

Summary
The search for novel targets in chronic myeloid leukaemia (CML) is ongoing, to improve treatment efficacy in refractory disease and increase eligibility for tyrosine kinase inhibitor (TKI) discontinuation. Increased frequency of Tregs and effector Tregs was evident at diagnosis, together with increased expression of T‐cell exhaustion markers, including in regulatory T cells at diagnosis and in patients with refractory disease. Plasma analysis revealed significantly increased levels of cytokines including tumour necrosis factor (TNF)‐a and interleukin (IL)‐6 at diagnosis, in keeping with a pro‐inflammatory state prior to treatment. We hence demonstrate T‐cell exhaustion and a pro‐inflammatory state at diagnosis in CML, likely secondary to leukaemia‐associated antigenic overload associated with increased disease burden.
Keywords: CML, immune checkpoint, inflammation, T cell, T‐cell exhaustion, Treg
INTRODUCTION
The search for new targets to improve outcomes in patients with chronic myeloid leukaemia (CML) is ongoing, with the aim of both improving outcomes for those with refractory disease and increasing the proportion of those eligible to attempt tyrosine kinase inhibitor (TKI) discontinuation. 1 CML is recognised as a particularly immune‐sensitive tumour and therapies that can enhance inherent immune surveillance mechanisms are an attractive option, as covered in a recent review article. 2 , 3 , 4 Recent studies have also shown that immune‐cell subsets, including natural killer (NK) cells and plasmacytoid dendritic cells, play a central role in maintenance of treatment‐free remission following TKI discontinuation, further highlighting this aspect. 5 , 6 , 7 , 8 Immune‐checkpoint inhibitors have demonstrated efficacy in solid tumours and remain under investigation in haematological malignancy, with variable results observed. 9 In CML, expression of the Programmed Death‐1 (PD‐1) inhibitory molecule on CD4+/CD8+ T cells has been shown to be increased in patients at diagnosis with restoration to normal levels in deep molecular response, suggesting a role for checkpoint molecules in response to treatment. 10 We hypothesised that expression of other checkpoint molecules including on regulatory T cells (Tregs) may also be significant, and play a role in resistance to treatment.
METHODOLOGY
Samples from patients with WHO‐defined diagnosis of chronic‐phase CML were obtained at diagnosis and at response or resistance to TKI therapy. Peripheral‐blood mononuclear cells (PBMCs) were isolated from venous blood samples using standard Ficoll (Cytiva, Uppsala, Sweden) density centrifugation and were cryopreserved in 30% dimethylsulphoxide (Sigma‐Aldrich, St. Louis, MO, USA) and fetal calf serum.
Flow cytometric analysis was performed for the expression of PD‐1, cytotoxic T‐lymphocyte‐associated antigen‐4 (CTLA‐4), T‐cell immunoglobulin and mucin domain‐containing protein 3 (TIM‐3) and lymphocyte activation gene 3 (LAG‐3) on T effectors and Tregs. T effectors included CD4+ and CD8+ subsets and a gating strategy of CD4+/CD25+/CD127lo/FOXP3+ cells for Tregs was employed (Figure 1Ai,ii). Treg subsets were defined based on expression of FOXP3 and CD45RA, with FOXP3hi/CD45RA− cells denoting effector Tregs, FOXP3lo/CD45RA+ cells denoting naive Tregs and FOXP3lo/CD45RA− denoting a non‐true Treg population (Figure 1Aiii). FMO controls were used to determine positive populations for each of the immune‐checkpoint molecules under investigation.
FIGURE 1.
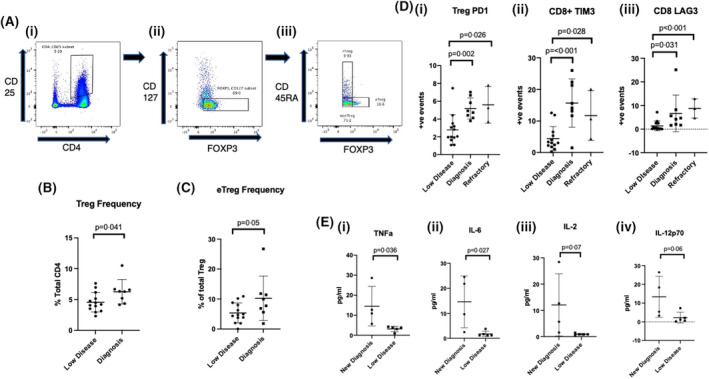
Comparison of Treg frequencies, T‐cell immune‐checkpoint expression and plasma cytokine levels between CML patient groups. (A) Regulatory T‐cell and effector Treg gating strategy. (i) CD4+CD25+ ➔ (ii) FOXP3+CD127lo ➔ (iii) FOXP3hi/CD45RA− (effector Tregs), FOXP3lo/CD45RA+ (naive Tregs), FOXP3lo/CD45RA− (non Treg population). (B) Treg and effector Treg (C) frequency showing higher proportion in patients at diagnosis compared to in those with low disease burden. (D) Increased proportion of immune‐checkpoint molecules in patients at diagnosis and with refractory disease compared to those with low disease burden. (i) Treg PD‐1 expression, (ii) CD8+ TIM3 expression and (iii) CD8+ LAG‐3 expression. (E) Increased levels of pro‐inflammatory cytokines observed in patients at diagnosis compared to those with low disease burden. (i) Tumour necrosis factor (TNF)‐a, (ii) interleukin (IL)‐6, (iii) IL‐2 and (iv) IL‐12p70.
Plasma cytokine levels were evaluated using the 9‐Plex ProcartaPlex Panel (Thermo Fisher Scientific) in view of the role of inflammation in the development and progression of myeloid malignancy. 11 Samples were incubated with magnetic capture beads with specific spectral properties, prior to streptavidin RPE conjugate and then biotinylated detection antibody being added. Data was acquired on the Luminex‐Flexmap3D.
Data are reported as mean values and p values were from independent‐sample t‐tests, with values <0.05 considered statistically significant. The distribution of the data was assessed using Levene's test for equality of variance. All reported p values are two‐sided.
Analyses were performed using SPSS version 24 (IBM Corp.) and Prism v8.
RESULTS
Samples from 22 patients were analysed, including sequential samples in two patients. Analysis included patients at diagnosis (n = 8), those with refractory disease, defined as less than complete cytogenetic response (CCyR; n = 3), and those with a molecular response of MR3 (n = 4) or greater (n = 9). The following TKIs were taken by the patients on treatment: imatinib (n = 5), nilotinib (n = 4), dasatinib (n = 5) and ponatinib (n = 2). None of the patients with refractory disease had evidence of ABL1 kinase domain mutation using next‐generation sequencing analysis or additional chromosomal abnormalities on karyotyping. Clinical characteristics are summarised in Table S1.
Patients at diagnosis had higher Tregs as proportion of the total CD4+ cells compared to those with low disease burden with a mean of 6.3 vs 4.6 (p = 0.041, Figure 1A,B). Similarly, effector Tregs, the most functionally suppressive subset, were higher at diagnosis at 10.3 vs 5.5 (p = 0.05, Figure 1A,C). No differences were observed in frequency of Tregs between refractory and low‐disease‐burden groups.
PD‐1 expression was higher at diagnosis in CD4+, CD8+ cells and Tregs when compared to those with a response greater than MR3, with results representing percentage expression (4.75 vs 2.75, 5.85 vs 2.43 and 5.2 vs 2.58, p = 0.034, p = 0.003 andp < 0.001 respectively, Figure 1Di). Similarly, TIM‐3 expression was higher at diagnosis compared to those with a response greater than MR3, in CD4+ (6 vs 1.9, p = 0.027) CD8+ (15.71 vs 4.41 p <0.001, Figure 1Dii) and Tregs (5.15 vs 1.88, p = 0.002). LAG‐3 expression was higher at diagnosis in CD8+ (6.67 vs 1.4, p = 0.031, Figure 1Diii) and Tregs (2.13 vs 0.68, p = 0.031). No significant differences were observed in CTLA4 expression between these groups although a trend towards significance was observed in Tregs (2.36 vs 0.67, p = 0.055).
Despite low sample size, PD‐1 expression was also higher in patients refractory to TKI treatment compared to those with a response greater than MR3, in CD4+ cells at 5.34, and in Tregs at 5.6 (p = 0.031 and p = 0.004 respectively, Figure 1Di). In addition, TIM‐3 expression was higher in the CD8+ subset from patients with refractory CML at 11.74 (p = 0.028, Figure 1Dii) while LAG3 showed higher expression in CD4+ cells at 1.56 vs 0.45 and in CD8+ cells at 8.81 (p = 0.004 and p < 0.001 respectively, Figure 1Diii). Finally, CTLA‐4 showed higher expression in CD4+ cells at 1.59 vs 0.47 and in Tregs at 1.99 vs 0.67 (p = 0.026 and p = 0.04 respectively).
Cytokine analysis was performed in four patients with plasma samples available at diagnosis and compared with five patients with a response greater than MR3. Tumour necrosis factor (TNF)‐a levels were higher in patients at diagnosis with mean levels of 14.5 pg/ml compared to 3.1 in those with a response greater than MR3 (p = 0.036, Figure 1Ei). Similarly, interleukin (IL)‐6 was higher at diagnosis at 14.6 vs 1.8 pg/ml at a response greater than MR3 (p = 0.027, Figure 1Eii). IL‐2 was 12.1 pg/ml at diagnosis versus 1.0 (p = 0.07, Figure 1Eiii), IL‐17a was 8.4 vs 1.7 pg/ml (p = 0.07) and IL‐12p70 was 13.4 vs 2.3 pg/ml (p = 0.06, Figure 1Eiv).
A patient analysed at diagnosis and again after achieving MR3 showed significant downregulation of Treg checkpoint expression following TKI treatment. Treg PD‐1 expression decreased from 7.03 to 4.86, TIM‐3 decreased from 2.57 to 0.83 and LAG‐3 decreased from 2.23 to 0.16 (Figure 2Ai–iii), while CTLA‐4 expression remained at a stable level. Another patient was analysed whilst in MR3 and following haematological relapse. In contrast, this patient showed upregulation of checkpoint molecules, particularly TIM‐3 expression, increasing from 9.2 to 22 in CD4+ and 12.6 to 20.1 in CD8+ cells (Figure 2Bi,ii).
FIGURE 2.
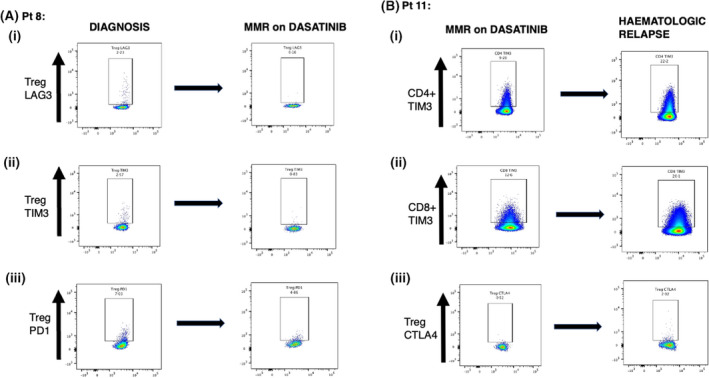
Longitudinal analysis of T‐cell immune‐checkpoint expression in paired samples from two CML patients evaluated at different disease stages. (A) Increased Treg expression of immune checkpoint‐molecules observed in patient at diagnosis (left column) compared with from the same patient after achieving MR3 (right column) following dasatinib treatment for 11 months. (i) LAG‐3 expression, (ii) TIM‐3 expression and (iii) PD‐1 expression. (B) Reduced expression of immune‐checkpoint molecules observed in another patient when in MR3 (left column) compared with samples from same patient at haematological relapse (right column). (i) CD4+ TIM‐3 expression, (ii) CD8+ TIM‐3 expression and (iii) Treg CTLA‐4 expression.
DISCUSSSION
Despite progress observed with TKI therapy in CML, a significant proportion of patients will have an inadequate response. 12 , 13 , 14 We show that immune‐checkpoint expression on T‐cell subsets correlates strongly with leukaemic disease burden. T‐cell exhaustion is defined by poor effector function with sustained expression of inhibitory receptors and is commonly observed in cancer and chronic infections. Functional T‐cell exhaustion has been previously suggested in CML patients at diagnosis, with reduced capacity for production of Th1 cytokines, when compared with patients in CCyR. 11 Hughes and colleagues noted a decrease in PD1 expression in CD4+ and CD8+ cells in CML patients responding to TKI therapy, with significantly higher PD1 expression in CD4+ and CD8+ cells at diagnosis and pre‐MR3 compared with healthy controls, with levels reducing to normal after achieving MR4.5. 6 Bruck and colleagues also recently reported on checkpoint expression in bone‐marrow samples from CML patients, showing decreased PD1 expression during TKI therapy. 10 , 15 , 16
We have expanded on these findings to show that a similar pattern is observed for other important checkpoint molecules, describing increased expression of TIM‐3 and LAG‐3, which is of significance given the recent development of inhibitors of these molecules. We demonstrate for the first time that increased expression of T‐cell exhaustion markers is observed in patients with refractory disease, although our sample size was small. We have also evaluated the expression of checkpoint molecules on Tregs, providing novel insight into their immunosuppressive mechanism in CML, with Tregs expressing checkpoint molecules including CTLA‐4 and TIM‐3 recognised to exert greatest suppressive activity. 17 , 18
Furthermore, we have found that the frequency of effector Tregs, the most functionally suppressive subset, is higher at diagnosis than with low disease burden. In addition, we observed significantly increased plasma levels of pro‐inflammatory cytokines in CML patients at diagnosis, when compared with those with low residual disease burden although this may be influenced by other variables. 19 Effector Tregs, which typically express Fas, are sensitive to myeloid‐derived inflammation and Fas‐L‐mediated cell death and as such switch to a Fas‐CD45RA+ phenotype. 20 The reduction in effector Tregs observed between diagnosis and achieving low disease burden is associated with a reduction in inflammatory cytokines and may be explained by these cells switching to a less proliferative state.
There is increasing interest in the role that inflammation plays in the progression of myeloid malignancies. It has also previously been shown that BCR‐ABL1 activity controls IL‐6 gene expression and plays a role in CML development and proliferation, with elevated IL‐6 levels also predictive of failure to obtain early molecular response. 21 , 22 Further, in‐vitro analysis has demonstrated that TNFa can regulate induction of PD1 expression in T cells, via activation of NF‐κB. 23
Finally, we have demonstrated a pro‐inflammatory state with resultant T‐cell exhaustion at diagnosis in CML, likely secondary to leukaemia‐associated antigenic overload associated with increased disease burden. These data support the future investigation of checkpoint inhibitors in patients with high‐risk disease at diagnosis as well as those with inadequate response. Analysis of the PD‐1 inhibitor nivolumab alongside dasatinib did not show meaningful clinical activity, although efficacy may have been limited by immunosuppressive effects of dasatinib. 9 , 24
Further studies evaluating alternative checkpoint targets and TKI combinations in CML are warranted and these findings should be validated in a larger cohort.
AUTHOR CONTRIBUTIONS
Patrick Harrington and Hugues de Lavallade designed the study; Patrick Harrington performed the research; Patrick Harrington and Hugues de Lavallade analysed the data and wrote the manuscript; Patrick Harrington, Richard Dillon, Deepti Radia, Donal McLornan, Claire Woodley, Susan Asirvatham, Kavita Raj, Natalia Curto‐Garcia, Jamie Saunders, Shahram Kordasti, Claire Harrison and Hugues de Lavallade helped with patient recruitment and reviewed the manuscript.
FUNDING INFORMATION
The authors have previously received research funding from Bristol Myers Squibb.
CONFLICTS OF INTEREST
Patrick Harrington: Research funding from Bristol Myers Squibb and speaker fees from Incyte. Donal McLornan: Speaker fees and advisory boards from Novartis, Celgene and Jazz pharmaceuticals. Shahram Kordasti: Research grants from Celgene and Novartis; Alexion speaker honorarium. Claire Harrison: Novartis; speaker fees from Novartis, Janssen, CTI, Celgene, Medscape; Advisory Board: Incyte, CTI, Sierra Oncology, Novartis, Celgene, Roche, AOP pharma, Geron and Astra Zenica. Hugues de Lavallade: Grants and speaker fees from Bristol Myers Squibb and Incyte; speaker fees from Novartis and Pfizer.
PATIENT CONSENT STATEMENT
Written informed consent was obtained from all patients on entry into the study.
Supporting information
Table S1
Harrington P, Dillon R, Radia D, McLornan D, Woodley C, Asirvatham S, et al. Chronic myeloid leukaemia patients at diagnosis and resistant to tyrosine kinase inhibitor therapy display exhausted T‐cell phenotype. Br J Haematol. 2022;198:1011–1015. 10.1111/bjh.18302
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Harrington P, Radia D, de Lavallade H. What are the considerations for tyrosine kinase inhibitor discontinuation in chronic‐phase chronic myeloid leukemia? Expert Rev Hematol. 2020;13(3):213–22. [DOI] [PubMed] [Google Scholar]
- 2. Kolb HJ, Schattenberg A, Goldman JM, Hertenstein B, Jacobsen N, Arcese W, et al. Graft‐versus‐leukemia effect of donor lymphocyte transfusions in marrow grafted patients . Blood. 1995;86(5):2041–50. [PubMed] [Google Scholar]
- 3. Talpaz M, Kantarjian HM, McCredie KB, Keating MJ, Trujillo J, Gutterman J. Clinical investigation of human alpha interferon in chronic myelogenous leukemia . Blood. 1987;69(5):1280–8. [PubMed] [Google Scholar]
- 4. Hsieh YC, Kirschner K, Copland M. Improving outcomes in chronic myeloid leukemia through harnessing the immunological landscape . Leukemia. 2021;35(5):1229–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ilander M, Olsson‐Stromberg U, Schlums H, Guilhot J, Bruck O, Lahteenmaki H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia . Leukemia. 2017;31(5):1108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rea D, Henry G, Khaznadar Z, Etienne G, Guilhot F, Nicolini F, et al. Natural killer‐cell counts are associated with molecular relapse‐free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study . Haematologica. 2017;102(8):1368–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Imagawa J, Tanaka H, Okada M, Nakamae H, Hino M, Murai K, et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial . Lancet Haematol. 2015;2(12):e528–35. [DOI] [PubMed] [Google Scholar]
- 8. Schutz C, Inselmann S, Saussele S, Dietz CT, Mu Ller MC, Eigendorff E, et al. Expression of the CTLA‐4 ligand CD86 on plasmacytoid dendritic cells (pDC) predicts risk of disease recurrence after treatment discontinuation in CML . Leukemia. 2017;31(4):829–36. [DOI] [PubMed] [Google Scholar]
- 9. Martinez‐Lopez J, Mustjoki S, Porkka K, Klisovic RB, Wolf D, Busque L, et al. The safety and efficacy of dasatinib plus nivolumab in patients with previously treated chronic myeloid leukemia: results from a phase 1b dose‐escalation study . Leuk Lymphoma. 2021;62(8):2040–3. [DOI] [PubMed] [Google Scholar]
- 10. Hughes A, Clarson J, Tang C, Vidovic L, White DL, Hughes TP, et al. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD‐1 and immune suppressors . Blood. 2017;129(9):1166–76. [DOI] [PubMed] [Google Scholar]
- 11. Chakraborty S, Shapiro LC, de Oliveira S, Rivera‐Pena B, Verma A, Shastri A. Therapeutic targeting of the inflammasome in myeloid malignancies . Blood Cancer J. 2021;11(9):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jabbour E, Saglio G, Hughes TP, Kantarjian H. Suboptimal responses in chronic myeloid leukemia: implications and management strategies . Cancer. 2012;118(5):1181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vigano I, Di Giacomo N, Bozzani S, Antolini L, Piazza R, Gambacorti PC. First‐line treatment of 102 chronic myeloid leukemia patients with imatinib: a long‐term single institution analysis . Am J Hematol. 2014;89(10):E184–7. [DOI] [PubMed] [Google Scholar]
- 14. Kalmanti L, Saussele S, Lauseker M, Muller MC, Dietz CT, Heinrich L, et al. Safety and efficacy of imatinib in CML over a period of 10 years: data from the randomized CML‐study IV . Leukemia. 2015;29(5):1123–32. [DOI] [PubMed] [Google Scholar]
- 15. Reuben JM, Lee BN, Johnson H, Fritsche H, Kantarjian HM, Talpaz M. Restoration of Th1 cytokine synthesis by T cells of patients with chronic myelogenous leukemia in cytogenetic and hematologic remission with interferon‐alpha . Clin Cancer Res. 2000;6(5):1671–7. [PubMed] [Google Scholar]
- 16. Bruck O, Blom S, Dufva O, Turkki R, Chheda H, Ribeiro A, et al. Immune cell contexture in the bone marrow tumor microenvironment impacts therapy response in CML . Leukemia. 2018;32(7):1643–56. [DOI] [PubMed] [Google Scholar]
- 17. Wing K, Onishi Y, Prieto‐Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA‐4 control over Foxp3+ regulatory T cell function . Science. 2008;322(5899):271–5. [DOI] [PubMed] [Google Scholar]
- 18. Gautron AS, Dominguez‐Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM‐3+ FoxP3+ regulatory T cells . Eur J Immunol. 2014;44(9):2703–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor . Immunity. 2009;30(6):899–911. [DOI] [PubMed] [Google Scholar]
- 20. Lim SP, Costantini B, Mian SA, Perez Abellan P, Gandhi S, Martinez Llordella M, et al. Treg sensitivity to FasL and relative IL‐2 deprivation drive idiopathic aplastic anemia immune dysfunction . Blood. 2020;136(7):885–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Reynaud D, Pietras E, Barry‐Holson K, Mir A, Binnewies M, Jeanne M, et al. IL‐6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development . Cancer Cell. 2011;20(5):661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nievergall E, Reynolds J, Kok CH, Watkins DB, Biondo M, Busfield SJ, et al. TGF‐alpha and IL‐6 plasma levels selectively identify CML patients who fail to achieve an early molecular response or progress in the first year of therapy . Leukemia. 2016;30(6):1263–72. [DOI] [PubMed] [Google Scholar]
- 23. Baxter AE, Kaufmann DE. Tumor‐necrosis factor is a master of T cell exhaustion . Nat Immunol. 2016;17(5):476–8. [DOI] [PubMed] [Google Scholar]
- 24. Harrington P, Dillon R, Radia DH, McLornan DP, Rousselot P, Rezvani K, et al. Inhibition of immune cell subsets is differentially affected by dasatinib dosage in patients with chronic phase CML . Blood. 2020;136:51–3. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.