

Abstract
The estimation of nuclear DNA content has been by far the most popular application of flow cytometry in plants. Because flow cytometry measures relative fluorescence intensities of nuclei stained by a DNA fluorochrome, ploidy determination, and estimation of the nuclear DNA content in absolute units both require comparison to a reference standard of known DNA content. This implies that the quality of the results obtained depends on the standard selection and use. Internal standardization, when the nuclei of an unknown sample and the reference standard are isolated, stained, and measured simultaneously, is mandatory for precise measurements. As DNA peaks representing G1/G0 nuclei of the sample and standard appear on the same histogram of fluorescence intensity, the quotient of their position on the fluorescence intensity axis provides the quotient of DNA amounts. For the estimation of DNA amounts in absolute units, a number of well‐established standards are now available to cover the range of known plant genome sizes. Since there are different standards in use, the standard and the genome size assigned to it has always to be reported. When none of the established standards fits, the introduction of a new standard species is needed. For this purpose, the regression line approach or simultaneous analysis of the candidate standard with several established standards should be prioritized. Moreover, the newly selected standard organism has to fulfill a number of requirements: it should be easy to identify and maintain, taxonomically unambiguous, globally available, with known genome size stability, lacking problematic metabolites, suitable for isolation of sufficient amounts of nuclei, and enabling measurements with low coefficients of variation of DNA peaks, hence suitable for the preparation of high quality samples.
Keywords: best practices, C‐value, flow cytometry, GC content, genome size, plant sciences, plant standard species, standardization
1. INTRODUCTION
Flow cytometry (FCM) allows rapid and accurate quantification of light scatter and fluorescence of microscopic particles during their movement at high speed in a narrow stream of liquid. As large plant populations can be measured, small subpopulations can be identified [1]. In plants, the most popular application of FCM has been the estimation of the nuclear DNA content (genome size, C‐values; [2, 3]). The assay is relatively straightforward and relies on staining nuclei by a DNA‐specific fluorescent dye and quantification of the amount of the bound fluorochrome [4, 5]. Thanks to the availability of affordable and easy‐to‐use flow cytometers, the method gradually replaced Feulgen densitometry, which is more laborious, has much lower throughput, and for which the production of new instruments was discontinued. Similar to Feulgen densitometry, FCM does not measure the nuclear DNA content in absolute units. In order to transform arbitrary to absolute units, mean peak position on the fluorescence intensity axis (PP) of the nuclei of the unknown sample is compared with that of a reference standard with known C‐value following the equation
Note: The resulting object's C‐level depends on the inserted C‐level of the standard organism—the C‐level is the number preceding the term “C‐value”. For instance, in the equation above it is 2. The equation with 2C is the typical case when working with mature vascular plants, while in some groups (e.g., bryophytes) one or both values may be 1C. The C‐level of a given nucleus depends on its stage within the courses of the cell cycle and the organism's life cycle (generative polyploidy is not discussed here, for nomenclature details, see [2, 3]). Nuclei are referred to according to the FCM relevant phases of the cell cycle as either G1/G0 or G2, whereas the alternation of generations (haplo‐diplontic life cycle) is connected to a change in the nuclear phases resulting a haplophasic generation (gametophyte/one holoploid chromosome set per nucleus) and a diplophasic generation (sporophyte/two holoploid chromosome sets per nucleus). As a consequence, regular gametes contain one holoploid chromosome set, each chromosome consisting of one chromatid. They are at the 1C‐level. Subsequent fertilization results in nuclei containing two holoploid chromosome sets with each one chromatid per chromosome or, in other words, two chromatids in each homologous chromosome pair, the 2C‐level. Replication of the DNA during the course of cell cycle doubles the number of chromatids from one chromatid per chromosome in the G1/G0 nuclei to two chromatids per chromosome within the G2 nuclei and, therefore, cause nuclei on the 4C‐level in the sporophytic generation (i.e., two chromatids per chromosome = 4 chromatids per homologous chromosome pair), but the 2C‐level in the gametophytic generation (i.e., two chromatids per chromosome, one chromosome set). In a mixed preparation containing different generations or cell cycle phases, the different C‐levels have to be compensated by an introduced factor. For example, given a gametophytic object (e.g., a moss or pollen) was prepared together with a sporophytic standard organism, the equation has the form 1C‐valueObject = 2C‐valueStandard × PPObject G1/G0 ÷ PPStandard G1/G0, and thus the object's 2C‐value = 2 × 2C‐valueStandard × PPObject G1/G0 ÷ PPStandard G1/G0. See References [2, 3] for a detailed overview including the distinction between holoploid (C‐levels) and monoploid genome size (Cx‐levels), which is particularly important in generative polyploids.
This relation makes the standardization the critical point in the whole procedure. This paper summarizes the information on standardization methods, reference standard selection, and the establishment of new standards.
2. TYPES OF STANDARDIZATION PROCEDURES
External standardization: The nuclei of the sample and the reference standard are isolated, stained, and analyzed separately. The positions of DNA peaks in both samples are recorded and the nuclear DNA content of the unknown sample is calculated based on the quotient of the PP. Even if the instrument is calibrated repeatedly, for example, each time after analyzing a few samples, this approach is not suitable to obtain precise data. Varying drifts intrinsic to the process of preparation (dye saturation of the DNA, preparation age or temperature‐driven nuclear disintegration, staining inhibition associated with the presence of secondary metabolites [6, 7]) and/or drifts inherent to the instruments (voltage‐dependent photomultiplier amplification, temperature‐dependent laser efficiency, sample flow shifts) may lead to variation in nuclear fluorescence intensity [8] and, therefore, to PP variation during the course of measurements. External standardization may be used for preliminary determination of genome size during the selection process of a suitable internal reference standard, as a tool for the standard peak identification during the course of measurement, or ploidy level estimation [9]. However, the method of external standardization is unacceptable for accurate measurements of nuclear DNA contents.
Pseudo‐internal standardization: In this type of standardization, suspensions of nuclei are prepared separately from the unknown sample and the reference standard and only afterward both are merged, stained, and measured simultaneously. The reason to do so may be the impossibility to isolate nuclei from both sources simultaneously, for example, when they are isolated by chopping plant tissues and by hypotonic lysis of animal or human cells. This approach does not enable precise measurements because the sample and the standard nuclei may be influenced by several factors in different ways (such as the effect of staining inhibitors or dye saturation, as described previously). Pseudo‐internal standardization may be acceptable when it is not possible to co‐process both sample and standard from the very beginning and high‐precision genome size measurement is not required. In the regard of requesting more precise approaches, caution is needed when interpreting such results.
Internal standardization: Nuclei of the unknown sample and the reference standard are isolated, stained, and measured simultaneously in the same suspension. This approach ensures identical treatment conditions for the nuclei of the standard and the unknown sample during the whole duration of sample preparation until measurement. Potential interfering factors, such as the presence of secondary metabolites [6, 7] are affecting both nuclei types in the same manner. This is the only standardization method acceptable for accurate measurement of genome size, detection of aneuploidy, and DNA base composition (not discussed in this paper—see Reference [9]).
One of the most serious mistakes in precise genome size measurement with FCM is the use of external standardization, which leads to artificial variation across individual samples (see References [10, 11, 12]). The extent of the peak shift that may occur even within a short time during the course of measurement must not be underestimated. The difference between internal and external standardization is demonstrated by the following experiment (Figure 1). Leaf tissues of Pisum sativum (P.s.) and Secale cereale (S.c.) were co‐chopped and the resulting nuclei suspension was split into two tubes. To each tube, staining buffer containing propidium iodide was added and five measurement runs per tube were performed at the same voltage settings of the instrument (laser equipped). The whole experiment took about 1 h. The positions of the corresponding G1/G0 nuclei peak pairs (S.c. and P.s.) were recorded. Peak position quotients (i.e., peak index) following the internal standardization approach from these pairs, showed 1.012‐fold variation among the 10 runs. The entire sample and instrument associated variation impacted all nuclei populations in the same way, hence the ratio between them was stable over all runs. However, resembling an approach with external standardization, the highest measured value for G1/G0 fluorescence (that of S.c.) and the lowest (from P.s.) were used to calculate the greatest possible S.c./P.s. quotient. Vice‐versa, the lowest value for the S.c. fluorescence together with the highest value from P.s. gave the smallest possible quotient within the underlying data set. The variation in this simulated approach of external standardization was up to 1.31‐fold in this data set, which was produced from, with regard to secondary substances, non‐problematic material. The peak shifts simply originated from the sample as well as instrument‐intrinsic drift during the course of measurement.
FIGURE 1.
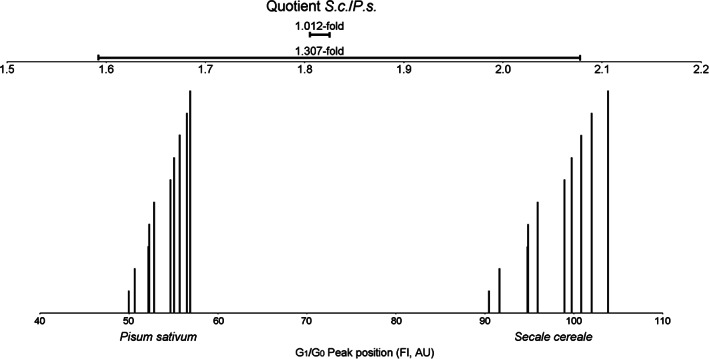
Lower scale: Illustration of the effect of G1/G0 peak position variation (in arbitrary units (AU)) on the fluorescence intensity (FI) axis of co‐chopped Secale cereale (S.c.) and Pisum sativum (P.s.) in two simultaneous preparations after five measurements each. The corresponding S.c./P.s. G1/G0 peak pairs share the same height and the peak shifts on the fluorescence intensity axis are clearly visible. The quotient accuracy is illustrated by the high congruency of the order of the columns (the height of the columns does not correspond to the time of measurement). Upper scale: The short (upper) horizontal bar represents the S.c./P.s. ratio range resulting from internal standardization (1.012‐fold), whereas the large (lower) horizontal bar represents the ratio range between the two extreme values (1.31‐fold) resulting from recalculation in order to reproduce external standardization. During the course of measurement, the peaks tended to shift to the right side on the x‐axis of the histogram
3. REQUIREMENTS ON A REFERENCE STANDARD
Genome size measurement with FCM relies on specific and stoichiometric binding of a fluorescence dye to DNA of intact nuclei in both the standard and the study object, in order to ensure linear proportionality of the resulting mean fluorescence intensity [13]. Unfortunately, there are many obstacles due to the properties of plant material, which may disturb the measurement in various ways. Considering this, it is imperative that the standard organism fulfills the requirements for accurate measurements. Some of the following items relate to mechanical and chemical issues directly influencing the measurements, while others regard taxonomic issues of the standard, availability, and cultivation.
Verified genome size stability within well‐delimited taxa is crucial for reproducibility of the results [9, 10, 14, 15, 16]. This includes also reproductive isolation (absence of cross‐hybridization) from the close relatives and any taxonomic ambiguity, as well as high genome stability in the course of cultivation and maintenance of the material (e.g., no stress‐induced ploidy level changes). This requirement excludes also the use of clones derived from in‐vitro tissue or cell suspension culture, since they frequently manifest cytological somaclonal variation, that is, polyploidy and aneuploidy [9, 17]. The genome size of a standard organism should be invariant. Any kind of different cytotypes, aneuploidy, B‐chromosomes, sex chromosome dimorphism, tendency to spontaneously form polyploids (in morphologically indistinguishable individuals), or any other genome size variation among the material of the standard is to be excluded. Material from guaranteed pure lines established by maintenance breeding or by natural clonal propagation is to be preferred. Special caution is needed in outcrossing species. All this information must be indicated in publications. In crops, the particular variety, cultivar, or lineage should be indicated.
Absence of anatomical structures and/or cell content affecting preparations in a mechanical or chemical way. First, the standard material should not possess mechanical properties that prevent easy sample preparation, such as thick cuticles, tough hairs, or silicate or carbonate incrustations, such as seen in samples of xerophytic species or other species with specialized life strategies. Second, the standard tissues should tolerate a sufficient time for interim storage in the refrigerator, indicated by retention of turgor pressure, and, additionally, should release a sufficient number of nuclei on homogenization. Third, a high content of specific plant products and secondary metabolites may decrease the yield of nuclei and, therefore, compromise the stability of the nuclear suspensions (these include mucilaginous compounds, latex, wax, resins, starch, oxalates, among other compounds [18]). Fourth, the standard should be free of DNA staining inhibitors, especially polyphenolics (such as tannins or anthocyanins; the latter particularly notable in colored tissues). This is critical, as it has been shown that phenolic compounds may influence nuclei of different species in different ways, even when co‐processed [7, 19, 20]. Although the effect of secondary metabolites may be partly reduced by selecting suitable buffer, or by adjusting buffer composition, and also by selecting a particular organ or tissue for nuclei isolation (for details see References [6, 12, 21]), the standard itself should not be a source of interfering compounds. Detailed tests for identification of secondary metabolites are described in References [6] and [22].
Texture homogeneity in different tissues/materials. Different tissues may require different conditions for proper nuclei isolation and quantitative staining.
Differences in chromatin accessibility to DNA fluorochromes may lead to nuclear fluorescence intensity variation among various tissues. Therefore, it is best to use similar tissues in the studied object and the standard for nuclei isolation. A simple test is advisable similar to that shown in Figure 2. A combined preparation of the tissue in question together with somatic tissues of the same species may yield two close peaks. Figure 2 shows nuclei from male sperms and somatic tissues from the liverwort Pellia epiphylla which were compared and a 1.115‐fold stainability variation was obtained. Although never systematically evaluated, Price et al. [23] also suggested to avoid the use of too distantly related organisms (e.g., animal vs. plant tissues).
The use of dry samples has become increasingly important, but they are almost always measured using fresh standard tissues. This may produce different results than using either fresh‐fresh or dry‐dry combinations. Bainard et al. [24] discussed the differences in DNA content estimation when dry versus fresh tissues were used and stated that “It is possible that increased fluorescence in dry tissue may be an indication of undetected self‐inhibition of staining in fresh tissue that is not present in dried tissue, or that nuclei from dried tissue tend to be closer to full staining saturation for other reasons.” Hence, a comparison of the PP between dried and fresh material is recommended and possibly informs about the presence of secondary metabolites.
Commercially available fixed cells or artificial beads are not suitable as standards for precise DNA content analysis, because they can only be applied as pseudo‐internal standards, that is, they are not entirely co‐processed, therefore not affected in the same way as the sample. Moreover, the accessibility of chromatin/DNA to a DNA fluorochrome differs between fixed and non‐fixed nuclei [25, 26]. Polystyrene and other type of beads are not suitable as they do not contain DNA and cannot resemble the kinetics of DNA staining of the nuclei in suspension.
The presence of sex chromosomes is another potential source of error. C‐values may differ between sexes by only a few percent, as in humans [27], or in Silene latifolia [28], but up to 35% in the liverwort Frullania dilatata [29].
FIGURE 2.
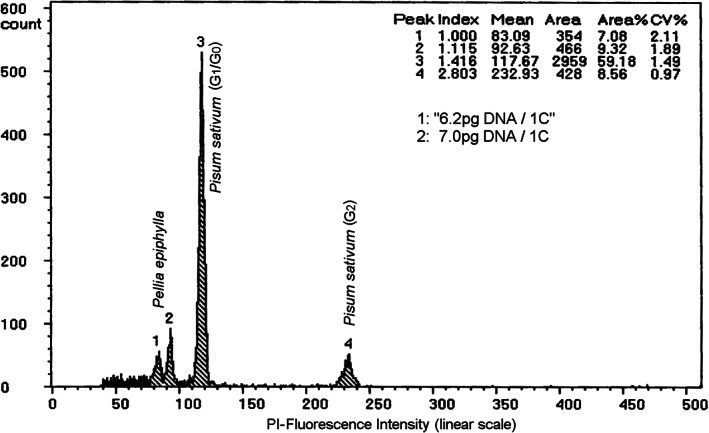
Fluorescence intensity variation among nuclei released from different cell types in the liverwort Pellia epiphylla (P.e.). Peak 1 contains male gamete nuclei emitting lower fluorescence intensity compared with the Peak 2, which contains somatic nuclei of the gametophyte. The variation of the fluorescence intensity is 1.115‐fold (index) and results from the stainability variation between the two different cell types. The Peaks 3 and 4 refer to the Pisum sativum (P.s.) G1/G0 and G2 nuclei respectively. The P. e. 1C‐value calculated from somatic tissues from both, P.e. and P.s. was 7.0 pg, whereas a false 1C‐value of 6.2 pg (given between quotation marks) resulted from the calculation, when the male gametes were used for C‐value calculation
However, in some cases, different tissues have to be used due to possible unavailability of the particular tissue in the standard species (such as when analyzing endosperm or pollen grains). In any case, the tissues used must be properly described in the publication.
High‐resolution histograms with peaks with low coefficients of variation (less than 3%) and low background noise, but a sufficient number of released nuclei should be obtainable from the reference standard [9].
Verified C‐level of the nuclei. The DNA content of a cell nucleus depends on the nuclear phase and the mitotic stage [2, 3]. In differentiated sporophytic organs (e.g., leaf), a majority of nuclei are in G1/G0 phase of cell cycle (2C‐level) and only a few in G2 (4C‐level), whereas in gametophytic tissues (e.g., bryophyte leaflets) the nuclei are at the 1C‐level or 2C‐level in G1/G0 or G2, respectively. If gametophytic and sporophytic tissues are mixed within one preparation, compensation has to be considered. Usually, the main mitotic cycle arrest is in the G1/G0 stage. Therefore, in a typical histogram of a single species one dominant (in terms of the peak area) G1/G0 peak and on the double position none, or a much smaller one in G2 appears (Figure 2). Rapidly proliferating tissues, for example, very young tomato leaves, may exhibit a larger fraction of G2 nuclei than fully developed tissues. Exceptions are rare, but do exist. In the moss Physcomitrella patens the vast majority of nuclei is arrested in G2 and G1/G0 nuclei, if existing at all, are beyond FCM detectability [30], or in cases of unidentifiable nuclear phases in organisms with isomorphic, but heterophasic life cycles [31]. Čerternová and Galbraith [31] suggest to refer to pg/cell without assignment to a 1C or 2C‐value exceptionally in those cases of unidentifiable nuclear phases. The knowledge of the C‐level in all tissues subjected to measurement is an indispensable prerequisite in FCM. It enables the correct calculation of the C‐values.
Presence of endopolyploidy is a matter of debate. Endopolyploidy is more abundant in certain plant groups (such as Brassicaceae, including A. thaliana), life strategy, and tissue types [32, 33, 34, 35, 36]. Provided complete replication of the genome, this phenomenon is tempting for use in standard organisms, since the same standard is suitable in a wider range of targeted samples, and additionally, may provide a simple, hence fast control of linearity depending on the instrumentation [37]. Galbraith [38] found almost perfect linearity among up to 32C endopolyploid A. thaliana nuclei populations (r 2 = 0.9999) and recommended the use of endopolyploid nuclei populations to stretch the range of one standard species. On the other hand, the peak with nuclei containing the least amount of DNA (1C in gametophytes and 2C in sporophytes) may be overlooked, since in endopolyploid tissues such nuclei are occasionally less frequent than the nuclei with higher C‐levels. The use of endopolyploid peaks of other species may introduce another error if incomplete endoreduplication occurs, such as observed in certain orchids [39] or chromatin structure and DNA accessibility vary between endopolyploid nuclei populations with varying architecture [40]. As a general rule, increased caution should be applied in regard of complete DNA replication or other factors affecting the fluorescence intensity proportionality among the nuclei populations, if endopolyploid nuclei are used as a standard.
Easy maintenance and availability of the standard material is an important precondition. The former includes sufficient and long‐lasting germination capacity of the seeds and practicable seed/diaspore storage conditions, short germination time in annuals, easy cultivation (no special conditions needed, e.g., provided by greenhouse pest resistance), and sufficient diaspore production (also in perennials as a backup, or for dissemination among colleagues). Perennial species are superior over annuals, as they provide a longer life‐span and permit utilization of the same individual (or ramets of one natural clone) for the preparation of all samples within an experiment or even for all samples studied in the laboratory over a certain period of time and without repeated germination efforts. A standard (either seeds or propagated plantlets from natural clones) should be globally available. In this respect, seed‐propagated standards (both annuals and perennials) are advantageous over vegetatively propagated ones, as seeds will survive more easily, when posted to other researchers upon request.
Usability of the standard organism in combination with various isolation buffers. Certain study objects may require special isolation buffers [6, 21, 41], and, as a matter of fact, scientists may also prefer a certain isolation buffer in their work. Therefore, a useful standard organism is subject to work well with several isolation buffers.
4. STANDARD SELECTION ACCORDING TO A GIVEN OBJECT
Since a linear response of a flow cytometer is an indispensable prerequisite for proper relative measurement, Suda and Leitch [42] strongly recommend locating the DNA peak of the target species between the G1/G0 and the G2 peaks of the standard as the ideal condition, and in no way should the difference between the target species and the standard genome size exceed fourfold. Sliwinska et al. [9] recommend threefold difference. This is a significant restriction compared with the potentially higher range provided by the linear scale measurement window of the instrument (Note: the logarithmic scale is not recommended for genome size measurement in instruments that employ logarithmic amplifiers for the logarithmic scale data collection). Modern instrumentation provides usually satisfactory linearity, which can be accurately verified according to Bagwell et al. [43]. Depending on the genome size variation within the targeted sample set, the use of more than one standard organism may be necessary.
The issue of maximum difference between genome size of the standard and unknown sample leads to the opposite question, concerning the minimum difference. Too close and hence “overlapping” DNA peaks on a histogram of relative fluorescence intensity lead to a kind of “additive spill over” resulting in an artificial approach of the two peak means, since the signals within the shared area will contribute mutually to the area of each other's peak. The closer the neighboring peaks come, the more signals will be added to channels in the periphery of the other peak. In Figure 3A individual peaks basing on a fictitious data set are shown as a superimposed projection. The tallest columns (the peak modes) in the center are columns no. 8 of the light gray peak, and 14 of the dark gray peak, respectively. Figure 3B display the data set in the same way as Figure 3A, but simulates combined measurement. Correspondingly, two strongly “overlapping” peaks with an unequal contribution of signals to the center columns of both peaks appear. Columns no. 9 and 13 are now the tallest, leading to a shift of both mean PP. In order to avoid such biased mean peak values, it is recommended to unambiguously determine at least 95% of the signals within each peak, which lies within ±2 times the SD from the mean value (or the coefficient of variation in % ‐ as provided by many flow cytometers). Subsequently, only about 2.5% of the signals will be shared in the area between the peaks. As a rule‐of‐thumb, the minimum distance between the standard (C1) and the study object (C2) C‐values (or peak positions on the histogram axis of relative fluorescence intensity) should not be less than two times the coefficient of variation given in % from the standard (CV%1) plus two times the study object (CV%2). The simplified inequality to calculate the actual (left side) and the essential (right side) distances between the genome sizes or the PPs on the fluorescence intensity axis is:
The actual distance has to be larger than the essential distance. C1 and C2 are either the C‐values or the PP. If provided by the flow cytometer, the CV% and the PP can be inserted directly from the histogram into this equation.
FIGURE 3.
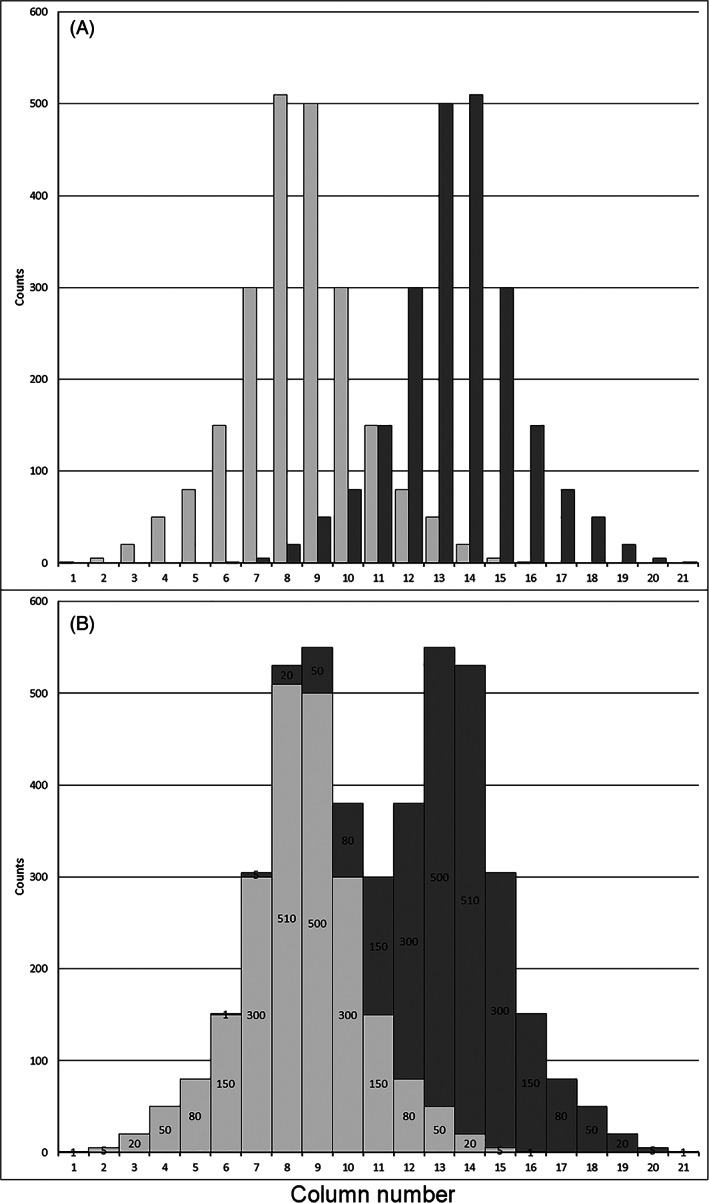
Illustration of minimum peak distance based on a fictitious data set. (A) Two visually superimposed, but independent individual peak histograms (light gray and dark gray). Columns 8 and 14 represent the modes of the light gray and dark gray peak, respectively; (B) the combined data set shows the “additive spill over.” Each column content mutually contributes to a column's content of the other peak. This resembles the situation of measurements, when nuclei with similar mean fluorescence intensity produce “overlapping” peaks in the histogram. For the left‐hand peak, the dark part on top of each column is the contribution of the right‐hand peak (dark gray). Since this contribution increases toward the right, the left mode is shifted to the middle between the two peaks. The modes of the two peaks approached and are now represented by the Columns 9 and 13, respectively
Example: Given, Hordeum vulgare (C1; PP = 113.57; 5.02 pg/1C) was measured against P. sativum (C2; PP = 100; 4.42 pg/1C) and the G1/G0 peak coefficients of variation were 2.5% and 1.5%, respectively. The genome size of H. vulgare is 1.136‐fold larger than that of P. sativum. Is the distance between them larger than the required minimum distance under the observed conditions? Provided, the H. vulgare C‐value was calculated from the histogram and the CV%s were directly taken from the respective histogram peaks, the equation is:
In this example, the actual difference between the PPs (13.57) is larger than the calculated minimum distance (8.68), thus under the precondition of the given CV%, the peak's overlapping area conforms with the above mentioned condition of nonoverlapping peaks.
5. PLANT GENOME SIZE VALUES RANGE AND THE BEST FITTING STANDARDS
Currently known genome sizes of Embryophyta vary about 2300‐fold between the smallest, found in Genlisea tuberosa (0.0624 pg/1C, [44]), and the largest, that of Paris japonica (152.23 pg/1C; [45]). The individual C‐values described for land plant species are not normally distributed between these two extremes, instead forming an extremely positively skewed distribution having a mode between 0.6 and 0.7 pg/1C (Figure 4). It is not likely that further findings will dramatically change this distribution pattern. Following the maximal distance between object and standard C‐value rule, standard species with a genome size between 0.15 and 2.76 pg/1C would be sufficient to cover these C‐values. Since 58% of all as yet known Embryophyta C‐values are in this range, it will be easy to select a suitable standard from the established standard species (Table 1) in most cases. Among the very low ranking C‐values, A. thaliana (0.16 pg/1C) is often used as a standard. Genlisea tuberosa, as the smallest known Embryophyte genome, was measured relative to A. thaliana, since their quotient is 2.56‐fold, which is still within an acceptable distance. In fact, standards in a range suitable for nuclear 1C‐values up to 118 pg (Haemanthus albiflos 29.5 pg/1C) are provided by the literature (see Table 1). Above 67 pg/1C, no sufficient standard had been established, when Pellicer et al. [45] published the genome size of P. japonica. In order to circumvent linearity problems created by the 9.1‐fold variation between P. japonica and A. cepa, they used the A. cepa 2C nuclei to calibrate two Trillium species to be used as further standards for P. japonica (152.23 pg/1C), together with 8C nuclei of A. cepa.
FIGURE 4.
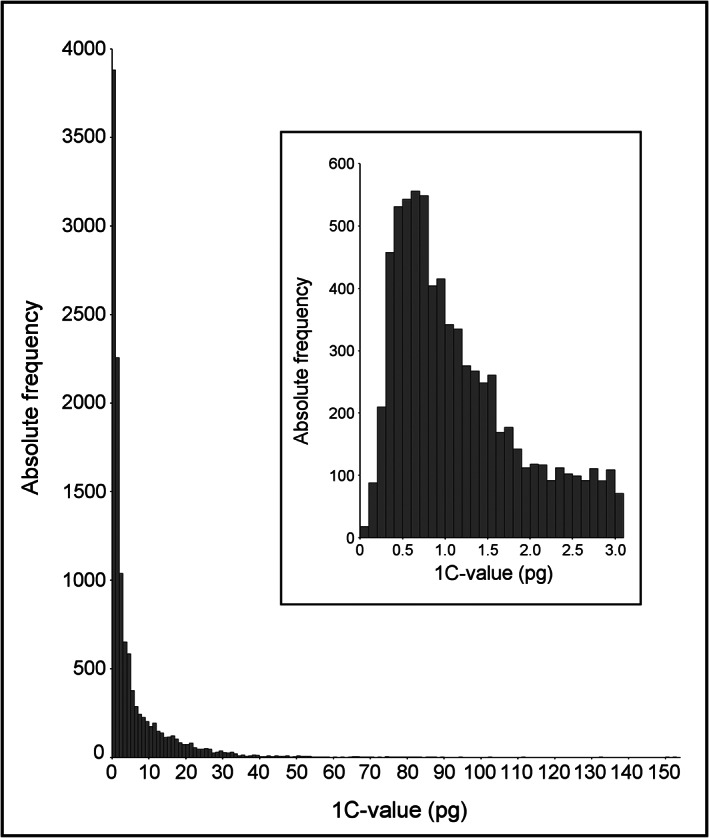
C‐values (prime estimates of 11,828 species) from the plant DNA C‐values database [63]. The smallest is 0.0624 pg/1C (Genlisea tuberosa; [44]), the largest is 152.23 pg/1C (Paris japonica; [45]). The insert shows an enlargement of the x‐axis section from 0 to 3.1 pg/1C in 0.1 pg classes
TABLE 1.
List of plant FCM standards for genome size and GC content measurements in current use
| Species | Variety/cultivar/clone | Life cycle | 2C | GC % | Ref. | Method | Primary std. (organism, GS, GC%, references) | Comments | |
|---|---|---|---|---|---|---|---|---|---|
| pg | Gbp | ||||||||
| Arabidopsis thaliana | ecotype Columbia | A | 0.321 | 0.314* | [67] | FCM; single std. | Caenorhabditis elegans Bristol N2, 100 Mbp/1C [66] | P: model plant; C: endopolyploidy a ; the species show intraspecific genome size variation, the value thus cannot be transferred to other ecotypes | |
| Verbena officinalis | P | 0.678 | 0.663 | [76] | FCM; single std. | Raphanus sativus ‘Saxa’, 1.11 pg/2C [13] | P: ‐; C: ‐ | ||
| Carex acutiformis | wild clone b | P | 0.818 | 0.800 | 36.4 | [48] c | FCM; single std. | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | P: clonally propagated; C: ‐ |
| Oryza sativa subsp. japonica | ‘Nipponbare’ | A/P | 0.795 | 0.778* | 43.6 | [69] | Whole‐genome sequencing | P: model plant, annual but can be kept as perennial using ratooning and can be propagated clonally when cultivated under laboratory conditions; C: ‐ | |
| Raphanus sativus | ‘Saxa’ | A | 1.11 | 1.09 | 39.1 | [13] d , e | FCM; cascade (human ‐ Z.m. ‐ S. l.) | human male leucocytes, 7.0 pg/2C [56], GC 40.5% [77] | P: ‐; C: endopolyploidy a |
| 0.997 | 0.976 | 40.3 | [46] | FCM; single std. | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6 [69] | ||||
| Solanum lycopersicum | ‘Stupické polní rané’ | P | 1.96 | 1.92 | 35.3 | [13] d , f | FCM; cascade (human ‐ Z.m. ‐ S. l.) | human male leucocytes, 7.0 pg/2C [56], GC 40.5% [77] | P: able to grow under humid conditions in tropical countries‐, C: |
| 1.735 | 1.697 | 38.7 | [48] c | FCM; single std. | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | ||||
| Glycine max | ‘Polanka’ | A | 2.50 | 2.45 | 36.4 | [75] e | FCM; single std. | human male leucocytes, 7.0 pg/2C [56], GC 40.5% [77] | P: ‐; C: relatively short seed longevity |
| 2.077 | 2.031 | 37.9 | [78] | FCM; cascade (O.s. ‐ S.l.) | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | ||||
| Solanum pseudocapsicum | commercial clone b | P | 2.590* | 2.533 | [29] | FCM; regression line | Raphanus sativus ‘Saxa’, 0.53 pg/1C; Zea mays ‘CE‐777’, 2.59 pg/1C; Pisum sativum ‘Kleine Rheinländerin’, 4.42 pg/1C; Hordeum vulgare ‘Ditta’, 4.83 pg/1C [49] g | P: perennial but self‐compatible and possible to propagate by seeds; C: ‐; | |
| 2.414 | 2.361 | 39.5 | P. Šmarda, unpublished h | FCM; cascade (S.l. ‐ O.s.) | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | ||||
| Petunia hybrida | ‘PxPc6’ | A | 2.85 | 2.79 | [79] | FCM; single std. | Gallus domesticus erythrocytes, 2.33 pg/2C [5] | P: ‐; C: ‐ | |
| Bellis perennis | Wild population | P | 3.38 | 3.31 | [80] | FCM; no details are given in the original publication, only “J. Suda, unpublished data” | P: ubiquitous plant, possible to propagate clonally; C: autotriploid individuals recorded (though very rare) | ||
| Wild clone b | 3.159 | 3.090 | 39.54 | [47]c | FCM; cascade (O.s. ‐ S.l.) | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | |||
| Petroselinum crispum | ‘Champion Moss Curled’ | B | 4.46 | 4.36 | [50] | FCM; regression line | Vigna radiata ‘Berken’, 1.06 pg/2C [81]; Solanum lycopersicum ‘Stupické polní rané’, 1.96 pg/2C [13]; Glycine max ‘Polanka’, 2.50 pg/2C [75]; Hordeum vulgare ‘Sultan’, 11.12 pg/2C [81] | P: ‐; C: ‐ | |
| Zea mays | ‘CE‐777’ | A | 5.433 | 5.313 | 45.4 | [82] i | FCM; single std. | human male leucocytes, 7.0 pg/2C [56], GC 40.5% [77] j | P: ‐; C: ‐ |
| Pisum sativum | ‘Ctirad’ | A | 9.09 | 8.89 | 38.5 | [71] e | FCM; single std. | human male leucocytes, 7.0 pg/2C [56], GC 40.5% [77] | P: able to grow under humid conditions in tropical countries; C: |
| 8.018 | 7.841 | 41.77 | [48] c | FCM; cascade k | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | ||||
| ‘Kleine Rheinländerin’ | 8.84*, l | 8.65 | [16] | Feulgen densitometry; single std. | Allium cepa ‘Stuttgarter Riesen’, 16.75 pg/1C [72] | ||||
| Hordeum vulgare | ‘Ditta’ | A | 10.43 | 10.20 | [71] | FCM; single std. | Pisum sativum ‘Ctirad’, 9.09 pg/2C [13] | P: ‐; C: ‐ | |
| ‘Hitchcock’ | A | 10.68 | 10.45 | [83] | No details given m | ||||
| Agave americana | Not given | P | 15.90 | 15.60 | [84] | FCM; two stds. | Pisum sativum, 8.75 pg/2C; Hordeum vulgare, 10.04 pg/2C [71] n | P: ‐; C: succulent plant, some isolation buffers may not work well with this species | |
| Secale cereale | ‘Daňkovské’ | A | 16.19 | 15.83 | 44.6 | [71] e | FCM; cascade (human ‐ P.s.) | human male leucocytes, 7.0 pg/2C [56], GC 40.5% [77] | P: ‐; C ‐ |
| Chlorophytum comosum | var. comosum, single clone b | P | 24.14 | 23.61 | [85] | FCM; single std. | Pisum sativum ‘Ctirad’, 9.09/2C [71] | P: easy to propagate clonally; C: mild endopolyploidy a | |
| Vicia faba | ‘Inovec’ | A | 26.90 | 26.31 | 38.1 | [13] d , e | FCM; cascade (human ‐ P.s.) | human male leucocytes, 7.0 pg/2C [56], GC 40.5% [77] | |
| 23.796 | 23.273 | 41.15 | [48] c | FCM; cascade k | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | ||||
| Allium cepa | ‘Alice’ | P | 34.89 | 34.12 | 34.7 | [71] e | FCM; cascade (human ‐ P.s.) | human male leucocytes, 7.0 pg/2C [56], GC 40.5% [77] | P: ‐; C: mucous, endopolyploidy a |
| 30.745 | 30.069 | 36.52 | [46] | FCM; cascade k | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | ||||
| not given | 33.55 | 32.81 | [57] | colorimetric measurement from known number of cells | |||||
| Haemanthus albiflos | commercial clone b | P | 59.143 | 57.842 | 38.76 | [48] c | FCM; cascade k | Oryza sativa subsp. japonica ‘Nipponbare’, 388.82 Mbp/1C, GC 43.6% [69] | P: ‐; C: succulent plant |
Note: For each standard, primary standards that were used to estimate the displayed genome size value are listed, along with the estimation method. For GC content, the same standards as for the genome were used, if not indicated otherwise. Specific features of each standard, such as notes on cultivation, propagation, or limitations for its use are also summarized. Various technical remarks (marked as superscript letters) are provided as footnotes to this table. It must be emphasized that the term ‘clone’ as used in the table refers to natural clones, since in vitro culture‐derived tissues or regenerates are excluded as described within the main text. Life cycle: A, annual; B, biennial; P, perennial. 2C: Holoploid genome size of the standard (2C‐value) given in pg and Gbp; the unit given in the original publications is displayed in bold and recalculation is provided, based on the usual equations 1 pg = 0.978 Gbp [65]; asterisks denote values published originally as 1C. Ref.: Reference. Methods: Calibration method used; in cascades, the species are abbreviated as follows: A.c. – Allium cepa, O.s. – Oryza sativa, P.s. – Pisum sativum, S.l. – Solanum lyopersicum, V.f. – Vicia faba, Z.m. – Zea mays. Primary standard: GS – Genome size, as shown in the original publication(s). Comments: P – Specific pros (not including the general requirements such as high‐quality peaks, low background, etc.), C – Specific cons.
Endopolyploidy is listed under “cons” here because of the possible overlap of the sample peak and higher‐ploidy peaks of the standard. However, it may be considered also as a pro as the higher‐ploidy peaks may be used as standard peaks to analyze samples with higher genome size (but see the main text for the specific comments).
The value refers only to a particular clone and should not be automatically transferred to clones or cultivars of different origin (possible intraspecific genome size variation should be considered).
Details on measurement methods are given in [46], Supplementary materials; for Bellis perennis, GC content is also published here.
GC contents indicated in Reference [13] were calculated with less reliable numeric methods. Here we use values re‐calculated by Barow and Meister [86], Table 1, based on measurements of Pisum sativum ‘Ctirad’ with human male leucocytes (GC content = 40.5%, 77) resulting in GC content of Pisum sativum ‘Ctirad’ = 38.5%. This value was used to calculate GC contents of other standards based on sample/standard ratios with DAPI and propidium iodide taken from Reference [71], tables 4 and 7, lab. No. 1.
Calculated in this paper in a cascade like manner based on measurements of Zea mays with human male leucocytes and Zea mays with Solanum lycopersicum ([13]: Table 1; note that the first and second row in this table are swapped), analogously to the calculation of GC content of other Doležel's standards in [86].
The values of the primary standards taken from Reference [49] are all inter‐related as they are all estimated using Feulgen densitometry with Pisum sativum ‘Kleine Rheinländerin’ 4.42 pg/1C [16]) as the only standard.
Unpublished value estimated by P. Šmarda, using similar methodology as in Reference [46].
Calculated in this paper based on measurements with Pisum sativum ‘Ctirad’ ([71]: tables 4 and 7), analogously to the calculation of GC content of other Doležel's standards in Reference [86]
The primary standard is not given in the original publication; the information is appended here by J. Doležel.
In References [48] and [46], the following cascade approach is used: S.l. is directly measured with O.s., while values of other standards are calculated from the estimated value of S.l. using the ratios of genome sizes taken from Reference [71] (A.c./V.f./P.s./S.l.); see table caption for abbreviations.
In Reference [16], the genome size of P. sativum is considered invariable and the reported value is the mean overall studied cultivars, incl. ‘Kleine Rheinländerin’
6. ESTABLISHMENT OF NEW STANDARDS
To date, a wide spectrum of standard organisms is available (Table 1) and their use should be preferred since it permits a comparisons of results from different researchers. But what if all suitable standard organisms elude flow cytometric sample measurement by conflicts like, for example, overlapping peaks, problems with germination, cultivation, or availability? Here, we describe methods that are used to establish new standards. Regardless of which approach was finally used, the method used and the primary standards together with their genome size and references have to be specified when the new standard is published. When establishing a new standard, researchers should be very stringent concerning the measurement precision (CVs of the peaks <3%, at least 1000 nuclei per peak, nonoverlapping and not too distant peaks, repeated measurements on different days; see Reference [9] for details).
Single calibration standard approach is the most often used approach. As for typical genome size measurements, a single calibration standard is used per new standard organism. This approach is more prone to error than approaches employing more than one primary standard, either derived already from an imprecise calibration standard C‐value selected or from imprecise measurements. Repeated measurements on different days (and different instruments, if available) partly compensate the latter problem. The main risk of this approach is that there is no other control. Thus if any unrecognized problems (e.g., cryptospecies or other) occur, these will not be noticed.
Cascade like approach: A set of new standards is calibrated in a cascade manner, that is, the C‐value of every species is compared with the C‐value of a species having a higher or lower C‐value. Starting the calculation from the calibration standard organism with known genome size (serving as an “anchor”; only one primary standard is thus needed), one after another species' C‐value is calculated in a cascade manner [46, 47, 48, 49]. All resulting genome sizes are supposed to be congruent within the new standard set. Therefore, the selection of the anchoring standard genome size is critical, since biased values will continuously impact all derived values over the subsequent steps. Highly precise measurements are essential for the cascade like approach.
Regression line approach [50]. This is one of the most robust approaches in terms of linearity problems and measurement errors, since the new standard species will be calibrated against multiple different calibration standards with known genome size and individual measurement errors will be eliminated. A new standard is measured against a set of other existing calibration standards, each preparation including the new standard together with one from the primary standard set. Subsequently, its genome size is calculated from the obtained quotients (x‐axis) and genome sizes (y‐axis) of the respective calibration standards using a linear regression (y = a × x + b, where a = slope, x = measured standard: object peak position quotient, b = intercept, y = C‐values). The C‐value (y) of the new standard is calculated by substituting x by 1. The reliability of a regression line decreases rapidly outside of the cloud of points. Therefore, the new standard should be within the range of primary calibration standards and outliers should be avoided.
Multiple standards approach: Nuclei of a new standard are isolated, stained, and analyzed together with two or more calibration standards as one sample (e.g., see References [51, 52]). Its C‐value is then calculated similarly as in the regression line approach by multiplying sample fluorescence by the slope (a) of the linear function y = a × x + b of x represented by G1/G0 peak fluorescence of standards and y their genome sizes. Compared with the regression line approach, this approach is faster (all quotients are obtained during a single measurement of sample and the mix of standard nuclei). A further advantage is that nuclei from all species are affected to the same extent during sample isolation and staining. A disadvantage is the presence of many peaks including those from G2 nuclei and occasional endopolyploid nuclei populations, hence resulting in a possible peak interference. Therefore, the number of calibration standards is limited by practicability.
7. THE STRUGGLE FOR EXACT ABSOLUTE GENOME SIZE DATA
All standards are calibrated against a primary reference standard. This leads to the question, where or when this chain of calibrations relative to standard organisms begun, or in other words, do we actually know the genome size of the reference standards?
Vendrely and Vendrely [53, 54] published the first estimate of absolute C‐values of humans in DNA extracts from known nuclei numbers by means of the diphenylamine reaction [55]. Other investigators followed, and a spectrum of human 1C‐values between 3.0 and 3.5 pg [56] was obtained. Van't Hof [57] evaluated the first 4C‐values in a set of plant species including A. cepa using a mathematical C‐level distribution model applied to average DNA contents estimated colorimetrically. He aimed to identify factors that influence the cell cycle duration and the genome size. Subsequent works [13, 27, 58, 59] proved the 1C‐value of A. cepa (16.75 pg) to be tolerably accurate and that the genome size is constant among several cultivars [60], and were recently be supported [61]. Basing on the genome size of Homo sapiens on the animal side and A. cepa on the plant side, further genome size measurements were less cumbersome, when relative methods were applied. The A. cepa genome is comparably large and, therefore covering a small range of all presumable plant C‐values, given the maximum distance between standard organism and study object for measurement (see chapter Standard Selection According to a Given Object). However, there is strong need to cover the whole range. Subsequently, in the following years, several thousands of plant species were measured either directly against the absolute values of A. cepa and H. sapiens or indirectly calibrated standard species [62, 63].
There is a small uncertainty left regarding the estimated numbers of 2C, 4C, and nuclei just passing DNA‐synthesis in the tissues, which were used to calculate the first absolute C‐values [57]. Therefore, more deliberate and highly precise absolute C‐values useful as “Gold”‐standards were awaited when the recently developed DNA sequencing method was applied [62]. Unfortunately, the size of the first genome assembly of a plant, that is, A. thaliana (0.13 pg/1C, [64]; calculated from Mb [65]) was most probably underestimated due to the presence of repetitive sequences, which were not included in the assembly. A FCM comparison with the animal Caenorhabditis elegans (0.10 pg/1C, [66]), whose genome contains only marginal amounts of repetitive DNA, revealed an actually 25% higher C‐value for A. thaliana (0.16 pg/1C; [67]). The A. thaliana genome, being one of the smallest of the angiosperms, and similar to the situation in A. cepa at the other extreme, covers only a part of all species values when used as a standard, but may be useful as the first “anchor” in order to calibrate the whole standard species network [62].
At present a complete set of truly “Gold” standards is not available, since there is no truly complete plant genome sequence or any other absolute measurement estimate in plant species available to date [68]. Among the most promising candidates is Oryza sativa 'Nipponbare' [69], which has been used to calibrate some standards with small genome size [46, 47, 48]. Meister [70] found the quotient between O. sativa and A. thaliana to be 2.545‐fold. His findings state a slight underestimation of the sequencing derived 1C‐value (0.398 pg, calculated from Mb [65]) if compared with A. thaliana 1C‐values (0.16 pg; [67]) resulting in a rice genome size of 0.407 pg/1C. Although it might not be really completely sequenced the corrected value may provide more realistic genome size estimates of plants with small genomes. However, Doležel and Greilhuber [62] suggested to continue with standard C‐values basing on the middle ranking 4.545 pg/1C of P. sativum [71] for plant objects and 3.5 pg/1C of H. sapiens [56] for the animal objects, and to recalculate all C‐values once a representative number of “Gold”‐standard C‐values is established. We expect that the uncertainties regarding the exact genome sizes of reference standards will disappear after the production of a representative number of complete plant genome assemblies by the initiatives similar to the human Telomere‐to‐Telomere (T2T) consortium (https://sites.google.com/ucsc.edu/t2tworkinggroup). Meanwhile, it is important to refer thoroughly the used reference standards together with its used C‐value in every publication, to allow re‐calculation of published values after exact genome sizes of the standards become available.
8. AVAILABLE STANDARDS, THEIR GENOME SIZE, AND GC‐CONTENT VALUES IN CURRENT USE
During the several decades of plant genome size measurement using FCM, a number of calibration standards has been established and published (to name only a selection: [13, 16, 29, 46, 47, 48, 49, 58, 67, 71, 72, 73]. Praça‐Fontes et al. [74] re‐investigated the FCM usability of a subset of standard species (Doležel's standard set [13, 71, 75] plus Drosophila melanogaster) by statistical comparison by means of cascade like calibration approach, and suggested to consider A. thaliana as a “Gold” primary standard, whereas Raphanus sativus and Glycine max could be exclusion candidates. However, the most currently used standards are summarized in Table 1, along with the calibration methods and primary standards. As mentioned above, there is some uncertainty about the exact genome sizes, and the listed values are thus to be considered as “values in current use” rather than true numbers. For many standards, we list two estimates, which can be considered as the likely upper (those based on human 3.5 pg/1C) and lower limit (those based on Oryza sativa 0.389 Mbp/1C). We strongly recommend using one or the other set of these values exclusively within a particular experiment and publication, to allow easy recalculations in the future. For this reason, we did not include the various published re‐calibrations of individual species or the whole set, which usually provide similar values subjected to the same uncertainty as the listed values.
Using base‐unspecific DNA fluorochromes in parallel with base‐specific DNA fluorochromes enables to calculate also the genomic GC content of a sample [9]. Because FCM measures relative fluorescence intensities of nuclei stained by a DNA fluorochrome, also the genomic GC content calculation in absolute units requires comparison to a reference standard of known GC content. The detailed instructions for preparation, measurement, and calculation of the GC content are given in [9], a set of recommended standard species is listed in Table 1.
All recorded standards were critically selected with a strong focus on our recommendations. However, it is recommended to examine at the beginning of each study, if the requirements of the proposed standard(s) are consistent with the targeted study objects in regard of the preparation and measurement conditions.
9. BEST PRACTICE RECOMMENDATIONS
The internal standardization approach must be used when estimating nuclear DNA content in absolute units.
Identify the standard peak position for the further measurements of the combined preparations unambiguously by running one preparation, which contains only the standard, on the flow cytometer at the same voltage setting as the combined preparation. The preparation with the standard alone must not be used for the result calculation!
Upon standard organism selection, consider the proper difference between the expected C‐values of the object and the potential standard (maximum difference to ensure the linearity of the measurement). On the other hand, avoid DNA peak overlap that happens at too close C‐values.
Assure the availability of the proper standard organism by timely or continuous cultivation of annuals or the use of perennial species (preferably natural clones), respectively.
Prefer established reference standard species (Table 1). Only if unavoidable, establish a new standard preferably by the regression line or multiple standards approaches. Select a species that fulfills all the standard organism requirements, hence are easy to identify and maintain, globally available, with verified genome size stability, lacking problematic metabolites, with tissues releasing a sufficient number of nuclei, and exhibiting low peak CV%. Avoid endopolyploid tissues when complete DNA replication within the endocycles or proportional fluorescence intensity is not guaranteed.
Use similar tissues to isolate nuclei from the sample and the standard to enable internal standardization and to secure similar dye accessibility to the DNA. Describe the tissue type of all material used in the publication. In species with chromosomal sex polymorphisms specify also the sex used.
Always specify the standard species/cytotype/cultivar/clone/line, its C‐value together with the reference in your publication.
In every study documentation, keep information (photographs, herbarium vouchers, or other) from your standard organisms together with the records of your objects.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1002/cyto.a.24495.
ACKNOWLEDGMENTS
The authors are grateful to the two anonymous reviewers for their helpful comments, to David Galbraith for editing the English language of the manuscript, and to Wilhelm Temsch for valuable mathematical suggestions.
Temsch EM, Koutecký P, Urfus T, Šmarda P, Doležel J. Reference standards for flow cytometric estimation of absolute nuclear DNA content in plants. Cytometry. 2022;101:710–724. 10.1002/cyto.a.24495
[Correction added on 25 August 2021, after first online publication: The layout of table 1 has been amended for clarity in this version.]
Funding information Czech Science Foundation, Grant/Award Numbers: GA19‐13231S, GA19‐18545S, GA20‐15989S; European Regional Development Fund, Grant/Award Number: CZ.02.1.01/0.0/0.0/16_019/0000827
REFERENCES
- 1. Vrána J, Cápal P, Bednářová M, Doležel J. Flow cytometry in plant research: a success story. In: Nick P, Opatrný Z, editors. Applied plant cell biology. Heidelberg Springer‐Verlag: Plant Cell Monographs. Berlin; 2014. [Google Scholar]
- 2. Greilhuber J, Doležel J. 2C or not 2C: a closer look at cell nuclei and their DNA content. Chromosoma. 2009;118:391–400. [DOI] [PubMed] [Google Scholar]
- 3. Greilhuber J, Doležel J, Lysák MA, Bennett MD. The origin, evolution and proposed stabilisation of the terms ‘genome size’ and ‘C‐value’ to describe nuclear DNA contents. Ann Bot. 2005;94:255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Doležel J, Greilhuber J, Suda J. Estimation of nuclear DNA content in plants using flow cytometry. Nat Protoc. 2007;2:2233–44. [DOI] [PubMed] [Google Scholar]
- 5. Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science. 1983;220:1049–51. [DOI] [PubMed] [Google Scholar]
- 6. Loureiro J, Kron P, Temsch E, Koutecký P, Lopes S, Castro M, et al. Isolation of plant nuclei for estimation of nuclear DNA content: overview and best practices. Cytometry A. 2021;99:318–27. [DOI] [PubMed] [Google Scholar]
- 7. Loureiro J, Rodriguez E, Doležel J, Santos C. Flow cytometric and microscopic analysis of the effect of tannic acid on plant nuclei and estimation of DNA content. Ann Bot. 2006;98:515–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doležel J. Flow cytometric analysis of nuclear DNA content in higher plants. Phytochem Anal. 1991;2:143–54. [Google Scholar]
- 9. Sliwinska E, Loureiro J, Leitch I, Šmarda P, Bainard J, Bureš P, et al. Application‐based guidelines for best practices in plant flow cytometry. Cytometry A. 2021;XX:XX–X. [DOI] [PubMed] [Google Scholar]
- 10. Doležel J, Bartoš J. Plant DNA flow cytometry and estimation of nuclear genome size. Ann Bot. 2005;95:99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Greilhuber J. Intraspecific variation in genome size: a critical reassessment. Ann Bot. 1998;82(Supplement A):27–35. [Google Scholar]
- 12. Greilhuber J, Temsch EM, Loureiro J. Nuclear DNA content measurement. In: Doležel J, Greilhuber J, Suda J, editors. Flow cytometry with plant cells. Analysis of genes, chromosomes, and genomes. Weinheim: Wiley‐VCH; 2007. p. 67–101. [Google Scholar]
- 13. Doležel J, Sgorbati S, Lucretti S. Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol Plant. 1992;85:625–31. [Google Scholar]
- 14. Baranyi M, Greilhuber J. Flow cytometric analysis of genome size variation in cultivated and wild Pisum sativum (Fabaceae). Plant Syst Evol. 1995;194:231–9. [Google Scholar]
- 15. Baranyi M, Greilhuber J. Flow cytometric and Feulgen densitometric analysis of genome size variation in Pisum . Theor Appl Genet. 1996;92:297–307. [DOI] [PubMed] [Google Scholar]
- 16. Greilhuber J, Ebert I. Genome size variation in Pisum sativum . Genome. 1994;37:646–55. [DOI] [PubMed] [Google Scholar]
- 17. Kaeppler SM, Kaeppler HF, Rhee Y. Epigenetic aspects of somaclonal variation in plants. Plant Mol Biol. 2000;43:179–88. [DOI] [PubMed] [Google Scholar]
- 18. Loureiro J, Rodriguez E, Gomes A, Santos C. Genome size estimations on Ulmus minor mill., Ulmus glabra Huds. and Celtis australis L., using flow cytometry. Plant Biol. 2007;9:541–4. [DOI] [PubMed] [Google Scholar]
- 19. Bennett MD, Price HJ, Johnston JS. Anthocyanin inhibits propidium iodide DNA fluorescence in Euoporbia pulcherrima: implications for genome size variation in flow cytometry. Ann Bot. 2008;101:777–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Noirot M, Barre P, Duperray C, Hamon S, de Kochko A. Investigation on the causes of stoichiometric error in genome size estimation using heat experiments: consequences on data interpretation. Ann Bot. 2005;95:111–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Loureiro J, Rodriguez E, Doležel J, Santos C. Two new nuclear isolation buffers for plant DNA flow cytometry ‐ a test with 37 species. Ann Bot. 2007;100:875–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Price HJ, Hodnett G, Johnston JS. Sunflower (Helianthus annuus) leaves contain compounds that reduce nuclear propidium iodide fluorescence. Ann Bot. 2000;86:929–34. [Google Scholar]
- 23. Price HJ, Bachmann K, Chambers KL, Riggs J. Detection of intraspecific variation in nuclear DNA content in Microseris douglasii . Bot Gaz. 1980;141:195–8. [Google Scholar]
- 24. Bainard J, Husband B, Baldwin S, Fazekas A, Gregory T, Newmaster S, et al. The effects of rapid desiccation on estimates of plant genome size. Chromosom Res. 2011;19:825–42. [DOI] [PubMed] [Google Scholar]
- 25. Darzynkiewicz Z. Critical aspects in analysis of cellular DNA content. Curr Protoc Cytom. 2010;7. 10.1002/0471142956.cy0702s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holtfreter H, Cohen N. Fixation‐associated quantitative variations of DNA fluorescence observed in flow cytometric analysis of hemopoietic cells from adult diploid frogs. Cytometry. 1990;11:676–85. [DOI] [PubMed] [Google Scholar]
- 27. Greilhuber J, Volleth M, Loidl J. Genome size of man and animals relative to the plant Allium cepa . Can J Genet Cytol. 1983;25:554–60. [DOI] [PubMed] [Google Scholar]
- 28. Doležel J, Göhde W. Sex determination in dioecious plants Melandrium album and M. rubrum using high‐resolution flow cytometry. Cytometry. 1995;19:103–6. [DOI] [PubMed] [Google Scholar]
- 29. Temsch EM, Greilhuber J, Krisai R. Genome size in liverworts. Preslia. 2010;82:63–80. [Google Scholar]
- 30. Schween G, Gorr G, Hohe A, Reski R. Unique tissue‐specific cell cycle in Physcomitrella . Plant Biol. 2003;5:1–9. [Google Scholar]
- 31. Čertnerová D, Galbraith D. Best practices in the flow cytometry of microalgae. Cytometry A. 2021;99:359–64. [DOI] [PubMed] [Google Scholar]
- 32. Bainard JD, Henry TA, Bainard LD, Newmaster SG. DNA content variation in monilophytes and lycophytes: large genomes that are not endopolyploid. Chromosom Res. 2011;19:763–75. [DOI] [PubMed] [Google Scholar]
- 33. Bainard JD, Newmaster SG. Endopolyploidy in bryophytes: widespread in mosses and absent in liverworts. J Bot. 2010;2010:7. [Google Scholar]
- 34. Barow M. Endopolyploidy in seed plants. BioEssays. 2006;28:271–81. [DOI] [PubMed] [Google Scholar]
- 35. Barow M, Meister A. Endopolyploidy in seed plants is differently correlated to systematics, organ, life strategy and genome size. Plant Cell Environ. 2003;26:571–84. [Google Scholar]
- 36. Sliwinska E. Flow cytometry – a modern method for exploring genome size and nuclear DNA synthesis in horticultural and medicinal plant species. Folia Hortic. 2018;30:103–28. [Google Scholar]
- 37. Galbraith D. Endoreduplicative standards for calibration of flow cytometric C‐value measurements. Cytometry A. 2014;85A:368–74. [DOI] [PubMed] [Google Scholar]
- 38. Galbraith DW. Simultaneous flow cytometric quantification of plant nuclear DNA contents over the full range of described angiosperm 2C values. Cytometry A. 2009;75A:692–8. [DOI] [PubMed] [Google Scholar]
- 39. Hřibová E, Holušová K, Trávníček P, Petrovská B, Ponert J, Šimková H, et al. The enigma of progressively partial endoreplication: new insights provided by flow cytometry and next‐generation sequencing. Genome Biol Evol. 2016;8:1996–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maluszynska J, Kolano B, Sas‐Nowosielska H. Endopolyploidy in plants. In: Leitch L, Greilhuber J, Doležel J, Wendel J, editors. Plant genome diversity. Volume physical structure, behaviour and evolution of plant genomes. Wien: Springer Verlag; 2013. p. 99–119. [Google Scholar]
- 41. Zonneveld B. Selected perennial plants do provide convenient standards for the determination of genome sizes with flow cytometry. Plant Syst Evol. 2021;307:28. [Google Scholar]
- 42. Suda J, Leitch IJ. The quest for suitable reference standards in genome size research. Cytometry A. 2010;77A:717–20. [DOI] [PubMed] [Google Scholar]
- 43. Bagwell CB, Baker D, Whetstone S, Munson M, Hitschcox S, Ault KA, et al. A simple and rapid method for determining the linearity of a flow cytometer amplification system. Cytometry. 1989;10:689–94. [DOI] [PubMed] [Google Scholar]
- 44. Fleischmann A, Michael T, Rivadavia F, Sousa A, Wang W, Temsch EM, et al. Evolution of genome size and chromosome number in the carnivorous plant genus Genlisea (Lentibulariaceae), with a new estimate of the minimum genome size in angiosperms. Ann Bot. 2014;114:1651–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pellicer J, Fay MF, Leitch IJ. The largest eukaryotic genome of them all? Bot J Linn Soc. 2010;164:10–5. [Google Scholar]
- 46. Šmarda P, Bureš P, Horová L, Leitch I, Mucina L, Pacini E, et al. Ecological and evolutionary significance of genomic GC content diversity in monocots. Proc Natl Acad Sci U S A. 2014;111:E4096–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Veselý P, Bureš P, Šmarda P. Nutrient reserves may allow for genome size increase: evidence from comparison of geophytes and their sister non‐geophytic relatives. Ann Bot. 2013;112:1193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Veselý P, Bureš P, Šmarda P, Pavlíček T. Genome size and DNA base composition of geophytes: the mirror of phenology and ecology? Ann Bot. 2012;109:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vilhar B, Greilhuber J, Dolenc‐Koce J, Temsch EM, Dermastia M. Plant genome size measurement with DNA image cytometry. Ann Bot. 2001;87:719–28. [Google Scholar]
- 50. Yokoya K, Roberts AV, Mottley J, Lewis R, Brandham PE. Nuclear DNA amounts in roses. Ann Bot. 2000;85:557–61. [Google Scholar]
- 51. Vindeløv L, Christensen I, Jensen G, Nissen N. Limits of detection of nuclear DNA abnormalities by flow cytometric DNA analysis. Results obtained by a set of methods for sample‐storage, staining and internal standardization. Cytometry A. 1983;3:332–9. [DOI] [PubMed] [Google Scholar]
- 52. Vindeløv L, Christensen I, Nissen N. Standardization of high‐resolution flow cytometric DNA analysis by the simultaneous use of chicken and trout red blood cells as internal reference standards. Cytometry A. 1983;3:328–31. [DOI] [PubMed] [Google Scholar]
- 53. Vendrely R, Vendrely C. La teneur du noyau cellulaire en acide désoxyribonucléique à travers les organes, les individus et les espèces animals. Experientia. 1948;4:434–6. [DOI] [PubMed] [Google Scholar]
- 54. Vendrely R, Vendrely C. La teneur du noyau cellulaire en acide désoxyribonucléique à travers les organes, les individus et les espèces animals. Experientia. 1949;5:327–9. [DOI] [PubMed] [Google Scholar]
- 55. Dische Z. Über einige neue charakteristische Farbreaktionen der Thymonukleinsäure und eine Mikromethode zur Bestimmung derselben in tierischen Organen mit Hilfe dieser Reaktionen. Mikrochemie. 1930;8:4–32. [Google Scholar]
- 56. Tiersch TR, Chandler RW, Wachtel SS, Elias S. Reference standards for flow cytometry and application in comparative studies of nuclear DNA content. Cytometry. 1989;10:706–10. [DOI] [PubMed] [Google Scholar]
- 57. Van't Hof J. Relationships between mitotic cycle duration, S‐period duration and the average rate of DNA synthesis in the root meristem cells of several plants. Exp Cell Res. 1965;39:48–58. [DOI] [PubMed] [Google Scholar]
- 58. Arumuganathan K, Earle ED. Nuclear DNA content of some important plant species. Plant Mol Biol Report. 1991;9:208–18. [Google Scholar]
- 59. Ulrich I, Fritz B, Ulrich W. Application of DNA fluorochromes for flow cytometric DNA analysis of plant protoplasts. Plant Sci. 1988;55:151–8. [Google Scholar]
- 60. Bennett MD, Johnston S, Hodnett GL, Price HJ. Allium cepa L cultivars from four continents compared by flow cytometry show nuclear DNA constancy. Ann Bot. 2000;85:351–7. [Google Scholar]
- 61. Finkers R, van Kaauwen M, Ament K, Burger‐Meijer K, Egging R, Huits H, Kodde L, Kroon L, Shigyo M, Sato S and et al. Insights from the first genome assembly of onion (Allium cepa). bioRxiv 2021. [DOI] [PMC free article] [PubMed]
- 62. Doležel J, Greilhuber J. Nuclear genome size: are we getting closer? Cytometry A. 2010;19:103–6. [DOI] [PubMed] [Google Scholar]
- 63. Leitch I, Johnston E, Pellicer J, Hidalgo O, Bennett M. Plant DNA C‐values Database. 2019. [DOI] [PubMed]
- 64. Initiative AG. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature. 2000;408:796–815. [DOI] [PubMed] [Google Scholar]
- 65. Doležel J, Bartoš J, Voglmayr H, Greilhuber J. Nuclear DNA content and genome size of trout and human. Cytometry A. 2003;51A:127–8. [DOI] [PubMed] [Google Scholar]
- 66. Consortium TCeS . Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–8. [DOI] [PubMed] [Google Scholar]
- 67. Bennett MD, Leitch IJ, Price HJ, Johnston JS. Comparisons with Caenorhabditis (~100 Mb) and Drosophila (~175 Mb) using flow cytometry show genome size in Arabidopsis to be ~157 Mb and thus ~25% larger than the Arabidopsis genome Initiative estimate of ~125 Mb. Ann Bot. 2003;91:547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Doležel J, Čížková J, Šimková H, Bartoš J. One major challenge of sequencing large plant genomes is to know how big they really are. Int J Mol Sci. 2018;19:3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Project IRGS. The map‐based sequence of the rice genome. Nature. 2005;436:793–800. [DOI] [PubMed] [Google Scholar]
- 70. Meister A. Calculation of binding length of base‐specific DNA dyes by comparison of sequence and flow cytometric data. Application to Oryza sativa and Arabidopsis thaliana . J Theor Biol. 2005;232:93–7. [DOI] [PubMed] [Google Scholar]
- 71. Doležel J, Greilhuber J, Lucretti S, Meister A, Lysák MA, Nardi L, et al. Plant genome size estimation by flow cytometry: inter‐laboratory comparison. Ann Bot. 1998;82:17–26. [Google Scholar]
- 72. Bennett MD, Smith JB. Nuclear DNA amounts in angiosperms. Philos Trans R Soc Lond Ser B Biol Sci. 1976;274:227–74. [DOI] [PubMed] [Google Scholar]
- 73. Johnston JS, Bennett MD, Rayburn AL, Galbraith DW, Price HJ. Reference standards for determination of DNA content of plant nuclei. Am J Bot. 1999;86:609–13. [PubMed] [Google Scholar]
- 74. Praça‐Fontes MM, Carvalho CR, Clarindo WR, Cruz CD. Revisiting the DNA C‐values of the genome size‐standards used in plant flow cytometry to choose the "best primary standards". Plant Cell Rep. 2011;30:1183–91. [DOI] [PubMed] [Google Scholar]
- 75. Doležel J, Doleželová M, Novák F. Flow cytometric estimation of nuclear DNA amount in diploid bananas (Musa acuminata and M. balbisiana). Biol Plant. 1994;36:351–7. [Google Scholar]
- 76. Martin S, Sauder C, James T, Cheung K, Razeq F, Kron P, et al. Sexual hybridization between Capsella bursa‐pastoris (L.) Medik (♀) and Camelina sativa (L.) Crantz (♂) (Brassicaceae). Plant Breed. 2015;134:212–20. [Google Scholar]
- 77. Shapiro HS. Distribution of purines and pyrimidines in deoxyribonucleic acids. In: Fasman G, editor. Handbook of biochemistry and molecular biology. Volume 2. Cleveland: CRC press; 1976. p. 241–81. [Google Scholar]
- 78. Veleba A, Šmarda P, Zedek F, Horová L, Šmerda J, Bureš P. Evolution of genome size and genomic GC content in carnivorous holokinetics (Droseraceae). Ann Bot. 2017;119:409–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Marie D, Brown SC. A cytometric exercise in plant DNA histograms, with 2C‐values for 70 species. Biol Cell. 1993;78:41–51. [DOI] [PubMed] [Google Scholar]
- 80. Schönswetter P, Suda J, Popp M, Weiss‐Schneeweiss H, Brochmann C. Circumpolar phylogeography of Juncus biglumis (Juncaceae) inferred from AFLP fingerprints, cpDNA sequences, nuclear DNA content and chromosome numbers. Mol Phylogenet Evol. 2007;42:92–103. [DOI] [PubMed] [Google Scholar]
- 81. Bennett MD, Leitch IJ. Nuclear DNA amounts in angiosperms. Ann Bot. 1995;76:113–76. [Google Scholar]
- 82. Lysák MA, Doležel J. Estimation of nuclear DNA content in Sesleria (Poaceae). Caryologia. 1998;51:123–32. [Google Scholar]
- 83. Tuna M, Vogel KP, Arumuganathan K, Gill KS. DNA content and ploidy determination of bromegrass germplasm accessions by flow cytometry. Crop Sci. 2001;41:1629–34. [Google Scholar]
- 84. Zonneveld BJM, Van Iren F. Flow cytometric analysis of DNA content in Hosta reveals ploidy chimeras. Euphytica. 2000;111:105–10. [Google Scholar]
- 85. Hornych O, Ekrt L, Riedel F, Koutecký P, Košnar J. Asymmetric hybridization in central European populations of the Dryopteris carthusiana group. Am J Bot. 2019;106:1477–86. [DOI] [PubMed] [Google Scholar]
- 86. Barow M, Meister A. Lack of correlation between AT frequency and genome size in higher plants and the effect of nonrandomness of base sequences on dye binding. Cytometry. 2002;47:1–7. [DOI] [PubMed] [Google Scholar]
- 87. Vogel KP, Arumuganathan K, Jensen KB. Nuclear DNA content of perennial grasses of the Triticeae. Crop Sci. 1999;39:661–7. [Google Scholar]
- 88. Bennett MD, Leitch IJ. Nuclear DNA amounts in angiosperms ‐ 583 new estimates. Ann Bot. 1997;80:169–96. [DOI] [PMC free article] [PubMed] [Google Scholar]