A. & Harper, J.W. (2020) Systematic quantitative analysis of ribosome inventory during nutrient stress. Nature, 583(7815), 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, R.G. & McDowell, J.M. (2015) A PCR assay for the quantification of growth of the oomycete pathogen Hyaloperonospora arabidopsidis in Arabidopsis thaliana . Molecular Plant Pathology, 16(8), 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batoko, H. , Zheng, H.Q. , Hawes, C. & Moore, I. (2000) A rab1 GTPase is required for transport between the endoplasmic reticulum and golgi apparatus and for normal golgi movement in plants. Plant Cell, 12(11), 2201–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley, B.D. , Chapman, A.M. & McNaughton, B.R. (2012) Split‐superpositive GFP reassembly is a fast, efficient, and robust method for detecting protein–protein interactions in vivo. Molecular BioSystems, 8(8), 2036–2040. [DOI] [PubMed] [Google Scholar]
- Bozkurt, T.O. & Kamoun, S. (2020) The plant–pathogen haustorial interface at a glance. Journal of Cell Science, 133(5), jcs237958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous, S. , Nguyen, H.B. , Pedelacq, J.‐D. , Koraïchi, F. , Chaudhary, A. , Ganguly, K. et al. (2013) A new protein‐protein interaction sensor based on tripartite split‐GFP association. Scientific Reports, 3, 2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous, S. & Waldo, G.S. (2006) In vivo and in vitro protein solubility assays using split GFP. Nature Methods, 3(10), 845–854. [DOI] [PubMed] [Google Scholar]
- Chalfie, M. (1995) Green fluorescent protein. Photochemistry and Photobiology, 62(4), 651–656. [DOI] [PubMed] [Google Scholar]
- Clough Steven, J. & Bent Andrew, F. (2008) Floral dip: a simplified method for agrobacterium ‐mediated transformation of Arabidopsis thaliana . The Plant Journal, 16(6), 735–743. [DOI] [PubMed] [Google Scholar]
- Heiman, M. , Schaefer, A. , Gong, S. , Peterson, J.D. , Day, M. , Ramsey, K.E. et al. (2008) A translational profiling approach for the molecular characterization of CNS cell types. Cell, 135(4), 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlihy, J. , Nora, R.L. , Guido van den, A. & John, M.M. (2019) Oomycetes used in Arabidopsis research. The Arabidopsis Book, 17, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hily, J.‐M. , Singer, S.D. , Yang, Y. & Liu, Z. (2009) A transformation booster sequence (TBS) from Petunia hybrida functions as an enhancer‐blocking insulator in Arabidopsis thaliana . Plant Cell Reports, 28(7), 1095–1104. [DOI] [PubMed] [Google Scholar]
- Ito, M. , Ozawa, T. & Takada, S. (2013) Folding coupled with assembly in split green fluorescent proteins studied by structure‐based molecular simulations. The Journal of Physical Chemistry B, 117(42), 13212–13218. [DOI] [PubMed] [Google Scholar]
- Kage, U. , Powell, J.J. , Gardiner, D.M. & Kazan, K. (2020) Ribosome profiling in plants: what is not lost in translation? Journal of Experimental Botany, 71(18), 5323–5332. [DOI] [PubMed] [Google Scholar]
- Kay, R. , Chan, A. , Daly, M. & McPherson, J. (1987) Duplication of CaMV 35S promoter sequences creates a strong enhancer for plant genes. Science, 236(4806), 1299–1302. [DOI] [PubMed] [Google Scholar]
- Kimberlin, A. , Holtsclaw, R.E. & Koo, A.J. (2021) Differential regulation of the ribosomal association of mRNA transcripts in an Arabidopsis mutant defective in jasmonate‐dependent wound response. Frontiers in Plant Science, 12, 637959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korndörfer, I.P. & Skerra, A. (2002) Improved affinity of engineered streptavidin for the strep‐tag II peptide is due to a fixed open conformation of the lid‐like loop at the binding site. Protein Science: A Publication of the Protein Society, 11(4), 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H.‐J. & Zheng, J.J. (2010) PDZ domains and their binding partners: structure, specificity, and modification. Cell Communication and Signaling, 8(1), 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell, J.M. , Hoff, T. , Anderson, R.G. & Deegan, D. (2011) Propagation, storage, and assays with Hyaloperonospora arabidopsidis: a model oomycete pathogen of Arabidopsis. In: McDowell, J.M. (Ed.) Plant immunity: methods and protocols. Totowa, NJ: Humana Press, pp. 137–151. [DOI] [PubMed] [Google Scholar]
- Meteignier, L.‐V. , El Oirdi, M. , Cohen, M. , Barff, T. , Matteau, D. , Lucier, J.‐F. et al. (2017) Translatome analysis of an NB‐LRR immune response identifies important contributors to plant immunity in Arabidopsis. Journal of Experimental Botany, 68(9), 2333–2344. [DOI] [PubMed] [Google Scholar]
- Mustroph, A. , Juntawong, P. & Bailey‐Serres, J. (2009) Isolation of plant polysomal mRNA by differential centrifugation and ribosome immunopurification methods. Methods in Molecular Biology, 553, 109–126. [DOI] [PubMed] [Google Scholar]
- Mustroph, A. , Zanetti, M.E. , Jang, C.J.H. , Holtan, H.E. , Repetti, P.P. , Galbraith, D.W. et al. (2009) Profiling translatomes of discrete cell populations resolves altered cellular priorities during hypoxia in Arabidopsis. Proceedings of the National Academy of Sciences, 106(44), 18843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedelacq, J.‐D. , Cabantous, S. , Tran, T. , Terwilliger, T.C. & Waldo, G.S. (2006) Engineering and characterization of a superfolder green fluorescent protein. Nature Biotechnology, 24(1), 79–88. [DOI] [PubMed] [Google Scholar]
- Reignault, P. , Frost, L.N. , Richardson, H. , Daniels, M.J. , Jones, J.D.G. & Parker, J.E. (1996) Four Arabidopsis RPP loci controlling resistance to the Noco2 isolate of Peronospora parasitica map to regions known to contain other RPP recognition specificities. Molecular Plant–Microbe Interactions, 9(6), 464–473. [DOI] [PubMed] [Google Scholar]
- Ron, M. , Kajala, K. , Pauluzzi, G. , Wang, D. , Reynoso, M.A. , Zumstein, K. et al. (2014) Hairy root transformation using Agrobacterium rhizogenes as a tool for exploring cell type‐specific gene expression and function using tomato as a model. Plant Physiology, 166(2), 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salih, K.J. , Duncan, O. , Li, L. , OLeary, B. , Fenske, R. , Trösch, J. et al. (2020) Impact of oxidative stress on the function, abundance, and turnover of the Arabidopsis 80S cytosolic ribosome. The Plant Journal, 103(1), 128–139. [DOI] [PubMed] [Google Scholar]
- Salih, K.J. , Duncan, O. , Li, L. , Trösch, J. & Millar, A.H. (2020) The composition and turnover of the Arabidopsis thaliana 80S cytosolic ribosome. Biochemical Journal, 477(16), 3019–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquidi Camacho, R.A. , Lokdarshi, A. & von Arnim, A.G. (2020) Translational gene regulation in plants: a green new deal. WIREs RNA, 11(6), e1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaghchhipawala, Z. , Rojas, C.M. , Senthil‐Kumar, M. & Mysore, K.S. (2011) Agroinoculation and agroinfiltration: simple tools for complex gene function analyses. Methods in Molecular Biology, 678, 65–76. [DOI] [PubMed] [Google Scholar]
- Van Damme, M. , Huibers, R.P. , Elberse, J. & Van den Ackerveken, G. (2008) Arabidopsis DMR6 encodes a putative 2OG‐Fe(II) oxygenase that is defense‐associated but required for susceptibility to downy mildew. The Plant Journal, 54(5), 785–793. [DOI] [PubMed] [Google Scholar]
- Vorst, O. , van Dam, F. , Weisbeek, P. & Smeekens, S. (1993) Light‐regulated expression of the Arabidopsis thaliana ferredoxin a gene involves both transcriptional and post‐transcriptional processes. The Plant Journal, 3(6), 793–803. [DOI] [PubMed] [Google Scholar]
- Walter, M. , Chaban, C. , Schütze, K. , Batistic, O. , Weckermann, K. , Näke, C. et al. (2004) Visualization of protein interactions in living plant cells using bimolecular fluorescence complementation. The Plant Journal, 40(3), 428–438. [DOI] [PubMed] [Google Scholar]
- Wang, T. , Cui, Y. , Jin, J. , Guo, J. , Wang, G. , Yin, X. et al. (2013) Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Research, 41(9), 4743–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Barnaby, J.Y. , Tada, Y. , Li, H. , Tor, M. , Caldelari, D. et al. (2011) Timing of plant immune responses by a central circadian regulator. Nature, 470(7332), 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, G. , Greene, G.H. , Yoo, H. , Liu, L. , Marqués, J. , Motley, J. et al. (2017) Global translational reprogramming is a fundamental layer of immune regulation in plants. Nature, 545(7655), 487–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo, H. , Greene, G.H. , Yuan, M. , Xu, G. , Burton, D. , Liu, L. et al. (2020) Translational regulation of metabolic dynamics during effector‐triggered immunity. Molecular Plant, 13(1), 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, S. & Pilot, G. (2014) Testing the efficiency of plant artificial microRNAs by transient expression in Nicotiana benthamiana reveals additional action at the translational level. Frontiers in Plant Science, 5(622). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanetti, M.E. , Chang, I.‐F. , Gong, F. , Galbraith, D.W. & Bailey‐Serres, J. (2005) Immunopurification of polyribosomal complexes of Arabidopsis for global analysis of gene expression. Plant Physiology, 138(2), 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Illustration of amino acid spacers placed between components of TRAP assemblies.
Figure S2. DMR6p:GUS activity in 3‐week‐old Arabidopsis leaves infected with Hpa.
Figure S3. Pathogen biomass assays for replicate selection.
Figure S4. Capillary electrophoresis analysis of RNA from transgenic Arabidopsis.
Table S1. Total yield (μg) of RNA extracted from SUP and IP Nicotiana benthamiana samples.
Table S2. RNA yield (μg) and quality from Arabidopsis TRAP conditions.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, GP, upon reasonable request.