

Abstract
Background
α‐Synuclein (αSyn) is believed to play a central role in Parkinson's disease (PD) neuropathology and is considered a target for disease modification. UB‐312 is a synthetic αSyn peptide conjugated to a T helper peptide and is expected to induce antibodies specifically against oligomeric and fibrillar αSyn, making UB‐312 a potential immunotherapeutic for synucleopathies.
Objective
To investigate the safety, tolerability, and immunogenicity of UB‐312 vaccination in healthy participants and to determine a safe and immunologically optimal dose for the first‐in‐patient study.
Methods
Fifty eligible healthy participants were enrolled in a 44‐week, randomized, placebo‐controlled, double‐blind study. Participants in seven cohorts were randomized to three intramuscular UB‐312 or placebo injections at weeks 1, 5, and 13 (doses ranging between 40 and 2000 μg). Safety and tolerability were assessed by adverse events, clinical laboratory, vital signs, electrocardiograms, and neurological and physical examinations. Immunogenicity was assessed by measuring serum and cerebrospinal fluid (CSF) anti‐αSyn antibody concentrations.
Results
Twenty‐three participants received all three vaccinations of UB‐312. Most adverse events were mild, transient, and self‐resolving. Common treatment‐emergent adverse events included headache, nasopharyngitis, vaccination‐site pain, lumbar puncture‐site pain, and fatigue. UB‐312 induced dose‐ and time‐dependent antibody production. Antibodies were detectable in serum and CSF of all participants receiving the 300/300/300 μg UB‐312 dose regimen. The average CSF/serum ratio was 0.2%.
Conclusions
UB‐312 was generally safe, well tolerated, and induced anti‐αSyn antibodies in serum and CSF of healthy participants. The 100 and 300 μg doses are selected for further evaluation in participants with PD. © 2022 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: Parkinson's disease, active immunotherapy, α‐synuclein, first‐in‐human, vaccine
1.
It is estimated that 7 to 10 million people worldwide are living with Parkinson's disease (PD). 1 PD evolves over many years with deterioration of motor, cognitive, behavioral, and autonomic functions caused by a progressive loss of synaptic function and neuronal death in selective brain regions. 2 The slow demise of dopaminergic and other neurotransmitter systems contributes to the spectrum of signs and symptoms in PD. 2 Although the mechanisms responsible for the dopaminergic cell loss in PD are not fully elucidated, several lines of evidence suggest that α‐synuclein (αSyn) plays a central role in the neurodegenerative process.
αSyn is a 14‐kDa (140‐amino acid) protein highly expressed in neurons, mostly at presynaptic terminals, suggesting a role in synaptic vesicle trafficking, synaptic functions, and regulation of neurotransmitter release at the synapse. 3 , 4 Duplications, point mutations, or single‐nucleotide polymorphisms in the gene encoding αSyn are known to cause or increase the risk for development of PD or dementia with Lewy bodies (LBs). Mutations have been shown to primarily alter the secondary structure of αSyn, resulting in misfolded and aggregated forms of aSyn (ie, “pathological” forms). 5 Although mutations in the αSyn gene are rare, aggregates of αSyn in the form of LBs and Lewy neurites are common neuropathological hallmarks of both familial and sporadic PD, suggesting a key role of αSyn in PD neuropathogenesis. Moreover, preformed fibrils of α‐Syn can induce the formation of LB‐like inclusions and cellular dysfunction in cell‐based assays, as well as in preclinical animal models. 6 , 7 Together, these data strongly suggest that targeting pathological forms of αSyn has therapeutic potential.
Immunotherapy approaches targeting α‐Syn have been shown to ameliorate αSyn pathology and functional deficits in mouse models of PD and were advanced into clinical development. 8 , 9 , 10 These include passive immunization therapy using humanized or human anti‐αSyn monoclonal antibodies (mAbs) or active immunization therapy aimed at inducing a humoral response against pathological αSyn. These approaches have thus far demonstrated good safety and tolerability profiles in phase 1 clinical trials. 8 , 9 , 11 Recently, a phase 2 clinical trial in patients with PD with prasinezumab, a mAb preferentially recognizing oligomeric and fibrillar forms of αSyn, showed a reduced decline in motor function and delayed time to clinically meaningful worsening of motor symptoms compared with placebo, 12 although the study did not meet its primary endpoint.
The success of a vaccine aimed at endogenous targets relies on its ability to overcome immune tolerance and generate a humoral antibody response against the desired target, while concomitantly avoiding T cell–mediated cytotoxicity. Toward that goal, a novel vaccine carrier platform was established that uses a library of proprietary synthetic UBITh® T helper peptides linked to the desired target epitopes. This synthetic peptide vaccine technology has demonstrated the feasibility of overcoming immune tolerance toward endogenous proteins, to induce a targeted B cell humoral response while avoiding T cell–mediated toxicity. 13 , 14 For instance, the UB‐311 vaccine directed against the β‐amyloid peptide and designed using this UBITh T helper peptide technology has demonstrated good safety and tolerability in the clinic, and it did not show any signs of immune response–related central nervous system adverse events (AEs). 13 , 14 , 15 , 16 Our clinical candidate for PD, UB‐312, was designed using the same UBITh technology and is expected to safely raise antibodies against pathological forms of αSyn. The 10‐residue C‐terminal epitope of αSyn contained in UB‐312 was selected after a comprehensive screening and optimization campaign of more than 60 peptide epitopes. Each epitope was conjugated to a UBITh T helper peptide via a small peptide linker and was tested for its immunogenicity in vivo. The vaccine candidate UB‐312 was selected for its ability to induce antibodies targeting specifically the pathological forms of αSyn. In preclinical studies, UB‐312 demonstrated high immunogenicity across species (unpublished data). Antibodies induced after UB‐312 immunization in guinea pigs were shown to selectively target oligomeric and fibrillar forms of αSyn, while showing little or no binding to monomeric αSyn. 17 Moreover, the antibodies specifically bound αSyn inclusions in postmortem brain sections from patients with PD, dementia with LBs, and multiple system atrophy. 17 Finally, UB‐312‐derived antibodies demonstrated neuroprotective effects in vitro, and UB‐312 immunization prevented motor function deficits in a transgenic mouse model of α‐synucleinopathy. 18 Here, we report the results of a first‐in‐human study aimed at determining the safety, tolerability, and immunogenicity of UB‐312 in healthy participants.
Subjects and Methods
Trial Design
This was a single‐center, randomized, double‐blind, placebo‐controlled study to assess safety and immunogenicity of seven treatment regimens of UB‐312 (ClinicalTrials.gov: NCT04075318). Eligible healthy participants were randomized within one of seven treatment cohorts. The first/lowest dose cohort, composed of eight participants, was randomized 6:2 (UB‐312/placebo) with sentinel dosing of the first two participants (randomized 1:1, UB‐312/placebo). The remaining cohorts were each composed of seven participants and randomized 6:1 (UB‐312/placebo). The planned cohorts (UB‐312 dose level at week 1/week 5/week 13) per protocol were as follows: cohort 1, 40/40/40 μg; cohort 2, 100/100/100 μg; cohort 3, 40/300/300 μg; cohort 4, 300/300/300 μg; cohort 5, 40/1000/1000 μg; cohort 6, 1000/1000/1000 μg; and cohort 7, 2000/2000/2000 μg. All eligible participants were enrolled in a 44‐week study period consisting of 12 weeks of dosing and up to 32 weeks of follow‐up. Between each cohort, blinded safety and tolerability data collected up to 7 days after the first dose of investigational product (IP) were reviewed before escalating to the next cohort. Vaccine description and administration, randomization, and dose escalation procedures are described in the Supporting Information. Supporting Information Table S1 shows UB‐312 vaccine components for each dose level.
This trial was performed at the Centre for Human Drug Research (CHDR, Leiden, the Netherlands) in compliance with Good Clinical Practice, the Declaration of Helsinki (2013), and local legal and regulatory requirements and applicable international regulations. Recruitment was done by CHDR's recruitment team by identifying individuals using its own database and by using advertisements to recruit new potential participants.
Participants
Male and female individuals aged 40 to 85 years, with a body mass index of 18 to 32 kg/m2, who were surgically sterile or using adequate contraception, and generally healthy with no clinically relevant abnormalities based on medical history, physical examination, clinical laboratory evaluations, and 12‐lead electrocardiograms (ECGs) were eligible to participate. Use of prohibited medications (ie, heparin or thrombolytic therapy, systemic corticosteroids, immune suppressants, immunomodulators, IPs other than this study's IP, and/or any vaccination) was not allowed from 30 days before screening until 8 weeks after last IP administration (week 21). Participants with a history of anergy or any confirmed significant allergic reactions were excluded.
Safety and Tolerability Assessments
Safety and tolerability were assessed by AEs, vital signs, neurological and physical examinations, laboratory tests, and ECGs. Brain magnetic resonance imaging (MRI) was collected for all participants at screening, and an unscheduled MRI could be requested per the investigator's judgment for safety evaluation. All participants were provided with an eDiary app for self‐recording of solicited local vaccination‐site reactions (ie, pain, tenderness, erythema/redness, induration/swelling) and systemic reactions (ie, fever, nausea/vomiting, diarrhea, headache, fatigue, myalgia, and illness) during a 7‐day period after each vaccination. Reactogenicity was assessed by the solicited AEs as reported in this eDiary. Cerebrospinal fluid (CSF) samples were collected and analyzed for leukocytes, erythrocytes, glucose, hemoglobin, albumin, and total protein, and stored for possible other assays in the event of unexpected findings in the clinical study.
Immunogenicity Assessments
Blood samples were collected at prevaccination baseline (week 1) and at all postvaccination visits (weeks 2, 5, 6, 9, 13, 14, 17, 21, 29, 37, and 45). CSF samples were collected at prevaccination baseline (week 1) and at postvaccination week 21. Immunogenicity was assessed by quantifying serum and CSF anti‐αSyn97−135 antibody concentrations using ELISA‐based methods qualified by United Neuroscience Ltd (Hsinchu, Taiwan). Antibodies against components of the vaccine (ie, CpG1 and UBITh1) were also assessed as an exploratory outcome. See detailed methods in the Supporting Information.
Exploratory Assessments
Additional exploratory measures included cytokine release in peripheral blood mononuclear cells (PBMCs), plasma and CSF inflammatory cytokines, total and free αSyn concentrations in plasma and CSF, and binding of antibodies to monomeric and oligomeric forms of αSyn. See detailed methods in the Supporting Information.
Statistical Analysis
The sample size was not based on statistical considerations but was considered adequate to initially characterize the safety, tolerability, and dose–response profile of UB‐312 immunogenicity. The trial was not powered for formal statistical comparisons between dose levels, and results presented for safety and immunogenicity analyses are descriptive. Seroconversion was defined as having anti‐αSyn97−135 antibody levels less than the lower limit of quantification before vaccination and having quantifiable levels postvaccination.
Safety and tolerability were analyzed based on the safety population defined as all participants randomized and exposed to at least one dose of the study drug, identical to the modified intent‐to‐treat population. The analyses of immunogenicity endpoints were performed by the treatment allocation and based on the per‐protocol (PP) population. The PP population was defined as all participants who received all planned doses of the study drug, completed the treatment period, fulfilled all entry criteria, and had no critical or major protocol deviations that required exclusion of the participant.
Results
Study Population
Screening of study participants took place between July and November 2019, and the study was conducted between August 2019 and August 2020. A total of 50 healthy participants were randomized to receive UB‐312 or matching placebo (Fig. 1). Baseline characteristics were similar across treatment groups (Table 1). None of the participants had a notable ongoing medical history that was deemed clinically relevant by the investigator (Supporting Information Table S2). Vascular risk factors, generally relevant in the parkinsonian population, were reported in only one subject.
FIG. 1.
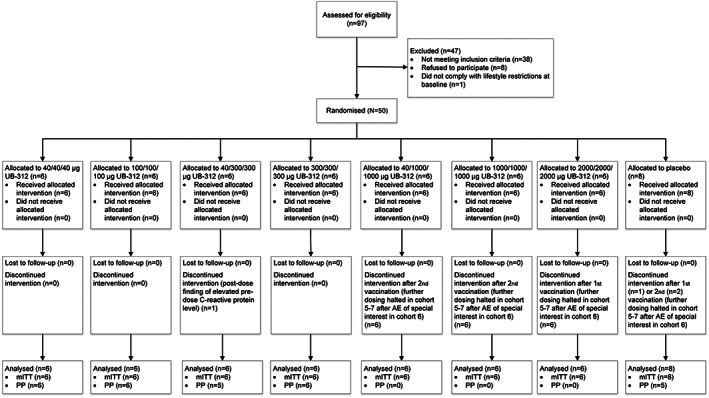
Consort flow diagram. AE, adverse event; mITT, modified intent‐to‐treat; PP, per protocol.
TABLE 1.
. Demographics of study participants
| Placebo (n = 8) | 40/40/40 μg UB‐312 (n = 6) | 100/100/100 μg UB‐312 (n = 6) | 40/300/300 μg UB‐312 (n = 6) | 300/300/300 μg UB‐312 (n = 6) | 40/1000 μg UB‐312 (n = 6) | 1000/1000 μg UB‐312 (n = 6) | 2000 μg UB‐312 (n = 6) | All Participants (N = 50) | |
|---|---|---|---|---|---|---|---|---|---|
| Age (y), median (range) | 70 (41–76) | 74 (62–79) | 69 (49–79) | 72 (49–77) | 60 (46–77) | 69 (44–85) | 69 (64–73) | 72 (64–80) | 70 (41–85) |
| Sex, n (%) | |||||||||
| Female | 4 (50.0) | 2 (33.3) | 4 (66.7) | 4 (66.7) | 4 (66.7) | 2 (33.3) | 1 (16.7) | 4 (66.7) | 25 (50.0) |
| Male | 4 (50.0) | 4 (66.7) | 2 (33.3) | 2 (33.3) | 2 (33.3) | 4 (66.7) | 5 (83.3) | 2 (33.3) | 25 (50.0) |
| Race, n (%) | |||||||||
| White | 7 (87.5) | 6 (100) | 5 (83.3) | 6 (100) | 6 (100) | 5 (83.3) | 6 (100) | 5 (83.3) | 46 (92.0) |
| Black or African American | 1 (12.5) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 2 (4.0) |
| Asian | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (2.0) |
| Hispanic | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 1 (16.7) | 0 (0) | 0 (0) | 1 (2.0) |
| BMI (kg/m2), median (range) | 25.7 (23.1–30.6) | 22.4 (20.3–26.9) | 25.1 (23.8–31.4) | 24.6 (19.9–31.0) | 24.2 (22.6–30.6) | 25.3 (24.1–27.3) | 24.9 (24.0–27.1) | 26.0 (24.2–29.7) | 24.7 (19.9–31.4) |
BMI, body mass index.
All participants in cohorts 1 to 4 received three vaccinations as per protocol, except for one subject in cohort 2 who had abnormal baseline laboratory values and was terminated from the study after receiving one dose of UB‐312. Dosing of cohorts 5 to 7 was halted after one subject in cohort 6 developed an AE of special interest, ie, grade 3 flu‐like symptoms. Due to the coronavirus disease 2019 (COVID‐19) pandemic and lockdown measures, the first two follow‐up visits were completed by a safety phone call instead of an on‐site visit for most participants. The last follow‐up visit was conducted on‐site for all participants with blood sampling and physical examination. One participant received tetanus vaccination because of a minor trauma to the head after a bicycle accident.
Safety
Vaccination with UB‐312 was generally safe and well tolerated up to a dose regimen of 300/300/300 μg UB‐312. No deaths and no serious AEs were reported. No clinically relevant or treatment‐emergent trends were observed in clinical laboratory data, physical examinations, neurological examinations, vital signs, and ECGs. No clinically significant trends or changes in T cell–mediated inflammatory response were observed (Supporting Information Fig. S1). Because there were no signs or symptoms of an adverse neuroinflammatory response, no postvaccination brain MRIs were performed. Further supporting the absence of central nervous system inflammation, CSF levels of glucose, albumin, and total protein remained within normal range (data not shown), while leukocytes (data not shown) and cytokines (Supporting Information Fig. S2) did not change from baseline after immunization with UB‐312.
Of the 50 healthy participants, 39 of 42 (92.9%) receiving UB‐312 and 8 of 8 (100%) receiving placebo experienced at least one treatment‐emergent AE (TEAE). The most common TEAEs (ie, reported by at least 10% of the participants), irrespective of study drug relatedness, were headache, nasopharyngitis, vaccination‐site pain, lumbar puncture‐site pain, and fatigue (Table 2). The vast majority (96%) of 193 TEAEs were mild in severity. There were 96 AEs classified as possibly or probably related to treatment by the investigator, of which 32 were vaccination‐site reactions and 31 were headaches. In the 7 days after each vaccination, tenderness and pain at the vaccination site were the most frequent local reactions reported in the eDiary. The most common systemic events that were self‐reported in the 7 days after each vaccination were headache, fatigue, and muscle pain. No increase in reactogenicity was observed after repeated vaccination.
TABLE 2.
Summary of treatment‐emergent adverse events occurring in at least 10% of participants
| AE | Placebo (n = 8) | 40/40/40 μg UB‐312 (n = 6) | 100/100/100 μg UB‐312 (n = 6) | 40/300/300 μg UB‐312 (n = 6) | 300/300/300 μg UB‐312 (n = 6) | 40/1000 μg UB‐312 (n = 6) | 1000/1000 μg UB‐312 (n = 6) | 2000 μg UB‐312 (n = 6) |
|---|---|---|---|---|---|---|---|---|
| Any TEAE, n (%) | 8 (100.0) | 5 (83.3) | 6 (100.0) | 5 (83.3) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 5 (83.3) |
| TEAEs in >10% of participants, n (%) | ||||||||
| Headache | 5 (62.5) | 2 (33.3) | 3 (50.0) | 3 (50.0) | 3 (50.0) | 3 (50.0) | 2 (33.3) | 3 (50.0) |
| Nasopharyngitis | 5 (62.5) | 1 (16.7) | 4 (66.7) | 2 (33.3) | 2 (33.3) | 3 (50.0) | 1 (16.7) | 2 (33.3) |
| Vaccination‐site pain | 2 (25.0) | 3 (50.0) | 1 (16.7) | 0 (0) | 4 (66.7) | 2 (33.3) | 2 (33.3) | 2 (33.3) |
| Lumbar puncture‐site pain | 2 (25.0) | 3 (50.0) | 1 (16.7) | 0 (0) | 1 (16.7) | 0 (0) | 3 (50.0) | 3 (50.0) |
| Fatigue | 1 (12.5) | 0 (0) | 0 (0) | 3 (50.0) | 2 (33.3) | 0 (0) | 2 (33.3) | 0 (0) |
Percentage is based on the number of subjects within the category in the column heading. Classifications are based on Medical Dictionary for Regulatory Activities (MedDRA) version 21.1.
TEAE, treatment‐emergent adverse event.
Two participants developed flu‐like symptoms with a temporal relationship to dosing. One participant (cohort 6, 1000/1000 μg UB‐312) developed symptoms of fever, malaise, nausea, and vomiting after administration of the second dose. The event was graded as severe (grade 3). The participant already had slightly elevated C‐reactive protein and erythrocyte sedimentation rate (ESR) before the second drug administration. Proinflammatory cytokines measured 24 hours after the second administration were within baseline values. Antibodies (both anti‐αSyn and anti‐CpG1) observed in this subject were high and developed rapidly but were not disproportionally high compared with the rest of the cohort. Another participant (cohort 4, 300/300/300 μg UB‐312) developed symptoms of moderate (grade 2) fever, malaise, and nausea after the third vaccination. Proinflammatory cytokines (ie, tumor necrosis factor α, interferon‐γ, interleukin [IL]‐2, IL‐6, and IL‐8) measured for this participant 6 hours after the third administration were elevated compared with baseline and reverted to normal limits within baseline values based on samples collected 1 week after administration. Paracetamol was given on both occasions for symptom relief. Both AEs were transient and resolved without intervention within 24 hours.
Most follow‐up visits during the COVID‐19 pandemic were performed remotely, and AE data were collected by phone; no safety trends were reported during the COVID‐19 pandemic.
UB‐312 Immunogenicity in Cohorts 1 to 4
Before UB‐312 vaccination, none of the participants in the UB‐312 groups had detectable anti‐αSyn antibodies against the synthetic αSyn97−135 peptide (target epitope). In contrast, one participant in the placebo group had detectable but low levels of naturally occurring anti‐αSyn97−135 antibodies (1.23 μg/mL) at baseline, which remained low and stable throughout the study. Overall, UB‐312 vaccination generated robust, dose‐ and time‐dependent serum antibodies against the target epitope (Fig. 2). Increases in antibodies were seen at week 9 (4 weeks after the second vaccination) for all cohorts, with anti‐αSyn97−135 antibody concentrations peaking at week 17 (4 weeks after the third vaccination) and subsequently declining during the follow‐up period. Mean serum anti‐αSyn97−135 antibody concentrations were still greater than baseline values at week 45 in the 100/100/100, 40/300/300, and 300/300/300 μg cohorts (concentration means 17.07, 2.86, and 19.12 μg/mL, respectively). The greatest increase was seen at week 17 in cohort 4 (300/300/300 μg), reaching a mean serum antibody concentration of 65.44 μg/mL (ranged from 10.31 to 166.71 μg/mL).
FIG. 2.
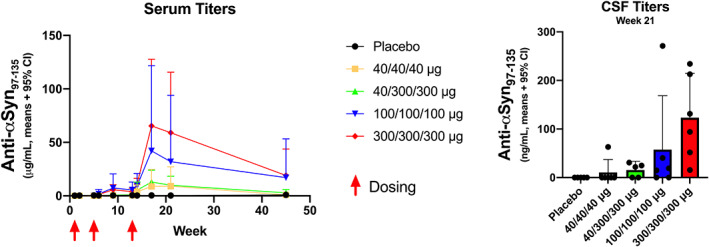
Serum and cerebrospinal fluid (CSF) concentrations of epitope‐specific antibodies (PP population). CI, confidence interval; α‐syn, alpha‐synuclein.
After three vaccinations, five of six participants in cohort 1 (40/40/40 μg), five of six participants in cohort 2 (100/100/100 μg), five of five participants in cohort 3 (40/300/300 μg), and six of six participants in cohort 4 (300/300/300 μg) met the definition of seroconversion. The overall seroconversion rate for 23 participants completing three vaccinations was 91.3% (n = 21).
Target epitope‐specific anti‐αSyn antibodies were detectable in the CSF sampled at week 21, while none were detectable in prevaccination CSF samples. For participants with detectable CSF antibodies (n = 1 in cohort 1, n = 4 in cohort 2, n = 3 in cohort 3, and n = 6 in cohort 4), the average CSF/serum ratio (CSF antibody concentration to serum antibody concentration at week 21) was 0.2%.
The immune response was specific to the target B cell epitope with no antibody response against the UBITh1 peptide and minimal response against CpG1 (Supporting Information Fig. S3). Analysis of cytokine secretion by PBMCs collected preimmunization and postimmunization and stimulated with the αSyn peptide epitope indicated minimal activation of T cells, if any (Supporting Information Fig. S1). The concentration of total and free αSyn in plasma and CSF did not change after vaccination throughout the study, as expected in healthy participants (Supporting Information Fig. S4). Preliminary dot blot analyses of the antibodies produced in healthy volunteers immunized with UB‐312 (Supporting Information Fig. S5) showed that the postimmune serum IgG fraction recognized all forms of αSyn, with a greater signal intensity for oligomers than monomers at each concentration tested.
Discussion
In this first‐in‐human study, UB‐312 was generally safe, well tolerated, and generated robust time‐ and dose‐dependent anti‐αSyn97−135 antibodies detectable in both serum and CSF. The overall safety profile showed no substantial difference between the UB‐312‐vaccinated groups and the placebo group, with the majority of TEAEs being transient and self‐resolving. Local vaccination‐site reactions did not increase after subsequent immunizations, and no severe local reactions were observed (as seen previously with other active immunotherapies targeting αSyn 19 ).
Dosing of cohorts 5 to 7 was deferred after observation of one AE of special interest in cohort 6, and eventually the decision was made not to resume dosing. A similar AE was observed in a participant in cohort 4, albeit less severe. The systemic AEs observed in these two participants were characteristic of an acute‐phase reaction associated with cytokine and chemokine activation after vaccination, which was consistent with the frequency of occurrence of acute‐phase reactions postvaccination. 20 , 21 , 22 , 23 This type of flu‐like reaction is commonly observed with active vaccines containing a potent adjuvant. Similar reactions have been observed for other nonreplicating vaccines, both approved and in clinical development. Effective vaccination requires induction of inflammatory cytokines and chemokines, 24 and these may be associated with systemic AEs similar to those observed in this trial. Interestingly, it has been reported that the extent of local and systemic AEs postvaccination may not be a meaningful predictor of antibody responses. 25 Our data support this observation because no significant differences were observed in anti‐αSyn antibody levels in subjects with and without flu‐like reaction, possibly precluding the correlation between the elevated systemic response and immunogenicity of UB‐312. Moreover, the flu‐like reactions and titer responses could not be clearly correlated to cytokine responses. Given the self‐limiting nature of the flu‐like reactions and their resemblance to such events occurring with many other vaccines, we believe the safety and reactogenicity of UB‐312 is acceptable and does not preclude further development of doses up to 300 μg.
All participants in the 300/300/300 μg group seroconverted. Achieving a 100% seroconversion rate is very encouraging and highlights the unique ability of the UBITh platform to overcome immune tolerance and target endogenous proteins of chronic disease, e.g., amyloid β and αSyn. The UBITh peptide linked to the C terminus of the αSyn peptide in UB‐312 did not elicit an antibody response against itself (ie, no anti‐UBITh1 antibodies). This approach also avoided the generation of T cell responses to the target epitope as assessed by T cell FluoroSpot. Results from this study are consistent with those previously reported with the UB‐311 UBITh anti‐amyloid ß vaccine, 13 , 14 further supporting the utility of the UBITh vaccine technology for targeting endogenous proteins. Indeed, the UBITh peptide vaccine technology has been optimized toward overcoming immune tolerance in a safe way and to trigger a B cell–mediated antibody production without activating the innate immune system or risking a T cell–mediated cytotoxic response. The αSyn epitope has been selected based on the following criteria: immunosilent on its own and immunogenic when conjugated to the T helper peptide (ie, the T helper peptide drives the type of immune response), present on pathological forms of αSyn, and accessible to antibodies. In this study, UB‐312 induced a strong IgG response (ie, specific anti‐αSyn antibody response) without signs of a systemic inflammatory response (ie, plasma cytokines), absence of antibody response against the T helper peptide, and very limited antibody response against CpG1. Furthermore, PBMCs collected after immunization had minimal interferon‐γ cytokine (Th1) release on stimulation with the antigen, confirming the lack of cellular immunogenicity of the αSyn antigen and the low risk for an autoimmune reaction.
In preclinical studies, UB‐312 induced antibodies against pathological αSyn as demonstrated by strong immunolabeling of αSyn inclusions in brain sections from patients with synucleinopathies. 17 Moreover, characterization of the binding profile of anti‐αSyn antibodies produced in guinea pigs after UB‐312 vaccination demonstrated strong affinity to a variety of fibrillar αSyn strains, as well as to naturally occurring αSyn oligomers. In contrast, these anti‐αSyn antibodies demonstrated poor binding to monomeric αSyn. 17 Preliminary dot blot analyses of the antibodies produced in healthy volunteers immunized with UB‐312 also suggest a preferential binding to oligomeric forms of αSyn. These antibodies and those produced in patients with PD will be further characterized.
In this first‐in‐human study, vaccination with UB‐312 induced αSyn‐specific serum antibody concentrations in the same range as those achieved in other active and passive immunotherapy studies (ie, typically requiring serum antibody concentrations of ~10–20 μg/mL for target engagement). 8 , 9 , 10 , 26 Minimal effects were observed on normal αSyn levels in plasma and CSF. This was expected because the UB‐312 vaccine was designed to induce antibodies recognizing pathological forms of αSyn and not normal αSyn. This is the first study to demonstrate target‐specific antibodies in the CSF after vaccination with a UBITh vaccine. The serum/CSF ratio was generally similar across doses and was consistent with ratios observed after mAb immunotherapy in healthy participants and participants with PD. 11 , 26 Whether the antibodies produced after immunization with UB‐312 can reach pathological αSyn located in neurons is unknown. However, αSyn can be released extracellularly and propagate PD pathology in animal models by its neuron‐to‐neuron spread. 27 , 28 , 29 , 30 Furthermore, “αSyn seeds” are present in the CSF of patients with PD. 31 This suggests that antibodies directed against pathological αSyn might bind extracellular forms of αSyn and neutralize their toxic effects. Furthermore, one should not neglect the potential role of antibodies at clearing peripheral αSyn toxic species, which might alleviate some of the nonmotor and/or peripheral symptoms, such as gastrointestinal symptoms, in patients. An ongoing clinical trial in patients with PD will aim to address whether antibodies produced after immunization reach sufficient expression levels to achieve target engagement, ie, reduce the amount of pathological forms of αSyn in CSF as analyzed using protein misfolding cyclic amplification.
Based on the safety and immunogenicity profile observed in healthy participants, both the 100 and 300 μg doses were selected for further evaluation of safety, tolerability, and immunogenicity in patients with PD. Results from the ongoing study of UB‐312 in patients with PD will provide more information about the pharmacokinetics of the antibodies and the potential boosting frequency in this patient population. High responder rates, reproducibility of response, and generation of antibodies directed to relevant toxic protein species are key elements of an effective therapeutic vaccine for neurodegenerative conditions. If UB‐312 continues to be safe, tolerable, and immunogenic with a selective humoral response toward pathological forms of αSyn, this vaccine approach may have significant advantages over other mAb immunotherapies; ie, it is convenient (intramuscular injection with less frequent dosing), cost‐effective (potentially fewer administrations needed), and suitable for early treatment and prevention of synucleinopathies.
Author Roles
1. Research project: A. Conception, B. Organization, C. Execution;
2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique;
3. Manuscript: A. Writing of the first draft, B. Review and Critique.
H.J.Y.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B.
E.T.: 1A, 1B, 1C, 2A, 2C, 3A, 3B.
E.v.B.: 1A, 1B, 1C, 2A, 2C, 3B.
J.L.v.d.P.: 1A, 1B, 1C, 2A, 2C, 3B.
I.R.: 1B, 1C, 2A, 2C, 3B.
M.M.: 1A, 2C, 3B.
E.H.: 1C.
G.J.G.: 1A, 1B, 1C, 2A, 2C, 3B.
J.‐C.D.: 1A, 1B, 1C, 2B, 2C, 3A, 3B.
Full Financial Disclosures for the Previous 12 Months
H.J.Y.: employee and hold stocks of Vaxxinity, Inc.
E.T.: employee of CHDR and LUMC, Leiden, the Netherlands.
E.v.B.: employee of CHDR, Leiden, the Netherlands.
J.L.v.d.P.: employee of CHDR and LUMC, Leiden, the Netherlands.
I.R.: employee of CHDR, Leiden, the Netherlands.
M.M.: employee of CHDR, Leiden, the Netherlands.
E.H.: employee of Vaxxinity, Inc.
G.J.G.: Chief Scientific Officer/Chief Medical Officer at CHDR and professorship at LUMC, Leiden, the Netherlands.
J.‐C.D.: employee and hold stocks of Vaxxinity, Inc.
Relevant conflicts of interest/financial disclosures
H.J.Y. and J.‐C.D. are employees and hold stocks in Vaxxinity.
Supporting information
Appendix S1. Supporting Information
Acknowledgments
The study was supported by Vaxxinity Inc. (formerly known as United Neuroscience Ltd). We thank the healthy volunteers who participated in this study.
Data Availability Statement
Authors have had full access to the data, have the right to publish all the data, and have had the right to obtain independent statistical analyses of the data. Authors take responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1. Yang W, Hamilton JL, Kopil C, Beck JC, Tanner CM, Albin RL, et al. Current and projected future economic burden of Parkinson's disease in the U.S. NPJ Parkinsons Dis 2020;6:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primer 2017;3:17013 [DOI] [PubMed] [Google Scholar]
- 3. Bartels T, Choi JG, Selkoe DJ. α‐Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011;477:107–110. 10.1038/nature10324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Valdinocci D, Radford RAW, Siow SM, Chung RS, Pountney DL. Potential modes of intercellular α‐synuclein transmission. Int J Mol Sci 2017;18:469 http://europepmc.org/abstract/MED/28241427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Melki R. How the shapes of seeds can influence pathology. Neurobiol Dis 2018;109:201–208. [DOI] [PubMed] [Google Scholar]
- 6. Stefanis L, Kholodilov N, Rideout HJ, Burke RE, Greene LA. Synuclein‐1 is selectively up‐regulated in response to nerve growth factor treatment in PC12 cells: synuclein induction by NGF. J Neurochem 2001;76:1165–1176. [DOI] [PubMed] [Google Scholar]
- 7. Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VMY. Intracerebral inoculation of pathological α‐synuclein initiates a rapidly progressive neurodegenerative α‐synucleinopathy in mice. J Exp Med 2012;209:975–986. 10.1038/nature10324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W, et al. First‐in‐human assessment of PRX002, an anti–α‐synuclein monoclonal antibody, in healthy volunteers. Mov Disord 2017;32:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Volc D, Poewe W, Kutzelnigg A, Lührs P, Thun‐Hohenstein C, Schneeberger A, et al. Safety and immunogenicity of the α‐synuclein active immunotherapeutic PD01A in patients with Parkinson's disease: a randomised, single‐blinded, phase 1 trial. Lancet Neurol 2020;19:591–600. [DOI] [PubMed] [Google Scholar]
- 10. Kuchimanchi M, Monine M, Kandadi Muralidharan K, Woodward C, Penner N. Phase II dose selection for alpha synuclein–targeting antibody cinpanemab (BIIB054) based on target protein binding levels in the brain. CPT Pharmacometrics Syst Pharmacol 2020;9:515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, et al. Randomized phase I clinical trial of anti‐α‐synuclein antibody BIIB054. Mov Disord 2019;34:1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prasinezumab slows progression on measures of Parkinson's disease in phase 2 study. Prothena Corporation plc. Accesed August 9, 2021.
- 13. Wang CY, Finstad CL, Walfield AM, Sia C, Sokoll KK, Chang T‐Y, et al. Site‐specific UBITh® amyloid‐β vaccine for immunotherapy of Alzheimer's disease. Vaccine 2007;25:3041–3052. 10.1016/j.vaccine.2007.01.031 [DOI] [PubMed] [Google Scholar]
- 14. Wang CY, Wang P‐N, Chiu M‐J, Finstad CL, Lin F, Lynn S, et al. UB‐311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer's disease. Alzheimers Dement 2017;3:262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. United Neuroscience Ltd (2020). A Randomized, Double‐blind, Placebo‐controlled, 3‐arm Parallel‐group, Multicenter, Phase IIa Study to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of UBITh® AD Immunotherapeutic Vaccine (UB‐311) in Patients with Mild Alzheimer's Disease. clinicaltrials.gov; (Report No. NCT02551809). https://clinicaltrials.gov/ct2/show/NCT02551809
- 16. United Neuroscience Ltd . (2021). An Extension Study of a Phase IIa Study in Patients with Mild Alzheimer's Disease to Evaluate the Safety, Tolerability, Immunogenicity, and Efficacy of UBITh® AD Immunotherapeutic Vaccine (UB‐311) . clinicaltrials.gov; (Report No: NCT03531710). https://clinicaltrials.gov/ct2/show/NCT03531710
- 17. Nimmo JT, Verma A, Dodart J‐C, Wang CY, Savistchenko J, Melki R, et al. Novel antibodies detect additional α‐synuclein pathology in synucleinopathies: potential development for immunotherapy. Alzheimers Res Ther 2020;12:159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nimmo JT, Smith H, Wang CY, Teeling JL, Nicoll JAR, Verma A, et al. Immunisation with UB‐312 in the Thy1SNCA mouse prevents motor performance deficits and oligomeric α‐synuclein accumulation in the brain and gut. Acta Neuropathol (Berl) 2021;143(1):55–73. 10.1007/s00401-021-02381-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meissner WG, Traon AP‐L, Foubert‐Samier A, Galabova G, Galitzky M, Kutzelnigg A, et al. A phase 1 randomized trial of specific active α‐synuclein immunotherapies PD01A and PD03A in multiple system atrophy. Mov Disord 2020;35:1957–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines 2011;10:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lal H, Cunningham AL, Godeaux O, Chlibek R, Diez‐Domingo J, Hwang S‐J, et al. Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N Engl J Med 2015;372:2087–2096. [DOI] [PubMed] [Google Scholar]
- 22. Adjuvants and vaccines | Vaccine Safety. CDC . 2020. Accesed April 16, 2021. https://www.cdc.gov/vaccinesafety/concerns/adjuvants.html
- 23. Scheiermann J, Klinman DM. Clinical evaluation of CpG oligonucleotides as adjuvants for vaccines targeting infectious diseases and cancer. Vaccine 2014;32:6377–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Talaat KR, Halsey NA, Cox AB, Coles CL, Durbin AP, Ramakrishnan A, et al. Rapid changes in serum cytokines and chemokines in response to inactivated influenza vaccination. Influenza Other Respir Viruses 2018;12:202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Christian LM, Porter K, Karlsson E, Schultz‐Cherry S. Proinflammatory cytokine responses correspond with subjective side effects after influenza virus vaccination. Vaccine 2015;33:3360–3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti–α‐synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol 2018;75:1206–1214. http://jamanetwork.com/journals/jamaneurology/fullarticle/2685097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jan A, Gonçalves NP, Vaegter CB, Jensen PH, Ferreira N. The prion‐like spreading of alpha‐synuclein in Parkinson's disease: update on models and hypotheses. Int J Mol Sci 2021;22:8338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stefanis L, Emmanouilidou E, Pantazopoulou M, Kirik D, Vekrellis K, Tofaris GK. How is alpha‐synuclein cleared from the cell? J Neurochem 2019;150:577–590. [DOI] [PubMed] [Google Scholar]
- 29. Hijaz BA, Volpicelli‐Daley LA. Initiation and propagation of α‐synuclein aggregation in the nervous system. Mol Neurodegener 2020;15:19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Upcott M, Chaprov KD, Buchman VL. Toward a disease‐modifying therapy of alpha‐synucleinopathies: new molecules and new approaches came into the limelight. Molecules 2021;26:7351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwon EH, Tennagels S, Gold R, Gerwert K, Beyer L, Tönges L. Update on CSF biomarkers in Parkinson's disease. Biomolecules 2022;12:329 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information
Data Availability Statement
Authors have had full access to the data, have the right to publish all the data, and have had the right to obtain independent statistical analyses of the data. Authors take responsibility for the integrity of the data and the accuracy of the data analysis.