

Abstract
To control infections phagocytes can directly kill invading microbes. Macrophage‐expressed gene 1 (Mpeg1), a pore‐forming protein sometimes known as perforin‐2, is reported to be essential for bacterial killing following phagocytosis. Mice homozygous for the mutant allele Mpeg1 tm1Pod succumb to bacterial infection and exhibit deficiencies in bacterial killing in vitro. Here we describe a new Mpeg mutant allele Mpeg1 tm1.1Pib on the C57BL/6J background. Mice homozygous for the new allele are not abnormally susceptible to bacterial or viral infection, and irrespective of genetic background show no perturbation in bacterial killing in vitro. Potential reasons for these conflicting findings are discussed. In further work, we show that cytokine responses to inflammatory mediators, as well as antibody generation, are also normal in Mpeg1 tm1.1Pib/tm1.1Pib mice. We also show that Mpeg1 is localized to a CD68‐positive endolysosomal compartment, and that it exists predominantly as a processed, two‐chain disulfide‐linked molecule. It is abundant in conventional dendritic cells 1, and mice lacking Mpeg1 do not present the model antigen ovalbumin efficiently. We conclude that Mpeg1 is not essential for innate antibacterial protection or antiviral immunity, but may play a focused role early in the adaptive immune response.
Keywords: dendritic cells, inflammation, macrophage expressed gene, monocytes and macrophages, mpeg1, perforin 2
Contrary to previous reports, Mpeg1 is not required for bacterial killing by phagocytes and antibacterial immunity. It also appears to affect antigen presentation.

INTRODUCTION
Macrophage‐expressed gene 1 (Mpeg1) was originally described in the mouse, 1 but is present in phagocytes of most metazoans. It is a type I transmembrane protein and the likely progenitor of the membrane attack complex/perforin (MACPF) family of proteins. 2
Vertebrate MACPF proteins include the terminal components of the complement system (the membrane attack complex proteins C6, C7, C8α, C8β and C9) and the key cellular immunity protein, perforin. Once associated with membranes, C9 and perforin oligomerize and form large transmembrane pores. C9 is directed to membranes by assembly of the other complement membrane attack complex proteins, while perforin binds via a dedicated calcium‐dependent C2 binding domain. 3 Mpeg1 also forms pores but possesses a dedicated yet different membrane‐binding domain that belongs to the multivesicular body of 12‐kDa‐associated β‐prism (MABP) family. 4 , 5
Macrophage‐expressed gene 1 is suggested to directly destroy phagocytosed bacteria as part of a key intracellular defense system analogous to the extracellular complement system. 1 , 6 , 7 Zebrafish with suppressed Mpeg1 expression show increased bacterial loads on Mycobacterium marinum or Salmonella typhimurium infection, but without deleterious effects on survival. 8 By contrast, mice lacking Mpeg1 reportedly do not survive infection with S. typhimurium or Staphylococcus aureus. 9 , 10
The current paradigm holds that in phagosomes Mpeg1 assembles on the surface of an internalized bacterium forming a lethal pore. 9 Support for this idea includes its formation of stable, circular oligomers and acquisition of lytic function at low pH in vitro. It is also asserted that Mpeg1 pore‐forming function is essential for immune defense against intracellular pathogenic bacteria. 6 , 10
In this study, we demonstrate that a new independently generated line of Mpeg1‐null mice has normal immune cell populations and displays no overt impairment during bacterial or viral infection. In vitro, macrophage from these mice kill bacteria equally as well as macrophage from wild‐type (WT) mice, and respond normally to inflammatory mediators. We demonstrate that Mpeg1 is processed to a two‐chain form, and is present in a CD68‐enriched endolysosomal compartment within both macrophages and cross‐presenting dendritic cells. Mice lacking Mpeg1 exhibit a subtle defect in antigen presentation. These findings contradict the current paradigm, and indicate the role of Mpeg1 is still to be uncovered.
RESULTS
Generation and validation of Mpeg1 knockout mice
As outlined in Figure 1, we produced embryonic stem (ES) cells and resulting mice carrying a conditional Mpeg1 mutant allele (Mpeg1 tm1Pib ). Mice arising from two independently generated ES cell lines were crossed with global Cre deleters to delete Mpeg1 from all tissues and the germ line. Genomic DNA analysis by PCR and sequencing confirmed deletion of the Mpeg1 coding sequence and selectable marker to generate the Mpeg1 tm1.1Pib (flox) allele (Figure 1c).
Figure 1.
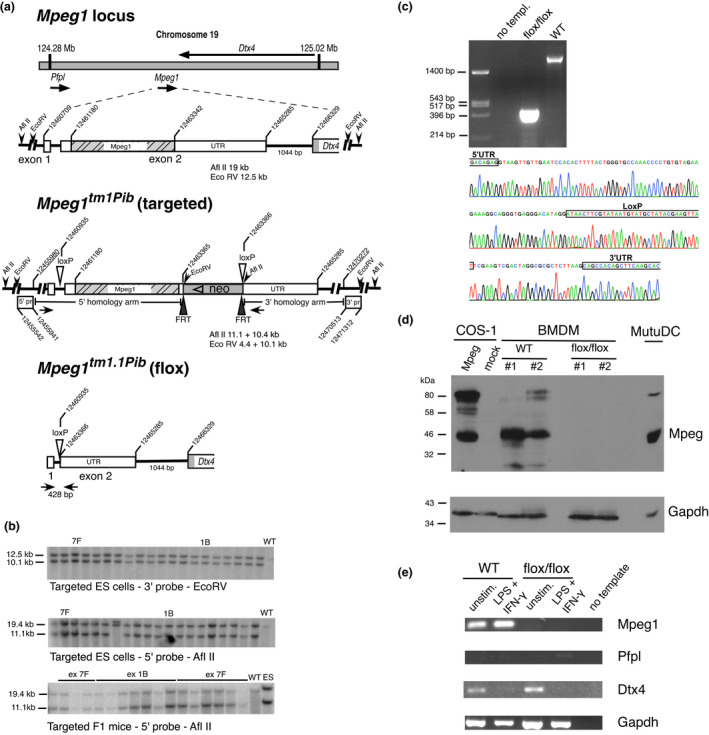
Gene targeting of Mpeg1. (a) The Mpeg1 locus, Mpeg1 structure and predicted allele structure after insertion of loxP into the 5′ UTR and the selectable marker (neo) and loxP into the 3′ UTR [Mpeg tm1Pib (targ)]. Subsequent removal of the Mpeg1 coding sequence and neomycin (neo) cassette by cre‐mediated recombination yields the knockout allele [Mpeg1 tm1.1Pib (flox)]. Sequences cloned and used as 5′ or 3′ flanking probes (pr) are indicated. Coordinates are from the mouse genomic reference sequence GRCm38.p3 C57/BL6. (b) Validation of ES cell and F1 mouse genotypes by Southern blotting. The predicted sizes of genomic fragments generated following EcoRV or AflII treatment and recognized by the indicated 5′ and 3′ probes are shown in a. (c) Validation of cre‐mediated Mpeg1 deletion in a floxed mouse. Shown is the relevant DNA sequence of the 428‐bp flox/flox PCR product amplified using the primers shown as closed arrowheads in the Mpeg1 tm1.1Pib allele shown in a. These primers yield a 2800‐bp product from WT mouse genomic DNA (upper panel). (d) Protein extracts from 106 BMDMs derived from WT or knockout (flox/flox) mice were separated by 10% SDS–PAGE and immunoblotted for Mpeg1 (1:2000 rabbit anti‐Mpeg), followed by immunoblotting for GAPDH (1:5000 anti‐GAPDH). These were compared with extracts from COS‐1 cells transiently expressing Mpeg1, and from the MutuDC line (104 cells). Image shown represents three independent experiments. (e) Total RNA from unstimulated or IFNγ‐ and LPS‐stimulated BMDMs derived from WT or knockout (flox/flox) mice was assessed via RT‐PCR for the presence of Mpeg1, Pfpl, Dtx4 and housekeeping Gapdh transcripts. ES cell, embryonic stem cell; BMDM, bone marrow‐derived macrophage; IFN, interferon; LPS, lipopolysaccharide; Mpeg1, macrophage‐expressed gene 1; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; RT‐PCR, reverse‐transcriptase PCR; UTR, untranslated region; WT, wild type. [Colour figure can be viewed at wileyonlinelibrary.com]
We next determined whether Mpeg1 protein is missing from Mpeg1 tm1.1Pib/tm1.1Pib mice (abbreviated from here onward as Mpeg1 −/−). Because Mpeg1 was first described in macrophages, 1 bone marrow‐derived macrophages (BMDMs) were prepared from two WT and two Mpeg1 −/− animals. As controls, we analyzed COS‐1 cells transfected with the Mpeg1 complementary DNA as well as the murine conventional dendritic cell 1 cell line, MutuDC, 11 given that proteomics analyses have identified Mpeg1 as an abundant DC protein (for example, see Luber et al. 12 ). Immunoblotting of cell extracts demonstrated the presence of the expected 80‐kDa full‐length Mpeg1 protein in the MutuDC and COS‐1 control cells (Figure 1d). In the COS‐1 cells there appeared to be a doublet at about 80 kDa: the slightly smaller species may represent an Mpeg1 isoform arising as a result of differential glycosylation or proteolysis. In both COS‐1 and MutuDC cells a major ~42‐kDa species was also observed. (As shown in Figure 2, this represents the processed two‐chain form of Mpeg1).
Figure 2.
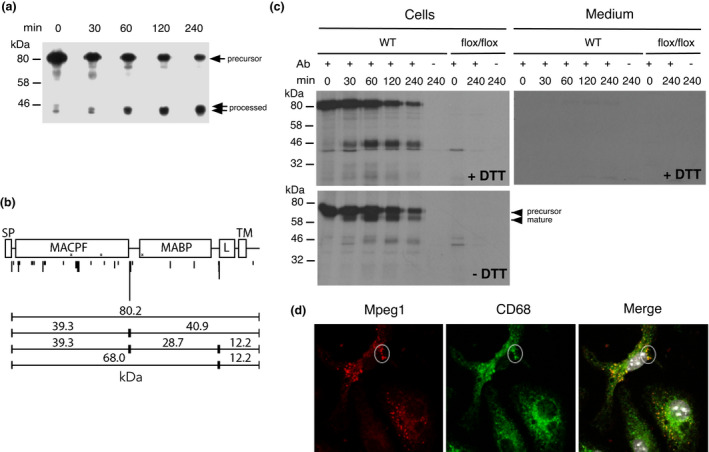
Processing of Mpeg1 during biosynthesis. (a) MutuDCs were pulse labeled with 50 μCi 35S‐met for 30 min, chased for the indicated times and then lysed in 0.5% (w/v) SDS. Samples were immunoprecipitated with protein G–Sepharose and rabbit anti‐Mpeg1, then resuspended in LSB/0.1 M DTT and separated by 10% SDS–PAGE. The experiment was performed three times. (b) Immunoprecipitated Mpeg1 was analyzed via proteomics. Scaled domain structure of Mpeg1 follows Pang et al. 5 Asterisks indicate N‐glycosylation site; Nontryptic cleavages identified are indicated beneath. The height of the mark reflects the number of times each cleavage was identified across all runs. Also shown are predicted molecular weights based on the major cleavage sites. (c) BMDMs were pulse labeled with 100 μCi 35S‐met for 60 min and chased for the indicated times. At each point, the medium was removed and separately analyzed for Mpeg1. Samples were immunoprecipitated using guinea pig Gp202 antiserum (antibody) and protein A–Sepharose. Before SDS–PAGE each sample was divided into two. LSB/DTT was added to one, and LSB to the other. The experiment was performed two times. (d) BMDMs were stained with 1:400 Gp202 and 1:800 anti‐Gp‐AF568 and 1:400 anti‐CD‐68‐AF647. Images are single optical slices representing five independent experiments. Ab, antibody; BMDM, bone marrow‐derived macrophage; DTT, dithiothreitol; LSB, Laemmli sample buffer; MABP, multivesicular body of 12‐kDa‐associated β‐prism; MACPF, membrane attack complex/perforin domain; L, L‐domain; Mpeg1, macrophage‐expressed gene 1; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; SP, signal peptide; TM, transmembrane domain. [Colour figure can be viewed at wileyonlinelibrary.com]
The different forms of Mpeg were evident in WT BMDMs but were completely absent from knockout (flox/flox) cells (Figure 1d). One of the WT samples exhibited essentially no full‐length Mpeg1 but substantially more of the 42‐kDa species This is consistent with conversion of the full‐length form to the smaller form, in this case possibly following lysis.
Because Mpeg1 is close to its placentally expressed paralog Pfpl, and overlaps the unrelated gene Dtx4, 13 we assessed the expression of these genes in BMDMs and in BMDMs stimulated with interferon‐gamma (IFN‐γ) and lipopolysaccharide (LPS; Figure 1e). There was no difference in the expression of Dtx4 when comparing WT with Mpeg1 −/− cells: transcripts were present in unstimulated BMDMs and were downregulated in stimulated BMDMs. Very low expression of Pfpl was evident in BMDMs from both WT or Mpeg1 −/− mice.
Together, these results confirmed that our knockout animals lack Mpeg1, and that there is no obvious impact on adjacent genes or evidence for paralog compensation in BMDMs.
Mpeg1 is a component of professional phagocytes and is processed to a two‐chain, disulfide‐linked form
Immunoblotting of samples from WT or Mpeg1 −/− mice determined which major immune cell populations express Mpeg1, and to what extent different Mpeg1 forms are evident. Mpeg1 was not detected in T, B or natural killer cells (data not shown). Splenic macrophages (Supplementary figure 1a), neutrophils (Supplementary figure 1b) and conventional dendritic cell 1 (Supplementary figure 1c, d) express Mpeg1 in both the expected full‐length (80 kDa) and smaller (about 42 kDa) forms (Figure 1, Supplementary figure 1), again suggesting that Mpeg1 is processed from a precursor to a mature form.
To demonstrate processing and rule out postlysis proteolysis, we carried out a pulse‐chase analysis of MutuDCs (Figure 2a). This clearly showed conversion of the 80‐kDa form to two ~42‐kDa species, with a t 1/2 of about 120 min. We mapped the processing sites on Mpeg1 using a proteomics approach. Immunoprecipitated Mpeg1 from MutuDCs was subjected to in‐gel tryptic digestion, and the resulting peptides were identified by liquid chromatography–tandem mass spectrometry with database matching restricted to Mpeg1 sequences. The major nontryptic cleavage point in Mpeg1 was identified at position 350. As shown in Figure 2b, cleavage of the full‐length Mpeg1 at this position is predicted to yield products of 39 and 41 kDa, consistent with the species we observed in macrophages and DCs. A minor cleavage point was observed at position 600.
Mapping these cleavage points onto the published Mpeg1 structures 4 , 5 showed that position 350 occurs in a surface loop between the MACPF domain and MABP domain (Figure 2b). Importantly, cleavage of Mpeg1 at this point yields subunits that remain associated via disulfide bonds. Position 600 occurs between the MABP and L domains and cleavage there would separate an Mpeg1 ectodomain from the transmembrane domain and potentially release Mpeg1 from the membrane.
To determine whether the processed Mpeg1 forms remain associated in the cell, we performed another pulse‐chase experiment, analyzing samples in the absence or presence of reductant dithiothreitol (DTT). As shown in Figure 2c, unreduced samples—which by definition are not fully denatured—showed a precursor (< 80 kDa) that was rapidly converted to a slightly smaller form, consistent with maturation into a folded, disulfide‐linked mature species. There was little indication of the ~42‐kDa species in the unreduced samples. By contrast, reduced samples from equivalent time points clearly showed the fully denatured precursor at 80 kDa and the presence of the ~42‐kDa species. This suggests that Mpeg1 is normally processed to a two‐chain form with subunits linked by disulfide bonds. We were unable to detect Mpeg1 release from cells into the medium (Figure 2c), suggesting that cleavage between the MABP and L domain (if it occurs) does not liberate a soluble, secreted form of Mpeg 1.
To identify the intracellular compartment in which Mpeg1 cleavage occurs, we repeated the pulse‐chase analysis in the presence of compounds that interfere with secretory protein trafficking and processing (Supplementary figure 2). We found that those that prevent post‐Golgi trafficking (brefeldin, golgicide A) or inhibit endolysosomal vesicle acidification (bafilomycin, monensin) reduced or abrogated Mpeg1 cleavage. This indicated that Mpeg1 processing occurs in a post‐Golgi, endolysosomal compartment.
Mpeg1 colocalizes with CD68 in a post‐Golgi compartment
We employed fluorescence microscopy to determine the location of Mpeg1 within macrophages and DCs. Pilot experiments indicated that the commercial rabbit anti‐Mpeg1 antibody employed by others 9 specifically detects Mpeg1 in sodium dodecyl sulfate (SDS)‐denatured cell extracts via immunoblotting (Figure 1; Supplementary figure 1) or immunoprecipitation (Figure 2a). However, when used in indirect immunofluorescence (Supplementary figure 3), it detected Mpeg1 only when overexpressed in transfected cells: it did not detect endogenous Mpeg1 in BMDMs. In addition, via immunofluorescence it cross‐reacted with an unknown protein/compartment in both WT and Mpeg1 −/− BMDMs (Supplementary figure 3). We therefore attempted to investigate Mpeg1 localization using forms of Mpeg1 fused to GFP (as previously reported) 6 , 9 , 14 or to small epitope tags (FLAG or V5) at either terminus (Supplementary figures 4 and 5). However, these derivatives localized predominantly in the endoplasmic reticulum (ER) of transfected cells, indicating impaired trafficking and localization. We concluded that the use of Mpeg1 fusion proteins as reporters in transfected cells is likely to be misleading.
Consequently, we raised new antisera to recombinant Mpeg1 in guinea pigs. As shown in Supplementary figure 6, these antisera detected recombinant Mpeg1 in COS cells primarily in a vesicular pattern. Importantly, they detected Mpeg1 via indirect immunofluorescence (Supplementary figure 6) or immunoprecipitation (Supplementary figure 6; Figure 2c) in WT but not in Mpeg1 −/− BMDMs. Using the guinea pig antiserum, we showed that Mpeg1 is present in a CD68‐positive vesicular compartment in BMDMs (Figure 2d) and DCs. Mpeg1 was not evident in early endosome antigen 1 (EEA1; early endosome) or lysosome associated membrane protein 1 (LAMP1; late endosome/lysosome) compartments (data not shown).
Mpeg1 is not required for immune system development
Mpeg1 −/− animals of both genders were morphologically indistinguishable from WT animals. No differences were noted in any of the nearly 40 organs and tissues examined (see the “Methods” section for the list); and red blood cell, platelet and differential white blood cell counts were normal (data not shown). Knockout animals were fertile, and dams produced and raised litters normally.
With its proposed role in immunity, we considered the possibility that Mpeg1 contributes to immune cell homeostasis. Splenocytes and thymocytes isolated from WT or Mpeg1 −/− animals were assessed for the proportion and numbers of B cells, T cells, macrophages and DCs. As shown in Supplementary figure 7, Mpeg1 −/− animals showed no evidence of compromised immune cell proportions in the spleen, and immune cell numbers were unaffected (data not shown). Similar results were observed for thymus populations (data not shown) and analysis of blood showed no differences in neutrophils (Supplementary figure 7), suggesting that the immune system develops normally in these animals.
Mpeg1 is not required for bacterial killing by BMDMs in vitro
McCormack et al. 9 concluded that Mpeg1 is a phagolysosomal component essential for bacterial killing following infection. They used a number of different bacteria, including Escherichia coli K12, S. aureus (pathogenic methicillin‐resistant S. aureus) and Mycobacterium smegmatis, to infect WT or Mpeg1 tm1Pod/tm1Pod mouse embryonic fibroblasts in vitro. They also used S. typhimurium to infect microglia cells. In all cases, absence of Mpeg1 resulted in a failure to control infection.
To replicate and extend these results, we examined the localization of Mpeg1 in BMDMs during infection by M. smegmatis via indirect immunofluorescence. As shown in Figure 3a and Supplementary figure 8a, we observed colocalization of Mpeg1 with phagocytosed bacteria within 4 h of infection, with the number of cells involved and the number of Mpeg1‐marked bacteria per cell increasing up to 18‐h after infection. At the times indicated, the survival of ingested M. smegmatis was determined. To our surprise, the data showed no significant differences in Mpeg1 −/− BMDMs bacterial killing compared with WT BMDMs (Figure 3a).
Figure 3.
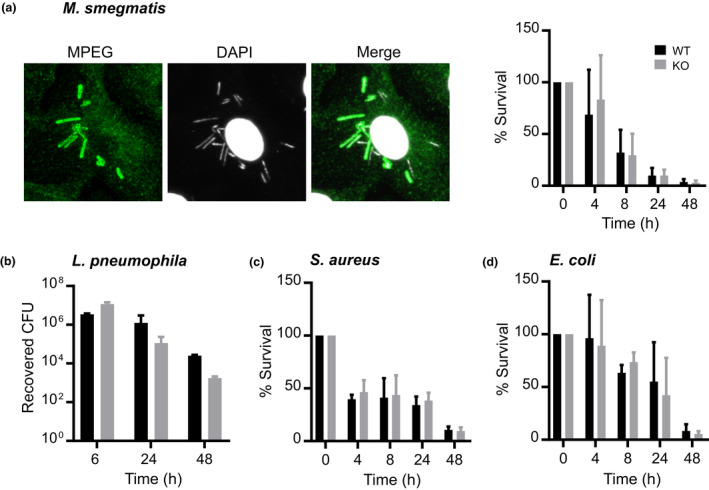
Mpeg1 is not essential for bacterial killing by BMDMs in vitro. BMDMs were infected with (a) Mycobacterium smegmatis (MOI = ~10–15), (b) Legionella pneumophila (MOI = ~10), (c) Staphylococcus aureus (MOI = ~5–10) and (d) Escherichia coli K12 (MOI = 30–50). Macrophages infected with M. smegmatis were permeabilized and stained for Mpeg1 and bacteria (see Supplementary figure 6a for details). At different times after infection, intracellular bacteria were released by lysing the cells in water and enumerated. Statistical significance was assessed using the Student's unpaired t‐test and showed no significant differences between groups. Data are mean ± standard deviation of the percentage of survival at each time point obtained from two (Legionella) or at a minimum of three independent experiments. BMDM, bone marrow‐derived macrophage; DAPI, 4′,6‐diamidino‐2‐phenylindole; KO, knockout; MOI, multiplicity of infection; WT, wild type. [Colour figure can be viewed at wileyonlinelibrary.com]
We then used three other bacterial species to infect BMDMs from WT or Mpeg1 −/− mice: an avirulent strain of the Gram‐negative organism Legionella pneumophila (Figure 3b); S. aureus (Figure 3c) or E. coli K12 (Figure 3d). In every case both WT and Mpeg1 −/− BMDMs cleared the bacteria equally well.
To rule out the possibility that genetic (strain) differences explain the discrepancy between these results and the observations of McCormack et al., we crossed our C57BL/6J Mpeg1−/− mice with 129X1/SVJ mice to produce Mpeg1 −/− animals with a genetically mixed background, similar to those reported by McCormack et al. 9 129X1/SVJ mice carry a naturally occurring inactivating mutation in the Caspase 11 gene, 15 so in case bacterial clearance is influenced by caspase 11 deficiency we derived Casp11 +/+/Mpeg1 +/+ ; Casp11 −/− /Mpeg1 +/+ and Casp11 −/− /Mpeg1 –/– mice. Bacterial killing assays were then repeated using BMDMs from the resulting 129X1/SvJ:C75BL/6 J animals. The results showed that both WT and Mpeg1 −/− BMDMs kill S. aureus or E. coli K12 equally well, irrespective of Caspase 11 status (Supplementary figure 8b). Taken together, these results showed that Mpeg1 is not required for bacterial killing by BMDMs in vitro.
Mpeg1 increases upon macrophage activation but does not affect the inflammatory response
Macrophage‐expressed gene 1 expression increases upon macrophage activation, and Mpeg1 messenger RNA increases in MEFs after activation with IFNγ, suggesting an association of this protein with inflammation. 9 By immunoblotting we evaluated the expression of Mpeg1 in BMDMs activated with LPS and/or IFNγ. Mpeg1 expression increased by approximately fivefold following stimulation with both LPS and IFNγ, with a greater proportion of the full‐length (80 kDa) form evident (Figure 4).
Figure 4.
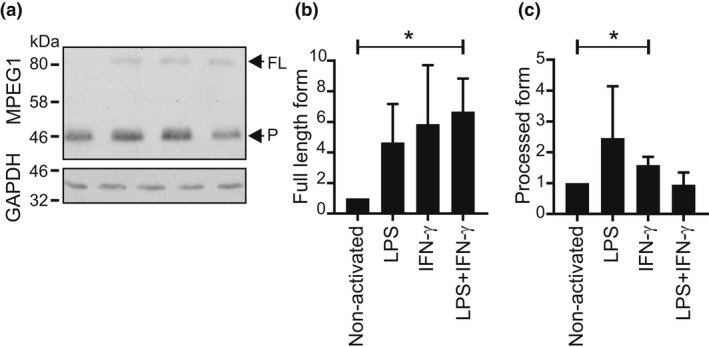
Increased full‐length Mpeg1 after BMDM activation. Day 9 BMDMs were activated overnight with LPS (10 ng mL−1) and/or IFNγ (100 ng mL−1). Extracts from 1 × 106 cells were resolved by 10% SDS–PAGE and transferred to nitrocellulose. (a) The membranes were sequentially probed with anti‐MPEG1 antibody and rabbit anti‐GAPDH. The figure shown is representative of two independent experiments. (b, c) The fold increase of Mpeg1 on activation was evaluated via densitometry. The Student's t‐test was used to compare outcomes. *P < 0.05. BMDM, bone marrow‐derived macrophage; FL, full length; IFN, interferon; LPS, lipopolysaccharide; Mpeg1, macrophage‐expressed gene 1; P, processed form; SDS–PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Pattern recognition receptors respond to pathogen‐associated molecular patterns and damage‐associated molecular patterns to invoke the innate immune response. Activation of pattern recognition receptor and Nod‐like receptors such as NLRP3 by cytosolic damage‐associated molecular patterns drives formation of the multiprotein inflammasome complex. Canonical activation of the inflammasome activates caspase 1, which initiates maturation and secretion of interleukin (IL)‐1β and IL‐18, and induces pyroptosis. Noncanonical activation of the inflammasome involves caspase 11. Given that Mpeg1 is a pore‐forming molecule present in the endolysosomal network, we speculated that it may play a role during infection in transferring moieties carrying pathogen‐associated molecular patterns or damage‐associated molecular patterns from the endosome to the cytosol to drive inflammasome activation.
WT, Mpeg1 −/− or inflammasome‐deficient BMDMs were infected with bacteria or treated with model activators known to induce inflammasome formation and cytokine release. Secretion of IL‐1β and IL‐18 was measured, and the extent of pyroptotic cell death evaluated via lactate dehydrogenase release. Impact on the general proinflammatory response was assessed by measuring tumor necrosis factor‐α secretion.
As shown in Figure 5, exposure of WT BMDMs to the canonical NLRP3 activators LPS/ATP; LPS/nigericin; Bacillus cereus or influenza A virus resulted in IL‐1β, IL‐18 and lactate dehydrogenase release. This response—but not tumor necrosis factor‐α release—was absent or significantly blunted in control BMDMs lacking Nlrp3. Mpeg1 −/− BMDMs showed comparable responses to WT BMDMs, indicating that Mpeg1 does not influence canonical NLRP3 inflammasome activation.
Figure 5.
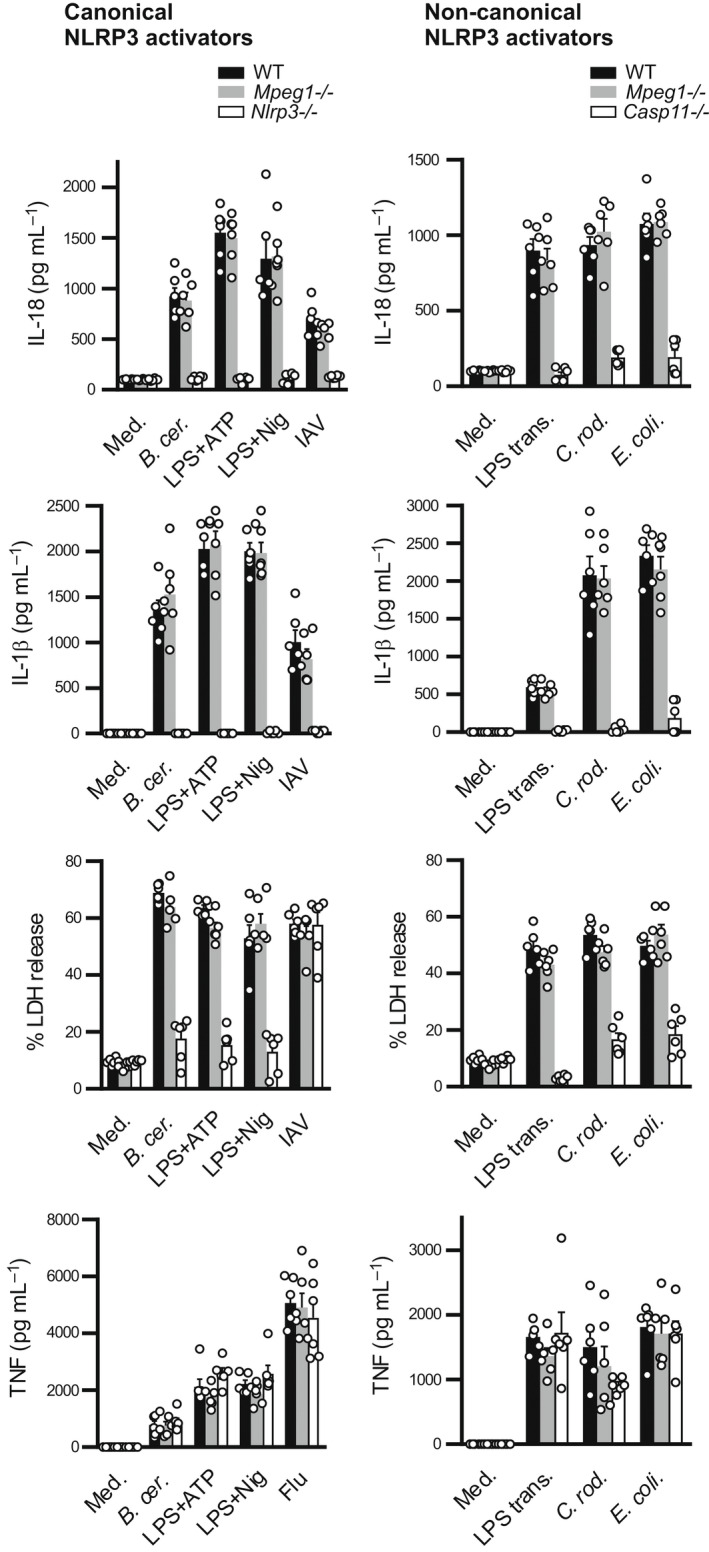
Mpeg1 is not required for inflammasome activation. BMDMs were treated with the indicated activators and cytokine release was measured by ELISA. Pyroptosis was evaluated via measurement of LDH release. B. cer., Bacillus cereus; BMDM, bone marrow‐derived macrophage; C. rod., Citrobacter rodentium; E. coli., Escherichia coli; Flu, influenza virus; IAV, influenza A virus; IL, interleukin; LDH, lactate dehydrogenase; LPS, lipopolysaccharide; LPS trans., transfected LPS; Med., medium; Mpeg1, macrophage‐expressed gene 1; Nig, nigericin; TNF, tumor necrosis factor. The experiment was carried out three times.
Activation of the inflammasome family members AIM2, NLRC4 and Pyrin was also tested using the appropriate synthetic [poly(dA:dT)], bacterial (Listeria monocytogenes, Francisella novicida, S. typhimurium or Clostridium difficile) and/or viral (murine cytomegalovirus) activators. No perturbation of these responses was evident in Mpeg1 −/− BMDMs (Supplementary figure 9).
Finally, noncanonical inflammasome activation was tested, using the following activators: transfected LPS; E. coli; or Citrobacter rodentium; and employing Casp11 knockout BMDMs as controls (Figure 5). Again, the Mpeg1 −/− response was indistinguishable from WT, indicating that Mpeg1 does not influence noncanonical inflammasome activation.
These results indicated that Mpeg1 does not play a role in immune signaling via inflammasome formation during bacterial or viral infection, or in response to stimulation with damage‐associated molecular patterns.
Mpeg1 is not essential for the control of bacterial infection
The in vitro results described so far were inconsistent with the current view that Mpeg1 is essential for the clearance of bacterial infections by direct killing of bacteria. 9 They also argued against a key role in immune signaling.
To further examine this issue, Mpeg1 −/− mice were infected with the human bacterial pathogens Mycobacterium tuberculosis (acid fast), methicillin‐resistant S. aureus (Gram positive) or Legionella longbeachae (Gram negative). It has been reported that humans carrying certain Mpeg1 gene polymorphisms are more susceptible to M. tuberculosis infection, 16 and that infection of Mpeg1‐deficient mice with S. aureus is lethal. 9
Mice were intranasally infected with M. tuberculosis under Physical Containment Level 3 laboratory conditions. Following infection, animals were periodically weighed (weight loss is a conventional indicator of the severity of disease progression) and bacterial loads in lungs and spleens were evaluated after about 8 weeks. There were no statistically significant differences in weight loss or bacterial load observed between WT or Mpeg1 −/− groups (Figure 6a).
Figure 6.
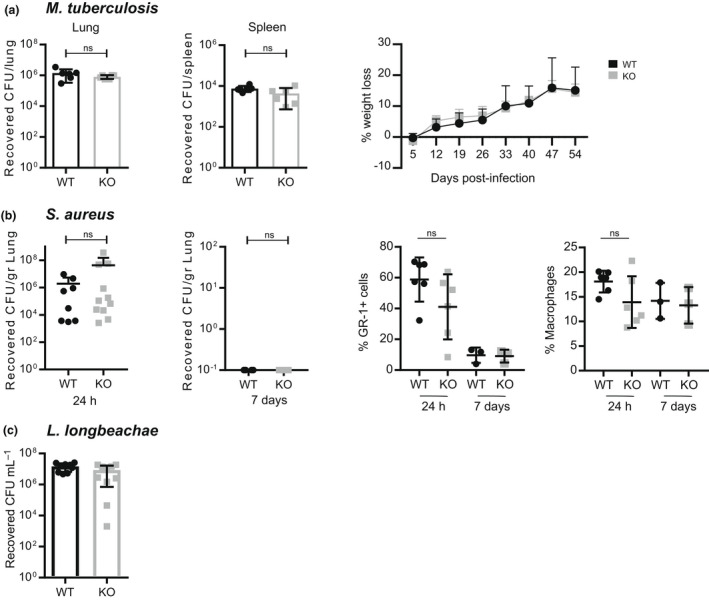
Mpeg1 is not essential for bacterial clearance. WT or Mpeg1 −/− mice were infected with the indicated pathogens. Tissue homogenates were serially diluted and aliquots plated on appropriate media. Colonies were counted and reported as CFU/gram of tissue. (a) Six mice of each genotype infected by aerosolized Mycobacterium tuberculosis (~100–200 CFU) were killed 8 weeks after the infection and CFU in lungs and spleen assessed. Weight loss was evaluated at the indicated times after the infection. The experiment was carried out once. (b) Ten mice of each genotype were infected with 1 × 107–5 × 107 CFU of Staphylococcus aureus USA 300 via intranasal inhalation. At the indicated times, CFU in lungs were evaluated, and the proportions of neutrophils (Gr+) and macrophages (CD11b+, F4/80+) in lungs compared. The experiment was carried out three times. (c) Eight mice of each genotype were inoculated intranasally with 4 × 104 CFU of Legionella longbeachae. CFU in lungs was assessed 3 days after the infection. The experiment was carried out once: each data point represents a single mouse. Statistical significance was assessed using the Student's t‐test. ns = P > 0.05. CFU, colony forming units; KO, knockout; Mpeg1, macrophage‐expressed gene 1; WT, wild type.
Mpeg1 −/− or WT mice were infected with S. aureus intranasally and the recovery of bacteria in the lungs was evaluated 24 h and 7 days after infection (Figure 6b). Comparable numbers of bacteria were recovered from the lungs of WT and Mpeg1 −/− mice after 24 h, indicating that Mpeg1 plays no role early in infection. At day 7 following the infection there were no detectable bacteria in either WT or Mpeg1 −/− mice, indicating that both cohorts successfully cleared the infection.
Finally, Mpeg1 −/− and WT mice were infected with L. longbeachae via intranasal inhalation. At day 3 after the infection, the bacterial load in the lungs was evaluated. The data showed no significant difference in the response to infection between the two genotypes (Figure 6c).
Taken together, these results showed that Mpeg1 is not essential to the response to bacterial infection.
Mpeg1 is not essential for antiviral immunity or antibody production
There is an open possibility that Mpeg1 functions in the adaptive immune response, for example, in viral clearance or antibody production. Supporting this notion, Mpeg1 was identified as a top hit in a proteomics screen of mouse CD11c+, CD8α+ DCs, cells that are important in antiviral responses. 12 Mpeg1 expression is also upregulated in abalone following challenge with hemorrhagic septicemia virus, 17 and in both abalone and mouse MZB cells in response to poly I:C (which simulates viral infections). 17 , 18
To test this idea, mice were infected intravenously with 2 × 106 focus‐forming units of lymphocytic choriomeningitis virus Docile strain, which induces chronic, persistent infection and exhaustion of CD8+ T cells. 19 The viral titer was evaluated in different organs at day 8 and day 28 after infection. As shown in Figure 7a, viral titers were comparable in both WT and Mpeg1 −/− groups of mice, in every organ tested, both at day 8 and at day 28. No differences were observed in the numbers or proportions of immune cells between genotypes during infection (Supplementary figure 10), and the generation of virus‐specific cytolytic T cells was not perturbed in the absence of Mpeg1 (Supplementary figure 11).
Figure 7.
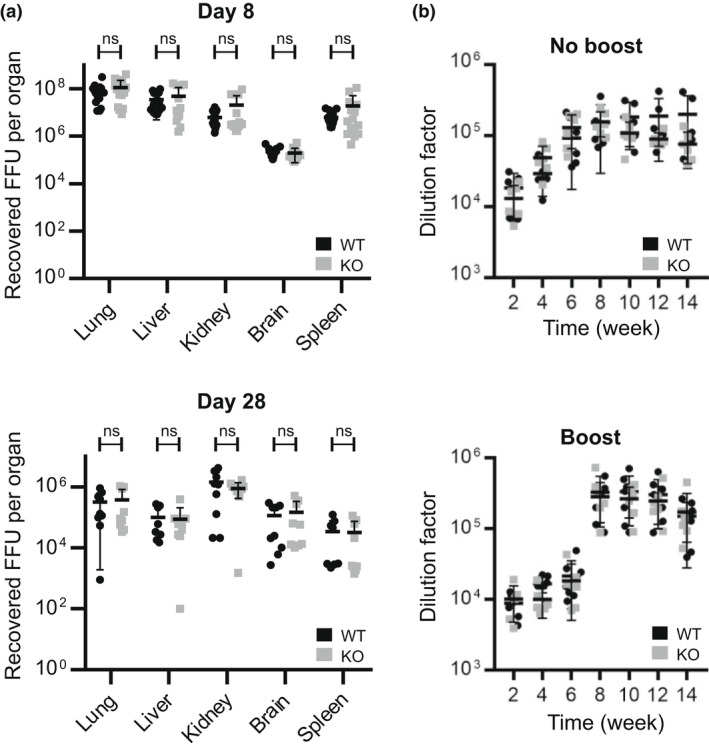
(a) Mpeg1 is not involved in the control of LCMV infection. Nine mice of each genotype were infected with 2 × 106 FFU of LCMV Docile strain intravenously and viral titers in tissues assessed at day 8 or 28. Each point indicates a single mouse. The experiment was carried out two times. (b) Mpeg1 does not influence the antibody response. Six mice of each genotype were injected with 10 μg ovalbumin–adjuvant mixture subcutaneously on day 0. In the experiment shown in the lower panel, mice received a boost at week 6. Sera were collected and checked for antibody level at the indicated times using an indirect ELISA assay. Data represent two independent experiments: each point indicates a single mouse. Statistical significance was assessed using the Student's t‐test with Holm–Šídák correction for multiple comparisons. ns = P > 0.05. FFU, focus‐forming unit; KO, knockout; LCMV, lymphocytic choriomeningitis virus; Mpeg1, macrophage‐expressed gene 1; WT, wild type.
To evaluate antibody production, WT or Mpeg1 −/− mice were injected with the model soluble antigen ovalbumin, and serum anti‐ovalbumin antibodies measured periodically out to 14 weeks. Immune cell populations in the spleen were assessed at weeks 6 and 14. In a separate study, animals were injected again at 6 weeks to assess the recall response. No differences in antibody levels or response were observed between genotypes (Figure 7b).
Antigen presentation is perturbed in Mpeg1‐deficient mice
Conventional dendritic cells 1 cross‐present extracellular antigen to CD8+ T cells, processed via either the vacuolar pathway or the endosome‐to‐cytosol pathway. Given endolysosomal location and pore‐forming ability of Mpeg1, we hypothesized that it facilitates antigen transfer from endosomes to cytosol. To test this, antigen presentation was evaluated in Mpeg1 −/− mice.
An in vivo cytotoxicity assay was used to investigate the potential role of Mpeg1 in cross‐priming. Splenocytes isolated from H2‐k bm1 (bm1) mice were coated with the model antigen ovalbumin to prepare ovalbumin‐coated splenocytes (OCSs), which were injected into WT or Mpeg −/− mice. bm1 mice carry a mutation in major histocompatibility complex (MHC)‐I and cannot activate CD8+ T cells by direct antigen presentation: thus, ovalbumin antigen from bm1 OCSs captured by host DCs must be cross‐presented to CD8+ T cells to induce generation of ovalbumin‐specific effector cytotoxic T cells. Such effector T cells lyse target cells coated with ovalbumin peptide 257–264 (SIINFEKL).
To evaluate cytotoxic T lymphocyte killing, mice primed intravenously with OCSs were injected later with Cell Trace Violet‐labeled target cells from C57BL/6J mice coated with SIINFEKL peptide and differentially labeled Cell Trace Violet‐labeled uncoated cells as nontargets. The proportion of peptide‐positive target cells remaining in the spleen was analyzed later by flow cytometry (Figure 8a). The results showed that Mpeg1 −/− mice can cross‐present extracellular antigen and elicit effective cytotoxic T lymphocyte killers.
Figure 8.
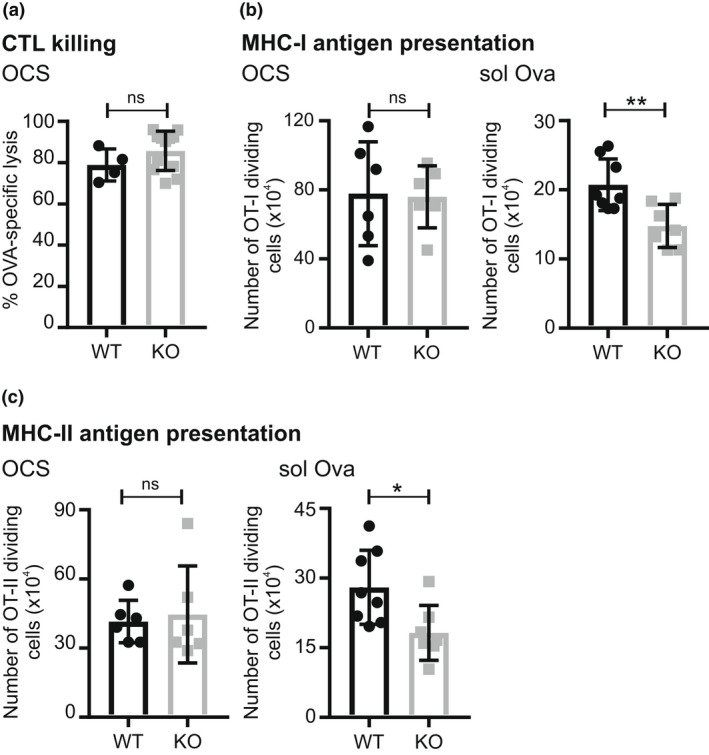
Role of Mpeg 1 in antigen presentation. (a) H‐2K bm1 OCSs were intravenously injected into WT or Mpeg1 −/− mice, followed by intravenous injection of equal numbers of CTVhigh cells labeled with ovalbumin257–264 peptide and unlabeled CTVlow cells after 6–8 days. CTVhigh and CTVlow cell numbers in recipients were determined 36–48 h later by flow cytometry. (b) Equal numbers of CTV‐labeled OT‐I T cells were intravenously injected into WT or Mpeg1 −/− mice, followed by intravenous injection of 20 × 106 OCSs or 25 μg ovalbumin after 24 h. Dividing OT‐I T cells in spleen were enumerated 72 h later by flow cytometry. (c) Equal numbers of CTV‐labeled OT‐II T cells were intravenously injected into WT or Mpeg1 −/− mice, followed by intravenous injection of 20 × 106 IA b−/− OCS mice or 50 μg ovalbumin 24 h later. About 72 h later the number of OT‐II dividing cells in spleen was assessed by flow cytometry. The experiment was performed two times. Each data point represents a single mouse. Statistical significance was assessed using the Student's t‐test. ns = P > 0.05; *P < 0.05; **P < 0.01. CTL, cytotoxic T lymphocyte; CTV, Cell Trace Violet; MHC, major histocompatibility complex; Mpeg1, macrophage‐expressed gene 1; OCS, ova‐coated splenocyte; WT, wild type.
To investigate the role of Mpeg1 in cross‐presentation of extracellular soluble antigen to CD8+ T cells, an in vivo T‐cell proliferation assay was performed. Cell Trace Violet‐labeled CD8+ T cells from OT‐I mice were adoptively transferred to WT or Mpeg1 −/− mice. OT‐I T cells express a transgenic T‐cell receptor that recognizes SIINFEKL peptide presented by H‐2Kb. Mice were subsequently injected with OCSs or ovalbumin protein. Splenocytes extracted later from the injected mice were analyzed by flow cytometry to assess proliferation of the labeled OT‐I cells. As shown in Figure 8b, OT‐I CD8+ T‐cell proliferation in response to OCSs was no different between the WT and Mpeg1 −/− mice. However, significantly lower proliferation of OT‐I CD8+ T cells was observed when ovalbumin protein alone was injected into Mpeg 1−/− mice. This suggested that cross‐presentation of ovalbumin protein is impaired in the absence of Mpeg1.
Dendritic cells also present extracellular antigen on MHC‐II to activate naïve CD4+ T cells. To test the involvement of Mpeg1 we used OT‐II mice, which are transgenic for a CD4+ T‐cell receptor that recognizes ovalbumin residues 323–339 (ISQAVHAAHAEINEAGR) when presented by I‐Ab. Labeled CD4+ T cells from OT‐II mice were adoptively transferred to WT or Mpeg1 −/− mice. Mice were then immunized with OCSs prepared from MHC‐II‐deficient mice or with ovalbumin protein, and the proliferation of splenic OT‐II T cells was analyzed. Given that OCSs from MHC‐II‐deficient mice cannot activate CD4+ T cells directly, CD4+ OT‐II T cells proliferate only if OCSs or soluble ovalbumin is processed by DCs as an extracellular antigen. The results showed that presentation of OCSs on MHC‐II is not significantly affected in the absence of Mpeg1. However, a significant decrease in the proliferation of CD4+ T cells was observed in Mpeg1 −/− mice in response to ovalbumin (Figure 8c).
Taken together, these results indicated that both MHC‐II and MHC‐I cross‐presentation of soluble antigen is perturbed in the absence of Mpeg1. To rule out a common deficit in internalization or proteolysis of ovalbumin by the antigen‐presenting cell, DCs and BMDMs were examined for their ability to take up CY5‐labeled ovalbumin, and to release fluorescence from DQ‐labeled ovalbumin. As shown in Supplementary figures 12 and 13, there was no difference in the uptake or processing of ovalbumin between WT and Mpeg −/− DCs or BMDMs.
DISCUSSION
We have shown here that Mpeg1 is not essential for either antibacterial or antiviral immunity, as Mpeg1 −/− mice successfully control S. aureus, L. longbeachae, M. tuberculosis and lymphocytic choriomeningitis virus infection. However, a pivotal role for Mpeg1 in the response to a specific, but as yet untested, bacterial pathogen, virus, or route of entry cannot be excluded. That Mpeg1 might be selective is suggested by the example of complement factor 9 (C9), the pore‐forming MACPF terminal protein of the classical complement pathway. Like Mpeg1, C9 forms lytic pores on membranes, and is thought to be crucial to controlling Gram‐negative bacteria. However, C9 genetic deficiency results in severe recurrent infections by Neisseria gonorrhoeae or Neisseria meningitidis, but not by other Gram‐negative species. Alternatively, given macrophages and neutrophils are also important in the control of parasite and yeast infections, Mpeg1 may play a role in the control of eukaryotic pathogens.
Notably, our conclusions contradict those of McCormack and colleagues who report that Mpeg1 is essential for antibacterial immunity. 6 , 9 , 10 How can these disparate findings be explained? The simplest view is that there are differences at the genetic level between mice carrying the published Mpeg1 tm1Pod allele, 9 and those carrying the Mpeg1 tm1.1Pib allele described here. One explanation is that the 129 ES cells used by McCormack et al. were mutated in an unidentified essential gene giving rise to the reported phenotype. This problem has been encountered in other gene‐targeting projects. 20 , 21
A second possibility is that the Mpeg1 tm1Pod allele perturbs expression of a neighboring gene essential for antibacterial immunity. Suppression of neighboring genes has been documented in other knockout models. 22 , 23 , 24 , 25 , 26 Perhaps the best example is the granzyme B (Gzmb) knockout line. 22 Here presence of a neomycin selection cassette in Gzmb suppresses expression of several other genes in the locus. An off‐target effect of neomycin cassette insertion also occurs in the Lta −/− (lymphotoxin A‐null) line, which exhibits perturbed expression of tumor necrosis factor‐α. 27 An obvious candidate for perturbation in the Mpeg1 tm1Pod/tm1Pod mice is Dtx4. This gene is immediately adjacent and antiparallel to the 3′ end of Mpeg1 in the mouse genome. It encodes a ubiquitin ligase expressed in macrophages that is implicated in cell differentiation and inflammation. 13 , 28 No information is available on Dtx4 expression in mice carrying the Mpeg1 tm1Pod allele. By contrast, Dtx4 expression is unaffected in mice carrying the Mpeg1 tm1.1Pib allele.
A third possibility is that the Mpeg1 tm1Pod allele is dominant negative, producing a truncated form of Mpeg1 that impairs and/or kills phagocytes. Truncated Mpeg1 could compromise phagocytes by misfolding and/or aggregating in the endoplasmic reticulum, inducing the unfolded protein response, which can initiate apoptosis. 29 Alternatively, the loss of C‐terminal domains downstream of the MACPF domain could result in an unregulated molecule that perforates membranes, eventually killing its host cell. A similar effect is seen in mutants of the unrelated pore‐forming protein, Gasdermin E. In humans, a dominant‐negative mutation produces truncated Gasdermin E that kills cells, leading to hearing loss. 30 If truncated MPEG1 is produced by the Mpeg1 tm1Pod allele, deletion of phagocytes would compromise the ability of the immune system to respond to bacterial infection, consistent with the phenotypes reported. 9 , 10
It is difficult to distinguish between these explanations because of insufficient information on the construction and validation of the Mpeg1 tm1Pod allele. 9 , 31 Specifically, (1) the insertion point and orientation of the selection cassette in Mpeg1 tm1Pod were not described, and genomic data confirming successful recombination and ruling out ectopic insertions were not provided; (2) whether the selection cassette is present or absent in the Mpeg1 tm1Pod allele was not reported, although its presence is implied by the allele nomenclature (i.e. it is designated tm1 not tm1.1); (3) the existence of truncated Mpeg1 protein in these knockout mice cannot be ruled out as only a cropped section of an immunoblot showing an undefined species without mass standards was presented (see Supplementary figure 1 in McCormack et al. 9 ). On the balance of probabilities, it is most likely that the Mpeg1 tm1Pod allele retains the neomycin selection cassette and, as has been reported in multiple studies, insertion or activity of the cassette generates a confounding phenotype. 26 , 32 , 33 , 34
It is also difficult to reconcile our results with in vitro experiments from the McCormack group showing that RNA interference‐based suppression of Mpeg1 in either IFNγ‐activated fibroblasts 6 , 9 or hemopoietic cells 9 apparently results in poorer killing of bacteria. The simplest explanation is that the small interfering RNAs used were toxic or had off‐target effects. However, if we accept the small interfering RNA results that Mpeg1 is an important player, then a more complicated explanation is that (1) genetic deletion of Mpeg1 tm1Pib in animals and cells derived from them results in upregulation of a protective compensating gene; and (2) this compensating gene is absent in Mpeg1 tm1Pod animals and cells. Even if correct, this explanation does not support the claim that Mpeg1 is required for immunity.
Our studies demonstrate for the first time that nascent Mpeg1 is processed from a single‐chain 80‐kDa precursor to a two‐chain, disulfide‐linked molecule after it has moved past the Golgi apparatus. Cleavage occurs between the MACPF and MABP domains. The consequences of this precise processing are unclear, but two possibilities can be suggested. First, processing is required for acquisition of biological function. Structural and biophysical studies of Mpeg1 have used single‐chain recombinant molecules, so the structure, function and pH requirements of a two‐chain molecule have yet to be investigated. Second, it can be envisaged that processing is required to terminate biological function, and that the cell continuously synthesizes full‐length Mpeg1 but inactivates it if not required. This maintains a functionally active pool that can be rapidly deployed but does not accumulate to a level that threatens the cell's integrity.
Mpeg1 is located in an endolysosomal compartment enriched in the LAMP family member CD68, and lacking the late endosomal/lysosome marker LAMP1 or the early endosomal marker EEA1. Limited insights can be gleaned from the colocalization of CD68 and Mpeg1 because of the paucity of information about the physiological role(s) of CD68. It has been implicated as a receptor for apoptotic cells with an undefined role in antigen presentation, and as a receptor for malaria sporozoites (but not bacterial or viral pathogens). 35
Absence of Mpeg1 significantly reduces both antigen presentation to CD4+ T cells and cross‐presentation to CD8+ T cells. The effect is specific to protein antigen with no impact on presentation of cell‐associated antigen, indicating that antigen cross‐presentation can occur in mice lacking Mpeg1. Conceivably, Mpeg1 may enhance the efficiency of cross‐priming when antigen is limiting, but it is clear that its role is not critical. Likewise, direct antigen presentation to CD8+ cytotoxic T lymphocyte via MHC‐I is unaffected as Mpeg1 knockout mice retain immunity to lymphocytic choriomeningitis virus, which directly infects DCs. Similar observations have been made previously in a study showing that lack of P38α affects both cross‐presentation in CD8+α DCs to CD8+ T cells and direct antigen presentation in CD8−α DCs to CD4+ T cells, and that proliferation of T cells decreases in response to soluble ovalbumin. 36 Like P38α, the lack of Mpeg1 might have different consequences depending on which cell type is primarily involved in the immune responses toward an antigen.
This phenotype may reflect the different routes and processing of soluble or cell‐associated antigens. Soluble ovalbumin is internalized by macropinocytosis or by receptor‐mediated endocytosis via CD206 (mannose receptor). Ovalbumin taken up by CD206 is processed for cross‐presentation. 37 , 38 Both DCs and macrophages have high levels of CD206, which binds to (among others) mannan of bacterial polysaccharide, ovalbumin and horse radish peroxidase. Mice lacking CD206 exhibit a decreased OT‐I proliferative response to soluble ovalbumin. 39 Other receptors implicated in processing of extracellular protein antigen include DEC205 and scavenger receptor. Scavenger receptor is expressed on DCs, macrophages and B cells and binds to negatively charged ligands such as ovalbumin, driving OT‐II proliferation or cross‐presentation. 40 Thus, it can be envisaged that Mpeg1 facilitates antigen endocytosis mediated by scavenger receptor or CD206.
Alternatively, Mpeg1 may affect endosomal components common to the vacuolar cross‐presentation pathway and the conventional MHC‐II pathway. Broadly speaking, cell‐associated antigens move through the phagocytic pathway, whereas soluble antigens are internalized via macropinocytosis or receptor‐mediated endocytosis. Cell‐associated antigens are directed to the late endosome/phagolysosome, whereas soluble antigens first move through the early endosome for processing. 39 , 41 , 42 , 43 Altered endolysosomal protease function affects antigen processing. 44 Perhaps Mpeg1 influences proteolysis by acting as a cofactor for one or more proteases; or by affecting endosomal formation or structure. If, for example, the absence of Mpeg1 altered the pH of the endosome, protease function would alter.
In summary, we conclude that Mpeg1 is not essential for antibacterial, antiviral or humoral responses. However, mice lacking Mpeg1 have a defect in the cellular immune response to soluble antigen. The physiological importance of Mpeg1 is yet to be established.
METHODS
Mouse strains
C57BL/6J, 129X1/SvJ and intercrossed mice with or without the Mpeg1 knockout allele were bred under specific pathogen‐free conditions at the Monash University Animal Breeding Facility (Clayton, Victoria, Australia). H2‐Kbm1 (bm1), MHC‐II knockout, OT‐I and OT‐II mice were maintained under specific pathogen‐free conditions at the Bio21 Molecular Science and Biotechnology Institute. All mice used were 6–14 weeks of age and handled according to the guidelines of the National Health and Medical Research Council of Australia. Experimental procedures were approved by Monash University and The Alfred Medical Research and Education Precinct Animal Ethics Committees (approvals MARP/2014/034/BC, MARP/2015/045, MARP 2017/166/BC, AMREP/E/1689/2016/M).
Generation and validation of Mpeg1−/− mice
The targeting vector and probes were designed and constructed by the Gene Recombineering Facility (Monash University). The vector was built by recombination‐mediated genetic engineering using the bacterial artificial chromosome clone RP23‐60G8 as a source of Mpeg1 DNA. This vector comprised about 4.6‐kb sequences upstream (5′ homology arm) and nearly 6.9 kb downstream (3′ homology arm) of Mpeg1, surrounding a neomycin transcriptional unit flanked by loxP elements (Figure 1a). In brief, the construction steps involved subcloning a 14.3‐kb fragment from the bacterial artificial chromosome clone into the plasmid pBluescript. The entire coding region of Mpeg1 in the resulting plasmid was replaced by a floxed PGK‐neomycin cassette amplified from a derivative of ploxPneo‐1.
Bruce 4 C57BL/6J‐derived ES cells were transfected with the linearized targeting construct, selected in G418 and 500 resulting clones analyzed by Southern blotting with the 5′ external probe on AflII‐cleaved genomic DNA (wt 19.4 kb; targeted 11.1 kb), and the 3′ external probe on EcoRV‐cleaved DNA (wt 12.5 kb; targeted 10.1 kb). Cells from 5 of 20 confirmed targeted ES clones containing the Mpeg1 tm1Pib allele were injected into BALB/c blastocysts, and 2 (clones 1B and 7F) yielded transmitting chimeric mice, which were crossed with C57BL/6J Cre‐deleter transgenic mice [Tg(CMV‐cre)1Cg] to remove the neomycin cassette from the targeted allele and produce “floxed” animals carrying the deleted Mpeg1 tm1.1Pib allele (MGI:6368993). To verify the lines, genomic DNA from spleen was isolated from F1 animals of different genotypes and analyzed by Southern blotting (see Figure 1b for details).
To routinely genotype mice, DNA from tail biopsies was analyzed by PCR. PCR used the primers 5′GGTTGCTACTAACTCCTGCGT3′ and 5′AGACTGGGGCAAACAGAAGG3′ to detect the WT allele (456‐bp product), and 5′GGTTGCTACTAACTCCTGCGT3′ and 5′CCACACCTCACTTCAAATGGCTCC3′ in a separate reaction to detect the floxed allele (428‐bp product). As shown in Figure 1c, the latter primer pair yields a 2800 bp on WT genomic DNA. Reactions comprised 200 μm deoxyribonucleotide triphosphates, 25 pmol each primer, 2.5 mm MgCl2 and Taq polymerase, at 95°C for 30 s, 58°C for 30 s, 72°C for 60 s, over 35 cycles.
RT‐PCR
Total RNA was purified from 1 × 107 cultured macrophages using TRI Reagent (Sigma Aldrich, Melbourne, VIC, Australia) according to manufacturer's instructions. Complementary DNA was reverse transcribed from 5 μg of RNA using the SuperScript III First‐Strand Synthesis System (Thermo Fisher, Melbourne, VIC, Australia) with oligo(dT)20. PCR was performed on 1 μL of complementary DNA with GoTaq Green Master Mix (Promega, Sydney, Australia) with 10 pmol of each primer. Cycle conditions were as follows: 94°C for 180 s followed by the indicated number of cycles of 30 s at 94°C, 30 s at 55°C and 60 s at 72°C. Dtx4 20 cycles; Gapdh 17 cycles; Pflp1, Mpeg1 25 cycles.
Primers: Gapdh 5′TGTGTCCGTCGTGGATCTGA3′; 5′TTGCTGTTGAAGTCGCAGGAG3′ yielding a 150‐bp product. Dtx4 5′TGGCTTCCCACGACATTGTTA3′; 5′ACTGCACCAAGGCATCCAAT3′ yielding a 788‐bp product. Pfpl 5′GGCAAAAGAATCTGGCACTGG3′; 5′TTCATGGTGCAGGTAGTCCC3′ yielding a 158‐bp product. Mpeg1 5′CAAAAGCCAGACAGAGCCTTC3′; 5′ACAAGACTGGGGCAAACAGAA3′ yielding a 153‐bp product.
Organ, cell and tissue isolation and analysis
Histopathological assessments and clinical hematological analysis of one male and one female 6‐week‐old Mpeg1 knockout mouse and matched WT controls were performed via the Australian Phenomics Network (http://www.australianphenomics.org.au/). The following organs were examined for macromorphological abnormalities: testes, epididymis, seminal vesicles, prostate glands, penis, preputial gland, mammary tissue, ovaries, oviducts, uterus, cervix, vagina, clitoral gland, bladder, liver, gall bladder, stomach, duodenum, jejunum, ileum, cecum, colon, mesenteric lymph node, spleen, pancreas, kidney, adrenal glands, salivary glands and regional lymph nodes, thyroids, trachea, lungs, thymus, heart, skin, tail, eyes, Harderian glands, brain, spinal cord and hind leg. For hematological analysis blood samples collected into ethylenediaminetetraacetic acid were run on the ADVIA 2120 Hematology system, which gives a red blood cell count (with indices), platelet count and a white blood cell differential by size, granularity and peroxidase absorption.
Mpeg antibodies
Rabbit polyclonal anti‐rat Mpeg1 was obtained from Abcam (ab25146; Cambridge, UK). Specificity was assessed as shown in Supplementary figure 3. Guinea pig antisera to recombinant mouse Mpeg1 was generated by Antibodies Australia (Melbourne, Australia). Specificity was assessed as shown in Supplementary figure 6.
Cells
MutuDC 11 and COS‐1 cells were maintained and transfected as described. The mouse Mpeg1 complementary DNA was cloned into the expression vector pSVTf described in Madison and Bird. 45
BMDMs were generated by culturing marrow cells for 7–12 days in Roswell Park Memorial Institute‐1640 (RPMI‐1640) and 15% (v/v) heat‐inactivated fetal calf serum, 2 mm glutamine and 20% (v/v) L929‐conditioned medium as a source of granulocyte macrophage colony‐stimulating factor. Cells were stimulated by addition of IFNγ (100 ng mL−1) and/or LPS (10 ng mL−1). Macrophage phenotype was confirmed using the markers CD11b (M1/70; BD Biosciences, Sydney, Australia) and F4/80 (BM8.1; Tonbo Biosciences, San Diego, CA, USA). Data from a minimum of 10 000 cells were collected on an LSRII instrument and analyzed using Cellquestpro software (BD Biosciences, Sydney, Australia).
Splenic macrophage marked with antibodies to CD11b and F4/80 were collected using a FACSAria Fusion Cell Sorter (BD Biosciences). DC‐enriched light‐density fractions were obtained by centrifugation of splenocytes in Nycodenz (Axis‐Shield, Oslo, Norway) of density 1.077 g cm3, stained for CD11c (mAb N418; eBioscience, Thermo Fisher Australia) and CD8 (BD Biosciences; mAb 53–6.7) and positively sorted on Influx or DIVA cell sorters (BD Biosciences). Alternatively, Nycodenz‐fractionated cells were further purified by negative selection using an antibody cocktail consisting of rat antibodies to CD3 (KT3‐1.1), Thy1‐1 (T24/31.7), CD45R (RA36B2), Ly6G (IA8) and erythrocytes (TER119) and BioMag anti‐rat IgG‐coated magnetic beads (QIAGEN, Sydney, Australia). The conventional dendritic cell‐enriched supernatant was assessed using antibodies to CD11c and MHC‐II (M5/114.15.2; eBioscience, Thermo Fisher Australia).
Neutrophils were purified from bone marrow by staining with anti‐LY6G (RB6‐8C5; Abcam) and separating positive and negative populations using a FACSAria Fusion cell sorter.
Pulse‐chase analysis
MutuDC or BMDM monolayers containing about 107 cells per 10‐cm dish were washed in phosphate‐buffered saline (PBS) and “starved” in RPMI lacking methionine (RPMI‐met−) for 30 min at 37°C. This was replaced by RPMI‐met− containing 50–100 μCi 35S‐methionine for 30–60 min. The labeling medium was then removed, monolayers washed once in PBS and 1 mL of complete medium was added for the indicated “chase” periods, terminated by placing dishes on ice. Medium was collected and clarified by centrifugation. MutuDC monolayers were washed once in PBS and lysed in 0.5% (w/v) SDS. Lysates were diluted into 50 mm Tris–HCI, pH 7.5; 150 mm NaCl; 1 mm ethylenediaminetetraacetic acid; 0.1% Nonidet P‐40; 0.25% gelatin and 0.02% sodium azide (NETGEL) to reduce the SDS concentration to 0.1% (w/v) and a needle and syringe were used to shear DNA and reduce viscosity. BMDM monolayers were washed once in PBS and lysed in 50 mm Tris–HCI pH 8.0, 10 mm NaCl and 1% Nonidet P‐40 (containing protease inhibitors aprotinin, pepstatin, leupeptin and 4‐(2‐aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF)).
Protein A– or protein G–Sepharose beads were added to each sample and incubated overnight at 4°C after which they were replaced with fresh Sepharose beads and 1–2 μL of the indicated antibody to Mpeg1. After overnight incubation at 4°C, the Sepharose beads were washed twice with NETGEL and once with 10 mm Tris–HCl, pH 8.0 and resuspended in LSB/0.1 m dithiothreitol. Following separation by SDS–polyacrylamide gel electrophoresis, the gel was fixed in 35% (v/v) methanol and 10% (v/v) acetic acid, treated with Amersham Amplify (GE Healthcare, Sydney, Australia) for 30 min at room temperature, dried and exposed to X‐ray film at –80°C.
Proteomics
About 107 MutuDCs washed in PBS were lysed in situ using 400 μL of 0.5% SDS. Then, 600 μL NETGEL was added and cellular DNA sheared by passaging it through a syringe. A further 1 mL of NETGEL plus 200 μL of protein G–Sepharose beads (10% w/v) were added, and the samples rocked gently at 4°C overnight. Beads were pelleted and the supernatant transferred to a new tube. Roughly 200 μL of fresh protein G–Sepharose beads plus 4 μL of rabbit anti‐Mpeg1 antibody (ab25146; Abcam, Cambridge, UK) were added. Following overnight incubation at 4°C, the beads were pelleted, washed twice for 15 min in NETGEL and then once in 10 mm Tris–HCl (pH 8.0). Beads were resuspended in 25 μL LSB/0.1 m dithiothreitol to release proteins for separation via 10% SDS–polyacrylamide gel electrophoresis. The gel section between the 58‐ and 30‐kDa markers was excised, and subjected to in situ tryptic digest. Then, liquid chromatography–tandem mass spectrometry analysis was performed at the Monash Proteomics platform.
In vitro bacterial killing
On Days 8–10, BMDMs were infected with the indicated bacteria. After 1 h, cells were washed three times with PBS and placed in a medium containing 50 μg mL−1 gentamicin for 1 h to kill noninternalized bacteria. Cells were washed and placed in medium with 10 μg mL−1 gentamicin for the indicated period. Cells were lysed using 1% (v/v) IGEPAL in water, and the released bacterial colony forming units determined.
Infection studies
Mice were infected intranasally with S. aureus or L. longbeachae as described in Chow et al. 46 and Speir et al. 47 Aerosol infection of mice with M. tuberculosis strain H37Rv via whole‐body inhalation exposure followed procedures described in Stutz et al. 48 Infection of mice with lymphocytic choriomeningitis virus Docile strain and subsequent tissue analysis followed procedures outlined in Pellegrini et al. 49 and Battegay et al. 50
Antigen presentation and proliferation assays
Cytotoxic T‐cell assays were performed as described in Wilson et al., 51 except 2.5 × 105 OCSs in 5 μg mL−1 LPS were injected into WT or Mpeg1 −/− mice. Target cells were labeled with 5 μm Cell Trace Violet (CTV; Thermo Fisher Scientific, Melbourne, Australia) and loaded with 300 ng OVA257–264 peptide (OVA+ CTVhigh); or labeled with 0.5 μM CTV alone (OVA− CTVlow). In vivo antigen presentation assays were performed as described in Wilson et al. 51
Inflammasome activators and activation assays
Bacteria, knockout mice lacking inflammasome components and detailed procedures for BMDM infection are described in Mathur et al. 52 Mefv −/− mice were sourced from the Australian National University. A clinical isolate of C. difficile positive for TcdA and TcdB toxin (ACT Pathology) was grown anaerobically in BHI media for 48 h at 37°C. The toxin‐containing supernatant was harvested by centrifugation. The murine cytomegalovirus Smith MSGV strain (ATCC VR‐1399) was propagated and expanded in M2‐10B4 murine bone marrow stromal cells (ATCC CRL‐1972) and concentrated via ultracentrifugation. To activate the pyrin inflammasome, BMDMs were primed with LPS from E. coli for 3 h and stimulated with 100 μL of C. difficile culture supernatant for 20 h.
Statistics
Statistical analysis used the GraphPad Prism software (GraphPad software, La Jolla, CA, USA). Unless otherwise noted, statistical significance was determined using the parametric Student's t‐test.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Phillip Ian Bird: Conceptualization; funding acquisition; project administration; supervision; writing – original draft; writing – review and editing. Salimeh Ebrahimnezhaddarzi: Investigation; writing – original draft. Catherina Bird: Investigation; supervision; writing – review and editing. Cody Allison: Investigation; writing – review and editing. Daniel Enosi Tuipulotu: Investigation; writing – review and editing. Xenia Kostoulias: Investigation; writing – review and editing. Christophe Macri: Investigation; supervision. Michael Stutz: Investigation; writing – review and editing. Gilu Abraham: Investigation. Dion Kaiserman: Investigation; supervision. Siew Siew Pang: Resources. Si Ming Man: Resources; supervision; writing – review and editing. Justine Mintern: Investigation; resources; supervision; writing – review and editing. Thomas Naderer: Resources; supervision; writing – review and editing. Anton Peleg: Resources; writing – review and editing. Marc Pellegrini: Resources; supervision; writing – review and editing. James Whisstock: Conceptualization; funding acquisition; resources; writing – review and editing.
Supporting information
ACKNOWLEDGMENTS
The Mpeg1 tm1.1Pib line may be obtained from the Australian Phenome Bank. We thank Jeanette Rientjes, Arianna Nenci and Jose Gonzalez (Monash Gene Targeting Facility) for contract production of the Mpeg1 tm1Pib mice; Qun Li and Iain Clarke (Antibodies Australia) for antibody production and Oded Kleifeld (Monash Proteomics Platform) for protein sequence analysis. This study used the Australian Phenomics Network Histopathology and Organ Pathology Service at the University of Melbourne (Australia). We thank Aimee Parker, Suzanne Emerton and Georgia Darley for contributions to the early phases of the study, and the Monash FlowCore staff for their assistance. The work was initially supported by the National Health and Medical Research Council Program Grant 490900 (Australia). Open access publishing facilitated by Monash University, as part of the Wiley – Monash University agreement via the Council of Australian University Librarians.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Spilsbury K, O'Mara M, Wu W, Rowe P, Symonds G, Takayama Y. Isolation of a novel macrophage‐specific gene by differential cDNA analysis. Blood 1995; 85: 1620–1629. [PubMed] [Google Scholar]
- 2. D'Angelo ME, Dunstone MA, Whisstock JC, Trapani JA, Bird PI. Perforin evolved from a gene duplication of MPEG1, followed by a complex pattern of gene gain and loss within Euteleostomi. BMC Evol Biol 2012; 12: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kondos SC, Hatfaludi T, Voskoboinik I, et al. The structure and function of mammalian membrane‐attack complex/perforin‐like proteins. Tissue Antigens 2010; 76: 341–351. [DOI] [PubMed] [Google Scholar]
- 4. Ni T, Jiao F, Yu X, et al. Structure and mechanism of bactericidal mammalian perforin‐2, an ancient agent of innate immunity. Sci Adv 2020; 6: eaax8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pang SS, Bayly‐Jones C, Radjainia M, et al. The cryo‐EM structure of the acid activatable pore‐forming immune effector macrophage‐expressed gene 1. Nat Commun 2019; 10: 4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCormack R, de Armas LR, Shiratsuchi M, Ramos JE, Podack ER. Inhibition of intracellular bacterial replication in fibroblasts is dependent on the perforin‐like protein (perforin‐2) encoded by macrophage‐expressed gene 1. J Innate Immun 2013; 5: 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCormack R, de Armas L, Shiratsuchi M, Podack ER. Killing machines: three pore‐forming proteins of the immune system. Immunol Res 2013; 57: 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Benard EL, Racz PI, Rougeot J, et al. Macrophage‐expressed perforins mpeg1 and mpeg1.2 have an anti‐bacterial function in zebrafish. J Innate Immun 2015; 7: 136–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McCormack RM, de Armas LR, Shiratsuchi M, et al. Perforin‐2 is essential for intracellular defense of parenchymal cells and phagocytes against pathogenic bacteria. Elife 2015; 4: e06508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bai F, McCormack RM, Hower S, Plano GV, Lichtenheld MG, Munson GP. Perforin‐2 breaches the envelope of phagocytosed bacteria allowing antimicrobial effectors access to intracellular targets. J Immunol 2018; 201: 2710–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fuertes Marraco SA, Grosjean F, Duval A, et al. Novel murine dendritic cell lines: a powerful auxiliary tool for dendritic cell research. Front Immunol 2012; 3: 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Luber CA, Cox J, Lauterbach H, et al. Quantitative proteomics reveals subset‐specific viral recognition in dendritic cells. Immunity 2010; 32: 279–289. [DOI] [PubMed] [Google Scholar]
- 13. Cui J, Li Y, Zhu L, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol 2012; 13: 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiong P, Shiratsuchi M, Matsushima T, et al. Regulation of expression and trafficking of perforin‐2 by LPS and TNF‐α. Cell Immunol 2017; 320: 1–10. [DOI] [PubMed] [Google Scholar]
- 15. Kayagaki N, Warming S, Lamkanfi M, et al. Non‐canonical inflammasome activation targets caspase‐11. Nature 2011; 479: 117–121. [DOI] [PubMed] [Google Scholar]
- 16. McCormack RM, Szymanski EP, Hsu AP, et al. MPEG1/perforin‐2 mutations in human pulmonary nontuberculous mycobacterial infections. JCI Insight 2017; 2: e89635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bathige SD, Umasuthan N, Whang I, Lim BS, Won SH, Lee J. Antibacterial activity and immune responses of a molluscan macrophage expressed gene‐1 from disk abalone Haliotis discus discus. Fish Shellfish Immunol 2014; 39: 263–272. [DOI] [PubMed] [Google Scholar]
- 18. Kleiman E, Salyakina D, De Heusch M, et al. Distinct transcriptomic features are associated with transitional and mature B‐cell populations in the mouse spleen. Front Immunol 2015; 6: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richter K, Perriard G, Oxenius A. Reversal of chronic to resolved infection by IL‐10 blockade is LCMV strain dependent. Eur J Immunol 2013; 43: 649–654. [DOI] [PubMed] [Google Scholar]
- 20. Westrick RJ, Mohlke KL, Korepta LM, et al. Spontaneous Irs1 passenger mutation linked to a gene‐targeted SerpinB2 allele. Proc Natl Acad Sci USA 2010; 107: 16904–16909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rawle DJ, Le TT, Dumenil T, et al. Widespread discrepancy in Nnt genotypes and genetic backgrounds complicates granzyme a and other knockout mouse studies. Elife 2022; 11: e70207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pham CT, MacIvor DM, Hug BA, Heusel JW, Ley TJ. Long‐range disruption of gene expression by a selectable marker cassette. Proc Natl Acad Sci USA 1996; 93: 13090–13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Olson EN, Arnold HH, Rigby PW, Wold BJ. Know your neighbors: three phenotypes in null mutants of the myogenic bHLH gene MRF4. Cell 1996; 85: 1–4. [DOI] [PubMed] [Google Scholar]
- 24. Maguire S, Estabel J, Ingham N, et al. Targeting of Slc25a21 is associated with orofacial defects and otitis media due to disrupted expression of a neighbouring gene. PloS One 2014; 9: e91807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pan Y, Zhang L, Liu Q, et al. Insertion of a knockout‐first cassette in Ampd1 gene leads to neonatal death by disruption of neighboring genes expression. Sci Rep 2016; 6: 35970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. West DB, Engelhard EK, Adkisson M, et al. Transcriptome analysis of targeted mouse mutations reveals the topography of local changes in gene expression. PLoS Genet 2016; 12: e1005691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liepinsh DJ, Grivennikov SI, Klarmann KD, et al. Novel lymphotoxin alpha (LTα) knockout mice with unperturbed tumor necrosis factor expression: reassessing LTα biological functions. Mol Cellular Biol 2006; 26: 4214–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. An T, Li S, Pan W, et al. DYRK2 negatively regulates type I interferon induction by promoting TBK1 degradation via Ser527 phosphorylation. PLoS Pathog 2015; 11: e1005179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lam M, Marsters SA, Ashkenazi A, Walter P. Misfolded proteins bind and activate death receptor 5 to trigger apoptosis during unresolved endoplasmic reticulum stress. Elife 2020; 9: e5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Op de Beeck K, Van Camp G, Thys S, et al. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis‐inducing protein. Eur J Hum Genet 2011; 19: 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCormack RM, Lyapichev K, Olsson ML, Podack ER, Munson GP. Enteric pathogens deploy cell cycle inhibiting factors to block the bactericidal activity of Perforin‐2. Elife 2015; 4: e06505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scarff KL, Ung KS, Sun J, Bird PI. A retained selection cassette increases reporter gene expression without affecting tissue distribution in SPI3 knockout/GFP knock‐in mice. Genesis 2003; 36: 149–157. [DOI] [PubMed] [Google Scholar]
- 33. Southwell AL, Smith‐Dijak A, Kay C, et al. An enhanced Q175 knock‐in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum Mol Genet 2016; 25: 3654–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Scacheri PC, Crabtree JS, Novotny EA, et al. Bidirectional transcriptional activity of PGK‐neomycin and unexpected embryonic lethality in heterozygote chimeric knockout mice. Genesis 2001; 30: 259–263. [DOI] [PubMed] [Google Scholar]
- 35. Chistiakov DA, Killingsworth MC, Myasoedova VA, Orekhov AN, Bobryshev YV. CD68/macrosialin: not just a histochemical marker. Lab Invest 2017; 97: 4–13. [DOI] [PubMed] [Google Scholar]
- 36. Zhou Y, Wu J, Liu C, et al. p38α has an important role in antigen cross‐presentation by dendritic cells. Cell Mol Immunol 2018; 15: 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zehner M, Chasan AI, Schuette V, et al. Mannose receptor polyubiquitination regulates endosomal recruitment of p97 and cytosolic antigen translocation for cross‐presentation. Proc Natl Acad Sci USA 2011; 108: 9933–9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kamphorst AO, Guermonprez P, Dudziak D, Nussenzweig MC. Route of antigen uptake differentially impacts presentation by dendritic cells and activated monocytes. J Immunol 2010; 185: 3426–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burgdorf S, Lukacs‐Kornek V, Kurts C. The mannose receptor mediates uptake of soluble but not of cell‐associated antigen for cross‐presentation. J Immunol 2006; 176: 6770–6776. [DOI] [PubMed] [Google Scholar]
- 40. Shakushiro K, Yamasaki Y, Nishikawa M, Takakura Y. Efficient scavenger receptor‐mediated uptake and cross‐presentation of negatively charged soluble antigens by dendritic cells. Immunology 2004; 112: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burgdorf S, Kautz A, Böhnert V, Knolle PA, Kurts C. Distinct pathways of antigen uptake and intracellular routing in CD4 and CD8 T cell activation. Science 2007; 316: 612–616. [DOI] [PubMed] [Google Scholar]
- 42. Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: downregulation by cytokines and bacterial products. J Exp Med 1995; 182: 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mintern JD, Macri C, Villadangos JA. Modulation of antigen presentation by intracellular trafficking. Curr Op Immunol 2015; 34: 16–21. [DOI] [PubMed] [Google Scholar]
- 44. Kim SJ, Schätzle S, Ahmed SS, et al. Increased cathepsin S in Prdm1 −/− dendritic cells alters the TFH cell repertoire and contributes to lupus. Nat Immunol 2017; 18: 1016–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Madison EL, Bird P. A vector, pSHT, for the expression and secretion of protein domains in mammalian cells. Gene 1992; 121: 179–180. [DOI] [PubMed] [Google Scholar]
- 46. Chow SH, Deo P, Yeung ATY, et al. Targeting NLRP3 and staphylococcal pore‐forming toxin receptors in human‐induced pluripotent stem cell‐derived macrophages. J Leuk Biol 2020; 108: 967–981. [DOI] [PubMed] [Google Scholar]
- 47. Speir M, Lawlor KE, Glaser SP, et al. Eliminating legionella by inhibiting BCL‐XL to induce macrophage apoptosis. Nat Microbiol 2016; 1: 15034. [DOI] [PubMed] [Google Scholar]
- 48. Stutz MD, Ojaimi S, Allison C, et al. Necroptotic signaling is primed in mycobacterium tuberculosis‐infected macrophages, but its pathophysiological consequence in disease is restricted. Cell Death Differ 2018; 25: 951–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pellegrini M, Calzascia T, Toe JG, et al. IL‐7 engages multiple mechanisms to overcome chronic viral infection and limit organ pathology. Cell 2011; 144: 601–613. [DOI] [PubMed] [Google Scholar]
- 50. Battegay M, Cooper S, Althage A, Bänziger J, Hengartner H, Zinkernagel RM. Quantification of lymphocytic choriomeningitis virus with an immunological focus assay in 24‐ or 96‐well plates. J Virol Meth 1991; 33: 191–198. [DOI] [PubMed] [Google Scholar]
- 51. Wilson KR, Jenika D, Blum AB, et al. MHC class II ubiquitination regulates dendritic cell function and immunity. J Immunol 2021; 207: 2255–2264. [DOI] [PubMed] [Google Scholar]
- 52. Mathur A, Feng S, Hayward JA, et al. A multicomponent toxin from Bacillus cereus incites inflammation and shapes host outcome via the NLRP3 inflammasome. Nat Microbiol 2019; 4: 362–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.