

Abstract
Tissue‐resident memory T cells (Trm) are retained in peripheral tissues after infection for enhanced protection against secondary encounter with the same pathogen. We have previously shown that the transcription factor Hobit and its homolog Blimp‐1 drive Trm development after viral infection, but how and when these transcription factors mediate Trm formation remains poorly understood. In particular, the major impact of Blimp‐1 in regulating several aspects of effector T‐cell differentiation impairs study of its specific role in Trm development. Here, we used the restricted expression of Hobit in the Trm lineage to develop mice with a conditional deletion of Blimp‐1 in Trm, allowing us to specifically investigate the role of both transcription factors in Trm differentiation. We found that Hobit and Blimp‐1 were required for the upregulation of CD69 and suppression of CCR7 and S1PR1 on virus‐specific Trm precursors after LCMV infection, underlining a role in their retention within tissues. The early impact of Hobit and Blimp‐1 favored Trm formation and prevented the development of circulating memory T cells. Thus, our findings highlight a role of Hobit and Blimp‐1 at the branching point of circulating and resident memory lineages by suppressing tissue egress of Trm precursors early during infection.
Keywords: Blimp‐1, CD8+ T‐cell differentiation, Hobit, LCMV, tissue‐resident memory CD8+ T cells
Tissue‐resident memory T cells (Trm) permanently lodge themselves in peripheral tissues to capture invading pathogens. We found that the transcription factors Hobit and Blimp‐1 already acted in Trm precursors to suppress tissue‐exit pathways. Our findings imply that establishment of the Trm lineage requires lodgment of their precursors early after infection.
Introduction
Immunity against intracellular pathogens relies on CD8+ T cells that can specifically eliminate infected cells through the production of cytotoxic molecules and proinflammatory cytokines. CD8+ T‐cell responses start with the priming of naïve CD8+ T cells by the recognition of pathogen‐derived antigens presented on APCs in the LNs. Antigen‐triggered CD8+ T cells proliferate and differentiate into effector T cells that migrate to the site of infection, where they eradicate the pathogen [1, 2]. The majority of effector CD8+ T cells undergoes a program of terminal differentiation and perish once the pathogen is cleared. These terminal effector cells are known as short‐lived effector cells (SLECs). A minor effector population, termed memory precursors effector cells (MPECs), survives and retains the capacity to develop into memory CD8+ T cells [3, 4]. Differences in the migration pattern and function classify memory CD8+ T cells into circulating and resident memory T cells. Circulating memory T cells consist of central memory (Tcm) and effector memory CD8+ T cells (Tem) that continuously patrol the body in search of invading pathogens [5]. In contrast, tissue‐resident memory CD8+ T cells (Trm) permanently reside in peripheral tissues and provide enhanced local protection against reinfection, given their strategic location at sites of pathogen entry and their ability to rapidly upregulate effector functions [6, 7].
Several transcription factors have been implicated in driving antigen‐triggered CD8 T cells toward either a terminal or a memory stage upon infection. The transcription factors Runx3, Blimp‐1, T‐bet, Notch, and Id2 have been described to mediate terminal effector CD8+ T‐cell differentiation and the acquisition of effector functions [3, 8–12]. In contrast, the transcription factors Eomes, Bcl‐6, and Id3 have been implicated in promoting memory T‐cell development [12, 13, 14, 15, 16]. Although several transcription factors have been shown to regulate the development of memory T cells, how and when antigen‐triggered CD8 T cells branch into resident and circulating memory lineages remains poorly understood.
We have recently described that the Trm lineage is already established during the effector stage of the immune response upon viral infection. We have characterized a subset of effector CD8+ T cells in tissues as Trm precursors, which are biased to form Trm rather than circulating memory T cells [17]. These committed Trm precursors were specifically identified by expression of the Trm‐restricted transcription factor Hobit [17]. Early commitment to the Trm lineage was also observed by others. During priming, signals from type 1 classical DCs in the draining LNs appear specifically required to favor the development of effector CD8+ T cells into Trm in the skin [18]. A recent study has shown that T‐cell clones with a higher propensity to form Trm are present in the circulation before tissue entrance after skin vaccination [19]. Furthermore, the transcription factor Id3 identifies effector CD8+ T cell with an elevated potential to form Trm in the small intestine (SI) [20, 21]. Altogether, these findings underline that the transcriptional program driving the separation of the Trm lineage from circulating memory T cells and terminal effector T cells takes place at early stages of the immune response.
Previously, we have demonstrated that Hobit and its homolog Blimp‐1 are essential for the formation of CD8+ Trm cells throughout tissues including skin, liver, kidneys, and the SI [22]. However, how and when Hobit and Blimp‐1 instruct the branching off of Trm from terminal effectors and circulating memory T cells upon infection remains unresolved. Expression of Hobit appears highly restricted to the Trm lineage and allows for the unequivocal identification of Trm precursors [17, 22]. In contrast, Blimp‐1 is broadly expressed in antigen‐experienced T cells and promotes terminal differentiation of effector CD8+ T cells besides instructing Trm formation [8, 9, 22]. Given that Blimp‐1 has a major impact in multiple processes involving effector CD8 T‐cell differentiation, it has not yet been possible to investigate the specific role of Blimp‐1 in the Trm lineage. Therefore, exploiting the specific expression pattern of Hobit, we developed mice with a conditional deletion of Blimp‐1 within the Trm lineage to specifically investigate the role of both Hobit and Blimp‐1 in Trm differentiation. In the absence of Hobit, Trm‐specific deletion of Blimp‐1 reduced Trm formation. Interestingly, Trm precursors formed in the absence of Hobit and Blimp‐1, but displayed impaired upregulation of CD69 expression and impaired downregulation of tissue exit receptors, indicating a dedicated role of both transcription factors in the early suppression of tissue egress. Thus, our results show that Hobit and Blimp‐1 cooperatively contribute to regulate expression of tissue exit pathways in Trm precursors during the effector phase of the immune response, which favors the subsequent formation of Trm cells.
Results
Blimp‐1 is widely expressed in antigen‐experienced CD8+ T cells in contrast to Trm‐restricted Hobit
To evaluate the expression of Hobit and Blimp‐1 in virus‐specific CD8+ T cells at different stages of the immune response, we infected HobittdTomato/WT × BlimpGFP/WT mice, which simultaneously report Hobit by tdTomato expression and Blimp‐1 by GFP expression, with LCMV Armstrong. The differentiation of virus‐specific CD8+ T cells into memory CD8+ T cells was monitored in spleen, liver, kidneys, and intraepithelial lymphocytes (IEL) of the SI at >30 days postinfection (p.i.) using Db tetramers loaded with the dominant LCMV peptide gp33‐41. Virus‐specific CD8+ memory T cells were classified based on the expression of CD62L and CD69 into TCM (CD62L+ CD69–), TEM (CD62L– CD69–), and TRM (CD62L– CD69+) (Supporting information Fig. S1A). In line with our previous studies [23], we found that Hobit (tdTomato) expression was confined to the Trm compartment (Fig. 1A and B). In contrast, Blimp‐1 (GFP) was widely expressed in Tcm, Tem, and Trm, although Blimp‐1 expression was lower in Tcm compared to the other memory fractions (Fig. 1C–E), as previously reported [9, 24]. Accordingly, transcriptional analysis showed that Blimp‐1 expression was increased in all memory CD8+ T‐cell subsets in comparison to the naïve compartment (Supporting information Fig. S1B). These data underline that Hobit expression is restricted to Trm while Blimp‐1 is broadly expressed in circulating and resident CD8+ T cells during the memory phase of infection.
Figure 1.
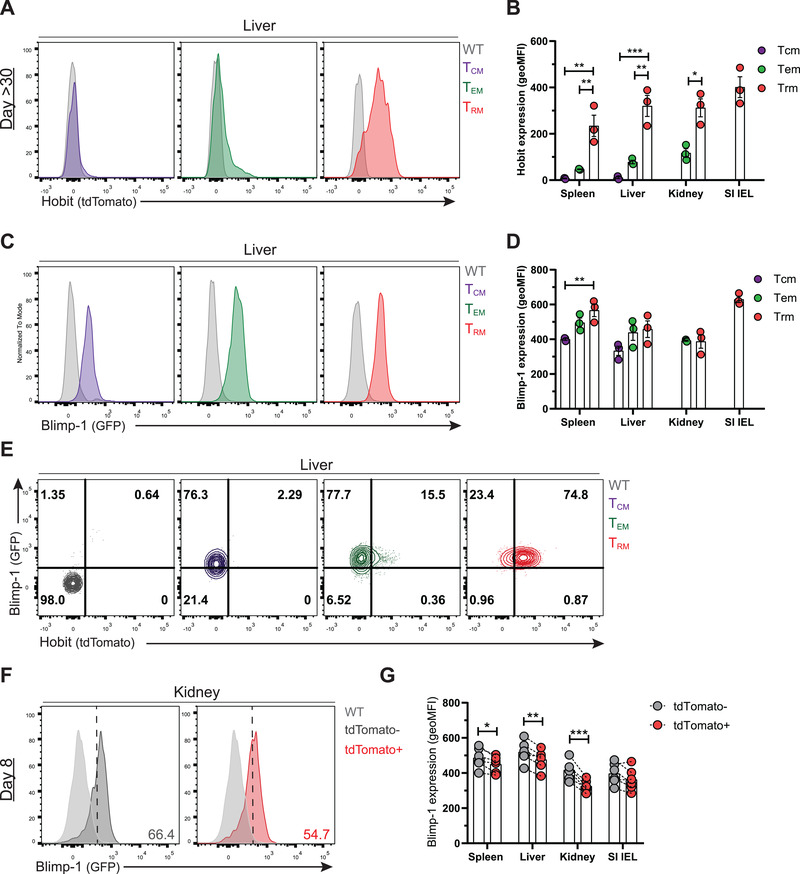
Hobit is restricted to the Trm lineage and Blimp‐1 is widely expressed in antigen‐experienced CD8+ T cells. Wild type control (WT) and HobittdTomato/WT × Blimp‐1GFP/WT mice were infected with LCMV Armstrong and virus‐specific (GP33+) CD8+ T cells were analyzed by flow cytometry at day 8 and day >30 post‐infection (p.i.). (A‐D) Representative histograms depict (A) tdTomato (Hobit) and (C) GFP (Blimp‐1) expression in GP33+ Tcm, Tem, and Trm from WT (grey) and HobittdTomato/WT × Blimp‐1GFP/WT mice at day >30 p.i. (B, D) The geometric MFI (geo MFI) of (B) tdTomato and (D) GFP expression was quantified in GP33+ CD8+ T cells from the indicated tissues. (E) Representative flow cytometry plots display expression of GFP and tdTomato in virus‐specific memory CD8+ T cells from the liver. (F) Representative histograms depict GFP expression in WT and tdTomato+ and tdTomato‐ GP33+ CD8+ T cells of HobittdTomato/WT × Blimp‐1GFP/WT mice at day 8 p.i. (G) The geo MFI of GFP expression was quantified in tdTomato+ and tdTomato‐ cells in the indicated tissues. (A‐G) Symbols represent individual mice. Error bars represent mean ± SEM. (A‐E) Representative data of one (n = 4) out of two independent experiments. Bars represent the mean. One‐way ANOVA. (F‐G) Combined data from two independent experiments (n = 6). Dotted lines connect paired samples. Paired t test. *p < 0.05; **p < 0.01; ***p < 0.001.
We have recently found that Hobit identifies effector CD8+ T cells that have committed to the Trm lineage [17]. Previous studies have shown that Blimp‐1 upregulation also takes place during the effector phase after viral infection [8, 9], but the expression of Blimp‐1 in Trm precursors has remained unknown. Analysis of LCMV‐specific CD8+ T cells at day 8 p.i. showed higher expression of Blimp‐1 in SLECs (CD127– KLRG1+) compared to MPECs (CD127+ KLRG1–) (Supporting information Fig. S1C–E), as previously reported [8, 9]. We found that Hobit+ virus‐specific effector CD8+ T cells consistently expressed slightly lower levels of Blimp‐1 compared to Hobit‐ cells in the spleen, liver, and kidneys, but not in the SI IEL (Fig. 1F,G; Supporting information Fig. S1F). Transcriptional analysis showed that Blimp‐1 is upregulated in total effector CD8+ T cells compared to naïve CD8+ T cells, but did not reveal significant differences in Blimp‐1 expression between Hobit‐ and Hobit+ subsets of effector CD8+ T cells (Supporting information Fig. S1G). Thus, these results show that Hobit expression is confined to the Trm lineage, whereas Blimp‐1 is broadly upregulated in effector CD8+ T cells, including Trm precursors, and maintained in all subsets of memory CD8+ T cells after viral clearance.
Hobit and Blimp‐1 instruct the separation between the Trm and Tcm lineages upon Hobit upregulation
We have previously addressed the role of Hobit and Blimp‐1 in memory CD8+ T‐cell differentiation using mice that completely lacked Hobit and Blimp‐1 in the T‐cell lineage [22]. These Hobit and Blimp‐1 double‐deficient mice have defects in both terminal effector and Trm differentiation, complicating the study of the specific role of these transcription factors in the Trm lineage. Here, we have used the newly developed reporter HobitKO/CRE × Blimpflox/flox mice to specifically address the role of Hobit and Blimp‐1 within the Trm lineage (Supporting information Fig. S2A). One of the Hobit alleles of the HobitKO/CRE × Blimpflox/flox mice is disrupted by a trapping cassette while the other allele is disrupted by insertion of the Hobit reporter construct, resulting in functional deficiency of Hobit, but permitting tracking of the transcriptional activity at the Hobit locus through tdTomato expression (Supporting information Fig. S2A). The Hobit‐driven Cre recombinase is designed to mediate the excision of exon 6 of the Blimp‐1 locus, which contains flanking LoxP sites, after upregulation of Hobit expression (Supporting information Fig. S2A). This experimental setup will result in dysfunctional Blimp‐1 in Hobit‐expressing cells of HobitKO/CRE × Blimpflox/flox mice. Given that Hobit is only consistently expressed in Trm precursors at day 8 after infection [17], these mice offer a unique opportunity to address the role of Blimp‐1 in the Trm lineage without affecting early effector CD8 T‐cell differentiation. To assess the efficiency of the Hobit‐driven Cre recombinase to delete Blimp‐1 in Trm, tdTomato+ and tdTomato‐ memory CD8+ T cells were isolated from the liver and SI IEL of LCMV‐infected HobitKO/CRE × Blimpflox/flox and control HobitWT/CRE mice at day >30 p.i. Using PCRs that distinguish the deleted from the nondeleted Blimp‐1 locus, we observed that the Blimp‐1 gene was largely deleted in tdTomato+ CD8+ T cells from both liver and SI IEL of HobitKO/CRE × Blimpflox/flox mice, but not in tdTomato‐ CD8+ T cells of these mice or in CD8+ T cells from these organs of control HobitWT/CRE mice (Supporting information Fig. S2B). Conversely, the nondeleted Blimp‐1 gene was detected more strongly in the tdTomato‐ fraction compared to the tdTomato+ fraction of CD8+ T cells of HobitKO/CRE × Blimpflox/flox mice (Supporting information Fig. S2C). Thus, the HobitKO/CRE × Blimpflox/flox mouse enabled us to specifically address the role of Hobit and Blimp‐1 during Trm development.
We followed the differentiation of virus‐specific (GP33+) memory CD8+ T cells in LCMV‐infected HobitKO/CRE × Blimpflox/flox and control HobitWT/CRE mice. The formation of GP33+ memory T cells was comparable between HobitKO/CRE × Blimpflox/flox and HobitWT/CRE mice in blood, peripheral LNs (pLN), and peripheral tissues including liver, kidneys and IEL, and lamina propria (LPL) of the SI at day >30 p.i (Fig. 2A and B). An increase in the percentage of GP33+ memory CD8+ T cells was observed in spleen of HobitKO/CRE × Blimpflox/flox mice compared to HobitWT/CRE mice (Fig. 2B). Analysis of memory CD8+ T cell subsets showed reduced formation of tdTomato+ Trm in spleen, liver, and kidneys (Fig. 2C and D), but not in the IEL and LPL compartment of the SI of HobitKO/CRE × Blimpflox/flox mice compared to HobitWT/CRE mice (Fig. 2C and D). In contrast, we observed increased formation of Tcm in spleen and pLN of HobitKO/CRE × Blimpflox/flox mice compared to HobitWT/CRE mice (Fig. 2E and F). These findings suggest that Blimp‐1 acts within the Trm lineage after upregulation of Hobit to instruct Trm formation together with Hobit. Moreover, our data indicate that Hobit and Blimp‐1 impair the development of Tcm. These findings are essentially similar as we previously observed in mice deficient in Hobit and Blimp‐1 in the complete T‐cell lineage, in which we showed that Hobit and Blimp‐1 synergistically instruct Trm development [22].
Figure 2.
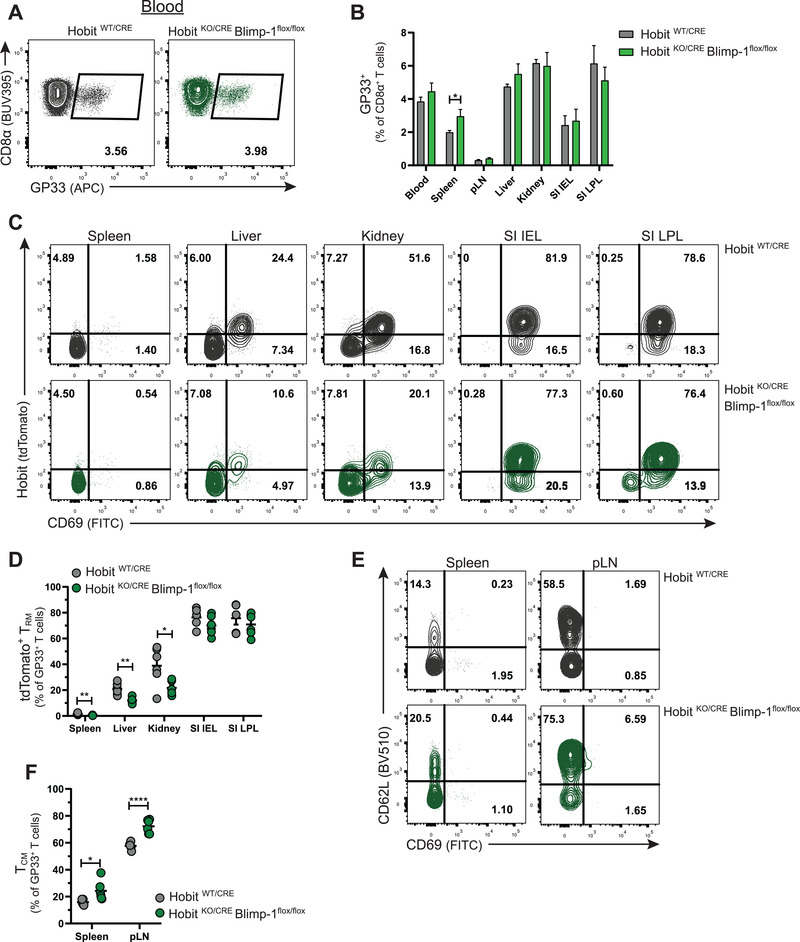
Hobit and Blimp‐1 instruct Trm formation and impair Tcm formation. HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice were infected with LCMV Armstrong and virus‐specific (GP33+) CD8+ T cells were analyzed by flow cytometry at day >30 p.i. (A) Flow cytometry plot displays the binding of Db GP33 tetramers to CD8+ T cells from the blood of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice. (B) The frequency of the LCMV‐specific CD8+ T cells as a percentage of total CD8+ T cells was quantified. (C‐F) Representative flow cytometry plots show (C) tdTomato and CD69 and (E) CD62L and CD69 expression in GP33+ CD8+ T cells within the indicated tissues. The frequency of (D) tdTomato+ Trm and (F) Tcm within GP33+ CD8+ T cells was quantified. Combined data from two independent experiments (n = 5–6). Symbols represent individual mice. Error bars represent mean ± SEM. Unpaired t test. *p < 0.05; **p < 0.01; ****p < 0.0001.
To exclude the potential impact of viral clearance on memory CD8+ T‐cell differentiation, we generated mixed BM chimeras with either WT and HobitWT/CRE compartments or WT and HobitKO/CRE × Blimpflox/flox compartments in a 1:1 ratio (Fig. 3A; Supporting information Fig. S3A). The HobitWT/CRE and HobitKO/CRE × Blimpflox/flox donor stem cells contributed equally compared to the WT donor stem cells to the establishment of CD8+ T cells (Supporting information Fig. S3B and C). The mixed BM chimeras were infected with LCMV Armstrong to analyze the virus‐specific memory CD8+ T‐cell response at day >30 after infection (Fig. 3A). The WT and HobitWT/CRE compartments contributed similarly to the formation of GP33+ CD8+ memory T cells in lymphoid and peripheral tissues of the BM chimeras (Fig. 3B and C). In contrast, the HobitKO/CRE × Blimpflox/flox compartment was overrepresented in GP33+ memory CD8+ T cells of lymphoid tissues, while this compartment was subtly underrepresented in virus‐specific memory cells in peripheral tissues including kidney and SI IEL of the mixed BM chimeras (Fig. 3B and D). Underlining the differences in the distribution of WT and HobitKO/CRE × Blimpflox/flox memory CD8+ T cells, we observed that Tcm in blood, spleen, pLN, and mesenteric LNs were relatively increased within the HobitKO/CRE × Blimpflox/flox compartment compared to the WT compartment (Fig. 3E and F), whereas Trm in liver, kidneys, and SI IEL were substantially decreased in the HobitKO/CRE × Blimpflox/flox compartment relative to the WT compartment (Fig. 3E and G). In WT and HobitWT/CRE mixed BM chimeras, no differences were observed in the contribution of the donor compartments to the formation of Tcm and Trm (Fig. 3E, H, I). Thus, these findings support that Hobit and Blimp‐1 instruct Trm formation and restrict Tcm development in antigen‐triggered CD8+ T cells that have already passed the initial differentiation steps and have upregulated Hobit expression.
Figure 3.
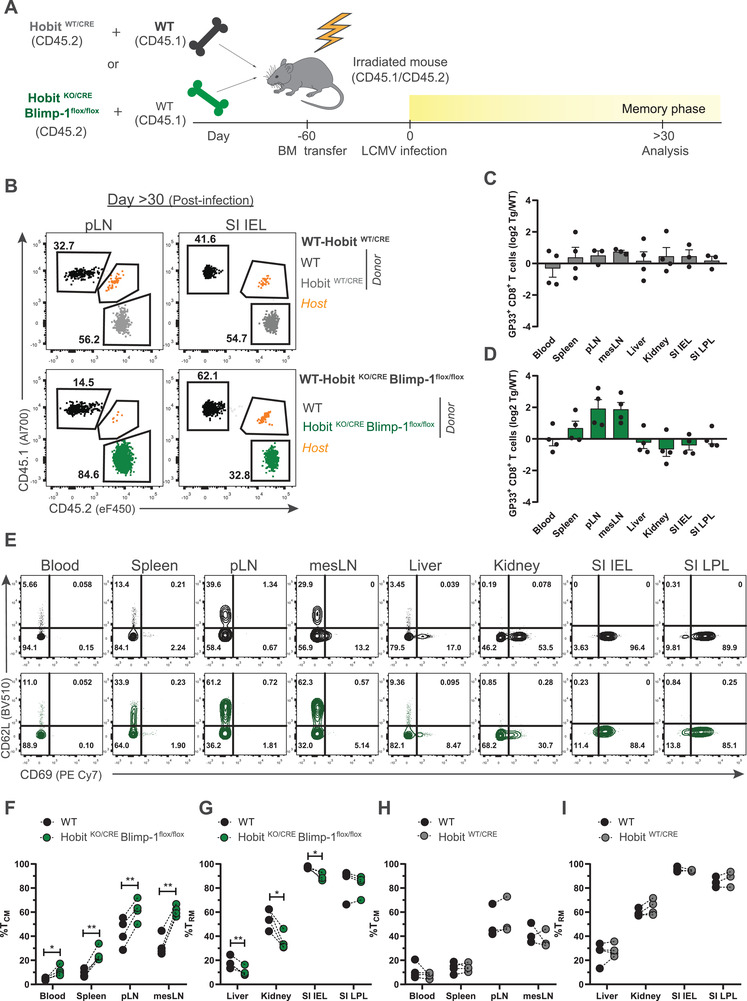
Hobit and Blimp‐1 instruct Trm formation after Hobit upregulation. (A) Scheme shows the setup of the LCMV infection experiment in mixed BM chimeras from WT and HobitWT/CRE mice or WT and HobitKO/CRE × Blimp‐1flox/flox mice (1:1 ratio) to analyze virus‐specific memory CD8+ T cells at day >30 p.i. by flow cytometry. (B) Representative flow cytometry plots display CD45.1 and CD45.2 to identify the contribution of WT (CD45.1+) and HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox (CD45.2+) compartments to the GP33+ CD8+ T‐cell fraction in the indicated tissues at day >30 p.i. (C, D) The log2 ratio of GP33+ CD8+ T cells of (C) HobitWT/CRE (transgenic, tg) and (D) HobitKO/CRE × Blimp‐1flox/flox (tg) relative to WT controls was quantified in the indicated tissues. (E) Representative flow cytometry plots display expression of CD62L and CD69 of LCMV‐specific memory CD8+ T cells of the WT (black) and HobitKO/CRE × Blimp‐1flox/flox (green) compartment in the indicated tissues of chimeric mice. (F‐I) The frequency of (F, H) Tcm and (G, I) Trm within GP33+ CD8+ T cells from WT and transgenic mice is displayed. Symbols represent individual mice. Dotted lines connect paired samples. Representative data of one (n = 3–4) out of two independent experiments. Paired t test. *p < 0.05; **p < 0.01.
Blimp‐1 does not regulate terminal differentiation upon establishment of the Trm lineage
Recently, we have identified Trm precursors through their specific expression of Hobit during the effector phase of infection [17]. To evaluate the impact of Hobit and Blimp‐1 on Trm precursor formation, virus‐specific effector CD8+ T cells of LCMV‐infected HobitWT/CRE and HobitKO/CRE × Blimpflox/flox mice were analyzed at day 8 p.i. We were able to identify tdTomato+ expression indicating upregulation of Hobit in a subset of effector CD8+ T cells located in peripheral, but not in lymphoid tissues (Fig. 4A and B), in line with previous findings [17]. The expression of Hobit was mainly, but not entirely, found in KLRG1‐ CD127+ MPECs rather than KLRG1+ CD127‐ SLECs of the peripheral tissues (Supporting information Fig. S4A–C). We did not observe differences in the presence of tdTomato+ effector CD8+ T cells between HobitKO/CRE × Blimpflox/flox and control HobitWT/CRE in blood, lymphoid tissues, such as spleen and pLN, or in peripheral tissues including liver, kidneys, SI IEL, and SI LPLs (Fig. 4A and B), suggesting that lack of Hobit and Blimp‐1 does not compromise the presence of Trm precursors within the tissues at day 8 after LCMV infection. Thus, Hobit and Blimp‐1 are relevant for the formation or maintenance of Trm, but appear redundant for the formation of Trm precursors.
Figure 4.
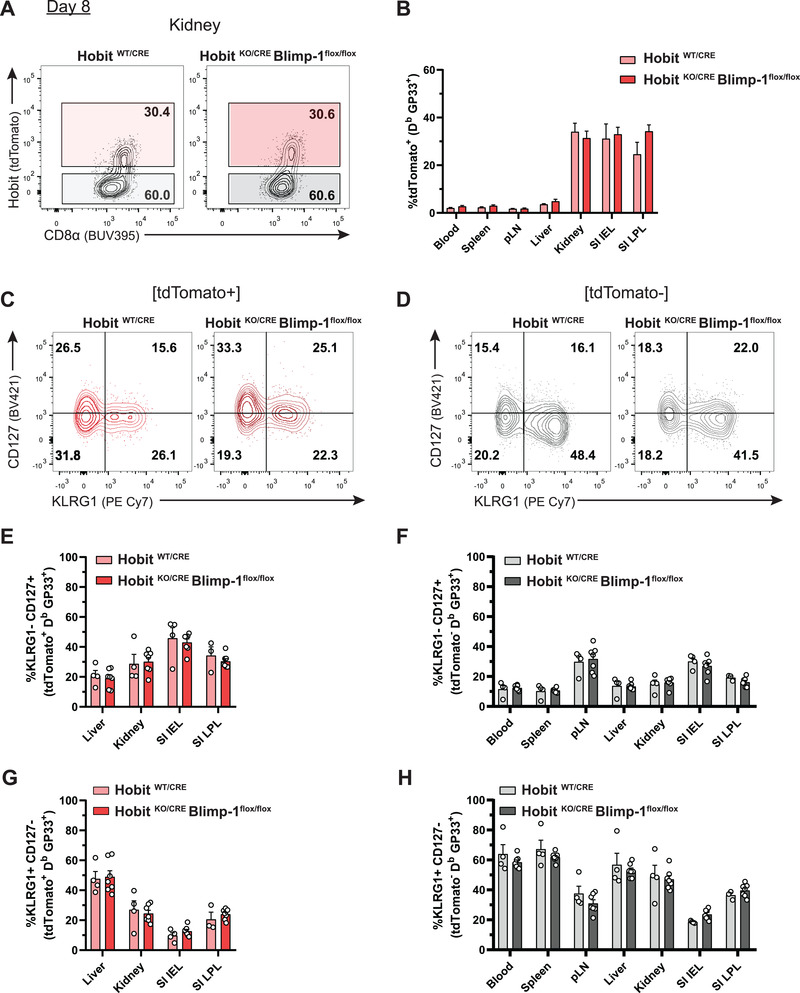
Blimp‐1 does not regulate terminal differentiation upon establishment of the Trm lineage. (A) Representative flow cytometry plots display tdTomato expression in GP33+ CD8+ T cells isolated from the kidney of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice at day 8 after LCMV infection. (B) Expression of tdTomato was quantified in GP33+ CD8+ T cells of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice in the indicated tissues at day 8 p.i. (C, D) Representative flow cytometry plots show expression of CD127 and KLRG1 within (C) tdTomato+ or (D) tdTomato‐ virus‐specific CD8+ T cells isolated from the kidneys of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice. (E‐H) The percentage of (E, F) MPECs (KLRG1‐ CD127+) and (G, H) SLECs (KLRG1+ CD127‐) was determined within the (E, G) tdTomato+ and (F, H) tdTomato‐ fraction of GP33+ CD8+ T cells. Combined data from two independent experiments (n = 4–7). Symbols represent individual mice. Error bars represent mean ± SEM. Unpaired t test.
Previously, Blimp‐1 has been shown to induce terminal effector differentiation [8, 9, 25]. However, whether Blimp‐1 instructs terminal differentiation in already established Trm precursors has not yet been addressed. We analyzed the distribution of MPECs and SLECs in the tdTomato+ and tdTomato‐ fractions of both HobitKO/CRE × Blimpflox/flox and control HobitWT/CRE mice (Fig. 4C–H). We found no differences in KLRG1 and CD127 expression in circulation, lymphoid, and peripheral tissues of HobitKO/CRE × Blimpflox/flox and control HobitWT/CRE mice, suggesting that Blimp‐1 does not affect terminal differentiation within the Hobit+ lineage (Fig. 4C–H). Therefore, these results show that Blimp‐1 does not drive terminal differentiation upon establishment of Trm precursors.
Hobit and Blimp‐1 promote CD69 expression on Trm precursors
We next evaluated whether Hobit and Blimp‐1 affected the phenotype of Trm precursors during the effector phase. For this purpose, we studied the expression of Trm‐associated molecules in tdTomato+ and tdTomato‐ virus‐specific effector CD8+ T cells at day 8 p.i. Interestingly, we found a substantial reduction of CD69 expression in the tdTomato+ fraction of liver, kidneys, SI IEL, and SI LPL of HobitKO/CRE × Blimpflox/flox compared to control HobitWT/CRE mice (Fig. 5A–C). In contrast, the tdTomato‐ population remained largely unaffected (Fig. 5A, D, E). To determine whether Hobit, Blimp‐1, or both transcription factors were responsible for the downregulation of CD69 expression, we monitored the development of LCMV‐specific effector CD8+ T cells from single HobitKO/CRE and HobitWT/CRE × Blimpflox/flox mice, in which either Hobit or Blimp‐1 were specifically deleted from the Trm lineage, respectively. Remarkably, we did not observe differences in CD69 expression in tdTomato+ effector CD8+ T cells in control HobitWT/CRE, HobitKO/CRE or HobitWT/CRE × Blimpflox/flox mice in contrast to the tdTomato+ fraction of HobitKO/CRE × Blimpflox/flox mice, which showed a reduction in CD69 expression (Supporting information Fig. S5A–D). In contrast, no differences were observed in the expression of CD103 or the Trm‐associated molecules, CD49a and CXCR6, [17] on tdTomato+ virus‐specific effector CD8+ T cells of control HobitWT/CRE, HobitKO/CRE, HobitWT/CRE × Blimpflox/flox or HobitKO/CRE × Blimpflox/flox mice (Supporting information Fig. S5E–J), indicating that neither Hobit or Blimp‐1 drive the expression of CD103, CD49a, and CXCR6 on Trm precursors. These findings indicate that Hobit and Blimp‐1 regulate CD69 expression on Trm precursors, but not the formation of Trm precursors. Frequencies of CD62L+ effector CD8+ T cells in pLNs tended to be higher in HobitKO/CRE × Blimpflox/flox mice compared to HobitWT/CRE mice (Fig. 5F and G), in line with the increased numbers of Tcm in the memory phase (Figs. 2 and 3). Altogether, these findings indicate that Hobit and Blimp‐1 cooperatively instruct upregulation of CD69 expression on Hobit+ Trm precursors.
Figure 5.
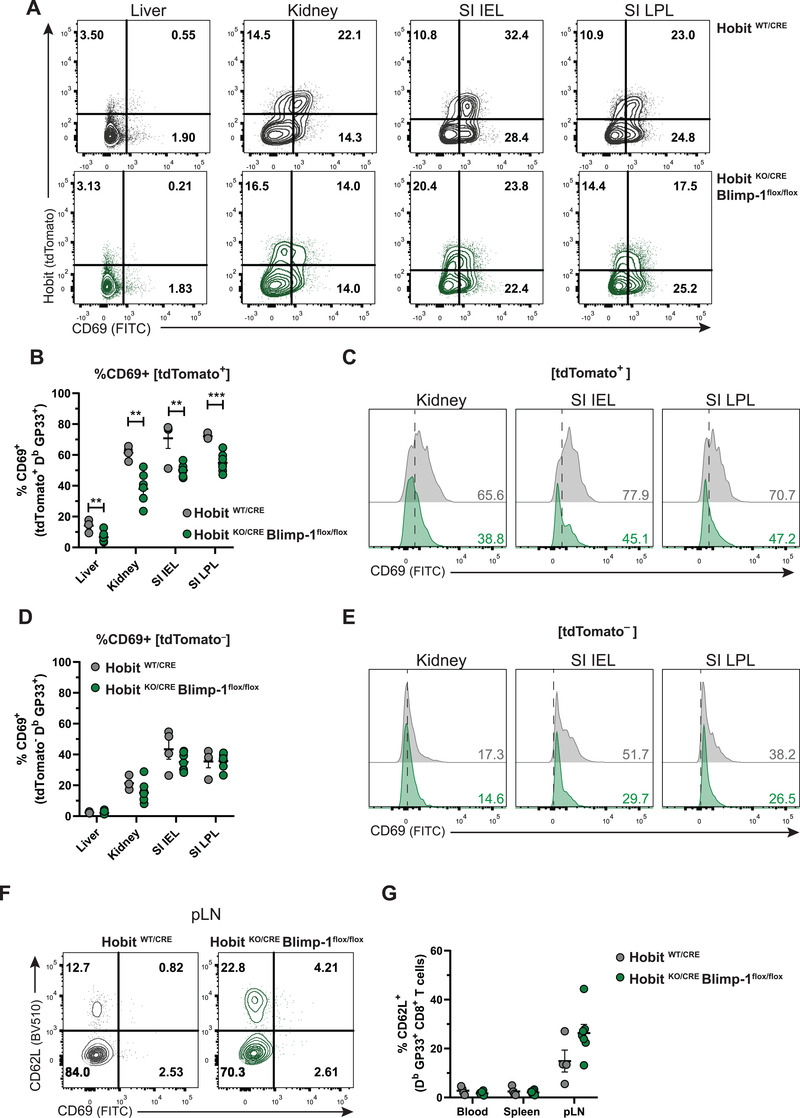
Hobit and Blimp‐1 promote CD69 expression on Trm precursors. (A) Representative flow cytometry plots display CD69 and tdTomato expression within GP33+ CD8+ T cells in the indicated tissues of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice at day 8 after LCMV infection. (B‐E) The percentage of CD69 expression was quantified in (B) tdTomato+ and (D) tdTomato‐ virus‐specific CD8+ T cells of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice. (C, E) Representative histograms display CD69 expression in (C) tdTomato+ and (E) tdTomato‐ virus‐specific CD8+ T cells in the indicated tissues of HobitWT/CRE (grey) and HobitKO/CRE × Blimp‐1flox/flox (green) mice. (F) Representative flow cytometry plots display CD69 and CD62L expression within GP33+ CD8+ T cells in the pLNs of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice at day 8 after LCMV infection. (G) The percentage of CD62L expression was quantified within GP33+ CD8+ T cells in the indicated tissues of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice at day 8 after LCMV infection. Combined data from two independent experiments (n = 4–7). Symbols represent individual mice. Error bars represent mean ± SEM. Unpaired t test. **p < 0.01, ***p < 0.001.
Next, we developed mixed BM chimeras with control HobitWT/CRE and HobitKO/CRE × Blimpflox/flox BM in a 1:1 ratio (Fig. 6A; Supporting information Fig. S6A) to exclude impact of viral clearance on the Hobit and Blimp‐1‐driven regulation of effector CD8+ T cells. The donor HobitWT/CRE and HobitKO/CRE × Blimpflox/flox stem cells contributed similarly to the establishment of the CD8+ T‐cell lineage (Supporting information Fig. S6B and C). Analysis of GP33+ T cells at day 8 after LCMV infection showed that the HobitKO/CRE × Blimpflox/flox compartment was at a disadvantage in populating peripheral organs, such as kidneys and SI, compared to the HobitWT/CRE compartment (Fig. 6B and C). The expression of tdTomato was equal in both HobitWT/CRE and HobitKO/CRE × Blimpflox/flox virus‐specific effector CD8+ T cells (Fig. 6D). In line with our previous observations, tdTomato+ effector CD8+ T cells, but not tdTomato‐ effector CD8+ T cells, from the HobitKO/CRE × Blimpflox/flox compartment displayed a reduction of CD69 expression compared to the HobitWT/CRE compartment (Fig. 6E–G). Moreover, CD62L expression appeared slightly increased on the HobitKO/CRE × Blimpflox/flox fraction compared to the HobitWT/CRE fraction of effector CD8+ T cells in the pLNs (Supporting information Fig. S6D, E). Taken together, these studies indicate that Hobit and Blimp‐1 instruct CD69 expression on effector CD8+ T cells favoring the subsequent development of Trm during the memory phase of the immune response. Thus, Hobit‐ and Blimp‐1‐driven CD69 upregulation appears an early event in the establishment of the Trm lineage.
Figure 6.
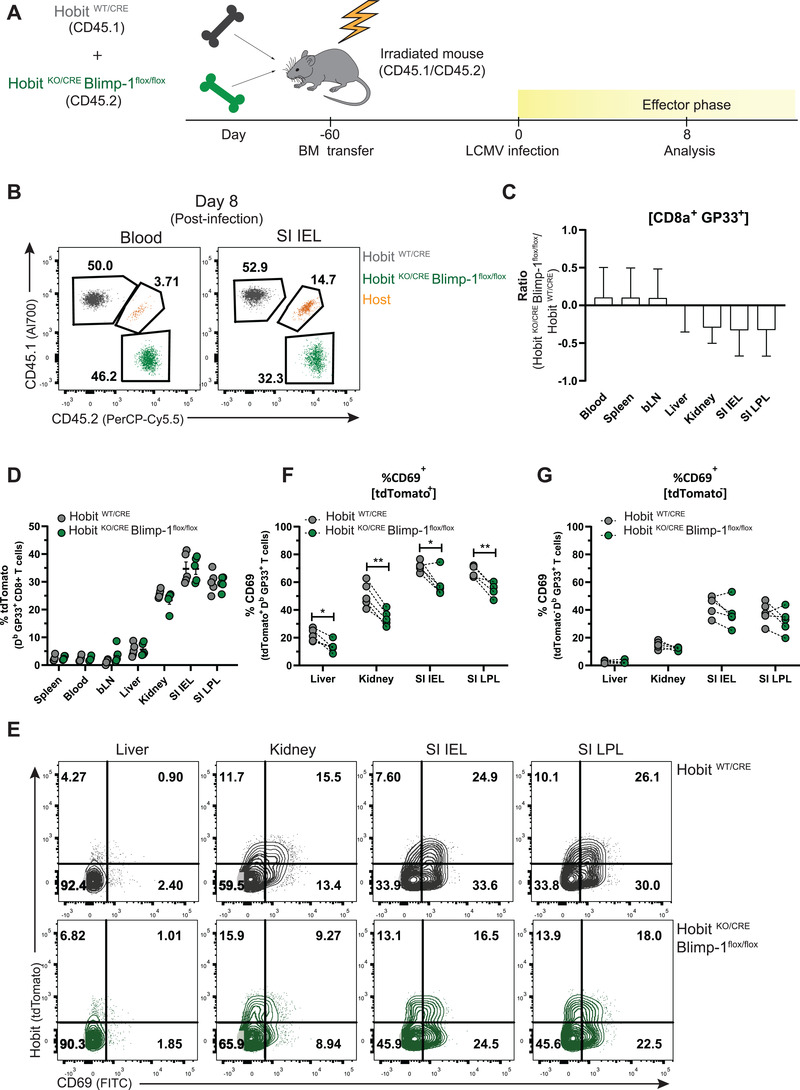
Hobit and Blimp‐1 are important for upregulation of CD69 on Trm precursors. (A) The experimental setup of LCMV infection in mixed BM chimeras from HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice (1:1 ratio) is shown. (B) Representative flow cytometry plots display CD45.1 and CD45.2 expression to identify the contribution of HobitWT/CRE (CD45.1+) and HobitKO/CRE × Blimp‐1flox/flox (CD45.2+) compartments to the GP33+ CD8+ T‐cell population of chimeric mice at day 8 p.i. (C) The log2 ratio of the percentage of GP33+ CD8+ T cells of HobitKO/CRE × Blimp‐1flox/flox mice relative to those of HobitWT/CRE mice was quantified in the indicated tissues. (D) The percentage of tdTomato expression was quantified in the HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox compartments of GP33+ CD8+ T cells in the indicated tissues of chimeric mice. (E) Representative flow cytometry plots display tdTomato and CD69 expression within the HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox compartment of GP33+ CD8+ T cells in the indicated tissues of chimeric mice. (F, G) The percentage of CD69 expression was quantified in (F) tdTomato+ and (G) tdTomato‐ virus‐specific CD8+ T cells of the HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox compartment of chimeric mice. Symbols represent individual mice. Dotted lines connect paired samples. Representative data of one (n = 5) out of two independent experiments. Paired t test. *p < 0.05; **p < 0.01.
Hobit and Blimp‐1 suppress the tissue exit receptors S1PR1 and CCR7 in Trm precursors
CD69 has an important role in the regulation of the tissue exit pathway S1PR1 [26], suggesting that Hobit and Blimp‐1 may impair tissue egress of Trm precursors through control of CD69 expression. To further address the regulation of tissue exit pathways in Trm precursors, we analyzed RNA sequencing data of virus‐specific effector CD8 T cells developing after LCMV infection for the expression of receptors driving migration. Analysis of a panel of S1P receptors showed that expression of S1PR1, S1PR4, and S1PR5 was reduced in tdTomato+ effector CD8 T cells of kidney and to a lesser degree in those of SI compared to their tdTomato‐ counterparts (Fig. 7A). In the panel of analyzed chemokine receptors, we observed that expression of the tissue exit receptor CCR7 and the SDF‐1 receptor CXCR4 was suppressed in tdTomato+ effector cells compared to tdTomato‐ effector cells (Fig. 7A). In contrast, expression of other chemokine receptors, including that of the Trm‐associated chemokine receptor CXCR6, was upregulated in tdTomato+ effector cells relative to tdTomato‐ effector cells, as previously noted [17]. We analyzed expression of S1PR1 and CCR7 in further detail, given that these tissue exit pathways have been shown direct targets of Hobit and Blimp‐1 [22]. Expression of S1PR1 was similarly downregulated in the virus‐specific and total fraction of tdTomato+ effector CD8 T cells compared to tdTomato‐ effector CD8 T cells after LCMV infection (Fig. 7B and C). To determine the role of Hobit and Blimp‐1 in the regulation of S1PR1 and CCR7 expression, we analyzed the expression of these tissue exit receptors in the tdTomato+ fraction of effector CD8 T cells in WT and in Hobit‐ and Blimp‐1‐deficient settings after LCMV infection. The expression of S1PR1 and CCR7 was elevated in tdTomato+ CD8 T cells of kidney and SI of HobitKO/CRE × Blimpflox/flox mice compared to those of HobitWT/CRE mice (Fig. 7D and E). Thus, Hobit and Blimp‐1 suppress the expression of S1PR1 and CCR7 in Trm precursors, which may facilitate long‐term lodgment of these effector CD8 T cells in the peripheral tissues to ensure development into tissue‐locked Trm.
Figure 7.
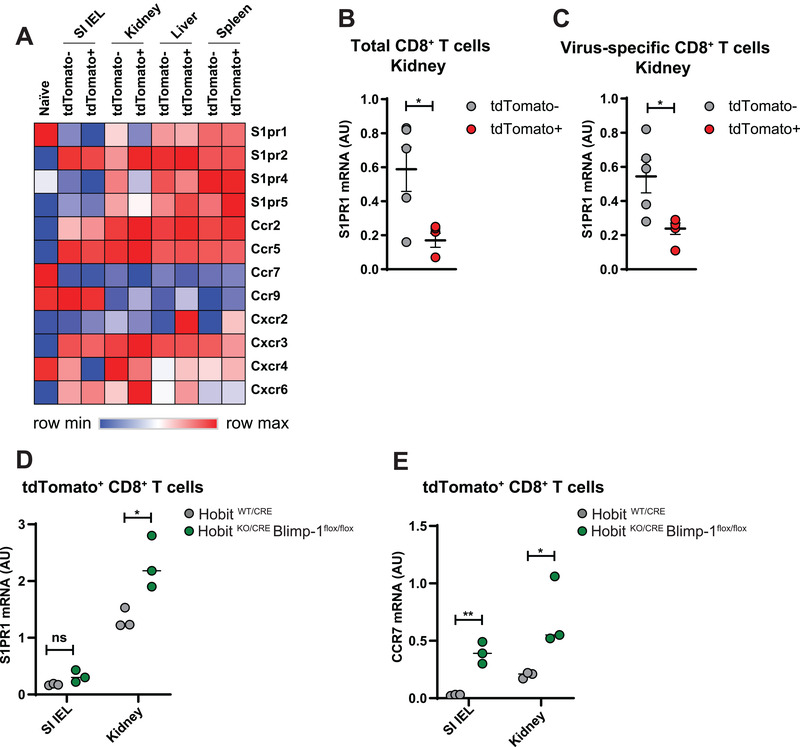
Hobit and Blimp‐1 suppress S1PR1 and CCR7 expression on Trm precursors. (A) The expression of the indicated panel of S1P receptors and chemokine receptors was determined in the tdTomato+ and tdTomato‐ fractions of virus‐specific effector CD8 T cells in the epithelial fraction of small intestine (SI IEL), kidneys, liver, and spleen of HobitWT/CRE mice at day 8 after infection with LCMV Armstrong using RNA sequencing. (B) S1PR1 expression was analyzed in tdTomato+ and tdTomato‐ fractions of (B) the total and (C) the virus‐specific fraction of effector CD8 T cells in kidneys of HobitWT/CRE mice using quantitative PCR. (D, E) The tdTomato+ fraction of CD8 T cells of SI IEL and kidney of HobitWT/CRE and HobitKO/CRE × Blimp‐1flox/flox mice was analyzed for expression of (D) S1PR1 and (E) CCR7 using quantitative PCR. (A) Representative data of three mice per group, (B,C) five mice per group, and (E,F) three mice per group. Symbols represent individual mice. Error bars represent mean ± SEM. Unpaired t test. *p < 0.05, **p < 0.01.
Discussion
The differentiation of CD8+ T cells into distinct subsets of memory CD8+ T cells is a tightly regulated process under the control of a network of transcription factors. Here, we studied the role of Hobit and Blimp‐1 during Trm development using mice that allowed tracking of the Trm lineage through the Hobit reporter and enabled the removal of these transcription factors from the Trm lineage. We found that Trm precursors could still be formed in the absence of Hobit and Blimp‐1, but both transcription factors were required to instruct the upregulation of the tissue‐retention molecule CD69 and the downregulation of the tissue exit receptors S1PR1 and CCR7 on Trm precursors. The impact of Hobit and Blimp‐1 on effector T cells favored Trm development and suppressed Tcm formation during the memory phase of the immune response. These findings underline an essential role of Hobit and Blimp‐1 at the branching point of the Tcm and Trm lineages, by promoting retention of Trm precursors, while restricting Tcm formation.
We have previously shown that Hobit and Blimp‐1 drive Trm differentiation [22], but the role of these transcription factors in early events of Trm development remained unresolved due to the inability to identify Trm precursors. We and others have shown that effector CD8 T cells destined to develop into Trm are already separate from other memory T‐cell lineages at early stages of the immune response [19, 20, 21]. Given that these committed Trm precursors uniformly expressed Hobit, we had the opportunity to interrogate the role of Hobit and Blimp‐1 in early events of Trm differentiation using Hobit‐ and Blimp‐1‐deficient Hobit reporter mice. We found that Hobit was not essential for the formation of Trm precursors as identified by expression of the Hobit reporter. Moreover, Hobit and Blimp‐1 did not have an effect on the distribution of Hobit+ effector T cells throughout peripheral organs, suggesting that these transcription factors do not instruct the trafficking of Trm precursors to peripheral tissues. In contrast, we found that Hobit and Blimp‐1 were essential for the expression of the tissue‐retention molecule CD69 on Trm precursors. Other Trm‐associated molecules, such as CD103, CD49a, or CXCR6, were not affected by the lack of Hobit and Blimp‐1. These observations underline an early role of Hobit and Blimp‐1 in the maintenance of Trm precursors in the peripheral tissues, given that CD69 is important for tissue retention of Trm [27]. CD69 suppresses surface expression of the tissue exit receptor S1PR1, thereby locking T cells into the tissues [26, 28]. In addition, we also observed that Hobit and Blimp‐1 directly suppress expression of S1PR1 and CCR7 in Trm precursors. Therefore, our findings suggest that Hobit and Blimp‐1 are responsible for the early retention of Trm precursors within the tissues through the regulation of pathways of tissue exit.
Even though Trm precursors from all studied peripheral tissues had defects in CD69 expression, Trm formation was mainly affected in liver and kidneys compared to the intestinal compartment. We have previously shown that both Hobit and Blimp‐1 drive Trm development in liver, kidneys, skin, and SI [22]. The mice from this previous study completely lacked Hobit and Blimp‐1 in the T‐cell lineage resulting in defects in both terminal effector differentiation and Trm differentiation. A recent study has shown the presence of terminal and memory‐resident populations in the SI, identified by Blimp‐1 and Id3, respectively [20]. Given that our newly developed mice are only Blimp‐1‐deficient after Hobit upregulation, it is possible that residual CD8 T cells in the SI represent Blimp‐1‐expressing terminal resident populations that develop independent of Hobit. In addition, in contrast to liver and kidneys, SI IEL Trm express the integrin CD103, which facilitates their binding to the epithelium and allow them to rely on additional retention mechanisms.
The classification of effector CD8+ T cells into SLECs and MPECs separates terminally differentiated cells from effector T cells with potential to differentiate into memory T cells [3, 4]. However, the MPEC population does not capture heterogeneity between precursors of the Tcm, Tem, and Trm lineages. The transcriptional regulation of effector T cells suggests that lineage specification of memory T cells may occur at early stages of the immune response. We have previously shown that Eomes suppresses Hobit expression and limits the development of Trm precursors to favor the development of the Tcm lineage [17]. Here, we showed that Hobit and Blimp‐1 specifically favored the generation of Trm while restricting the formation of Tcm. Our results suggest that Trm precursors that are unable to persist within the tissues develop into Tcm in circulation. We have previously demonstrated that Hobit and Blimp‐1 via direct binding to the TCF‐1 encoding Tcf7 locus suppress the expression of TCF‐1 [22], which is an essential transcription factor for the development of Tcm cells [29]. Thus, Hobit and Blimp‐1 may impair Tcm development through suppression of the Tcm‐inducing transcription factor TCF‐1. Taken together, these findings suggest that Eomes, Hobit, Blimp‐1, and TCF‐1 control a branching point in effector T‐cell differentiation resulting in the early separation of the Tcm and Trm lineages.
As previously observed [22], the combined action of Hobit and Blimp‐1 was required to regulate Trm differentiation. In contrast to double deficiency of both transcription factors, single deficiency in Hobit or Blimp‐1 within the Trm lineage did not affect the differentiation of Trm precursors. The synergy between Hobit and Blimp‐1 was not unexpected, given that these transcription factors are homologous with high similarity in the functional Zinc Finger domains [30, 31], which are important for the recognition and binding of DNA sequences. Indeed, both Hobit and Blimp‐1 bind to common motifs in the DNA and share many target genes in CD8+ T cells [22, 31]. These similarities in DNA binding suggest that both transcription factors act in a cooperative manner to regulate the formation of Trm precursors. Importantly, the shared target genes of Hobit and Blimp‐1 include transcription factors that regulate tissue exit, such as KLF2 and TCF‐1, and tissue exit receptors themselves such as S1PR1 and CCR7 [22]. Here, we observed that Hobit and Blimp‐1 already regulated these tissue exit pathways in the effector phase in Trm precursors. It is possible that the regulation of CD69 expression is downstream of the collaborative Hobit‐ and Blimp‐1‐driven transcriptional regulation of S1PR1 expression.
In contrast to their overlapping targets, Hobit and Blimp‐1 have divergent expression patterns in CD8+ T cells. In comparison to Hobit, which is confined to the Trm lineage, Blimp‐1 has a much wider expression pattern. Blimp‐1 is upregulated in CD8+ T cells after encountering cognate antigen [32, 33] and it acquires its maximum expression in terminal effector CD8+ T cells to promote their terminal differentiation [8, 9, 20]. In contrast to these findings, we did not observe that Blimp‐1 impacted terminal differentiation upon establishment of the Trm lineage despite expression of KLRG1 and CX3CR1 on a subset of Hobit+ effector CD8+ T cells [17]. These observations suggest that Blimp‐1 may acquire distinct roles during the process of T‐cell differentiation, driving terminal differentiation upon T‐cell priming and promoting tissue retention upon establishment of the Trm lineage.
Hobit and Blimp‐1 are an integrated part of a larger transcriptional program governing the differentiation of effector CD8+ T cells into different lineages of memory precursors and terminal effectors. Transcription factors, such as Bcl‐6, Id3, and Eomes, have been implicated in favoring the differentiation of memory precursors, in particular those upstream of Tcm [12, 13, 14, 15, 16]. Other transcription factors, such as Tbet, Id2, Runx3, and Notch appear to preferentially drive terminal differentiation of effector CD8+ T cells as well as the development of Trm [34, 35, 36]. Thus, all of these transcriptional regulators appear important to regulate distinct aspects of Trm differentiation. In line with our previous findings in Trm [22], we have shown that Hobit and Blimp‐1 have an important early role for the retention of Trm precursors within the tissues through sustained suppression of tissue exit pathways and upregulation of CD69, which counteracts tissue exit. Thus, Hobit and Blimp‐1 appear to be an essential pair of transcription factors in the transcriptional network regulating the developmental pathway of Trm.
Materials and methods
Mice
Wild‐type CD45.2+ (C57BL/6JRj) mice were purchased from Janvier, WT CD45.1+ (B6.SJL‐Ptprca Pepcb/BoyJ) mice were purchased from the Jackson Laboratory and both of these lines were crossed to obtain CD45.1 × CD45.2 mice. HobitWT/CRE (B6‐Tg [Zfp683‐tdTomato‐P2A‐CRE‐P2A‐DTR]) mice were developed at Ozgene (Perth, Australia) as previously described [23]. HobitKO/CRE × Blimp‐1flox/flox, HobitKO/CRE and HobitWT/CRE × Blimp‐1flox/flox were generated by crossing HobitWT/CRE with HobitKO/KO [30] and/or Blimp‐1flox/flox [8] mice. The Hobit‐driven CRE recombinase of HobitWT/CRE × Blimp‐1flox/flox and HobitKO/CRE × Blimp‐1flox/flox mice will act on exon 6 of the Blimp‐1 locus that contains flanking loxP sites to create Blimp‐1 deficiency exclusively in Hobit‐expressing cells. HobittdTomato/WT × Blimp‐1GFP/WT were generated by crossing of HobitWT/CRE and Blimp‐1GFP/WT mice [37]. CD45.1 × CD45.2 mice were used as recipients for the generation of mixed BM chimeric mice. Following irradiation (2 × 5 gray), recipient mice were reconstituted by intravenous transfer of 2 × 107 BM cells. Recipients were used in experiments 8 weeks after reconstitution. Chimerism was analyzed in the blood prior to experiments using the congenic markers CD45.1 and CD45.2 to establish the relative size of host and donor compartments. All mice were maintained under specific pathogen‐free conditions in the animal facility of the Netherlands Cancer Institute. Experimental mice were age matched and between 8–12 weeks at the start of the experiment. Both female and male mice were used for this study. For BM chimeras, donor and recipient mice were sex matched. Animal experiments were conducted according to institutional and national guidelines. The permission of the national authorities for animal experiments (Centrale Commissie Dierproeven, CCD) has been obtained under number AVD3010020172205.
LCMV infection
Mice were infected i.p. with 1 × 105 plaque‐forming units of LCMV Armstrong obtained from the European Virus Archive (EVAg). Infected mice were sacrificed and organs were collected for analysis of CD8+ T‐cell responses at the indicated time points after infection.
Tissue preparation
SI LPL and IEL preparations were obtained from the SI. After removal of residual fat tissue, Peyer's patches, and feces, the SI was cut into pieces of 1 cm and incubated in HBSS (Gibco) with 10% FCS, 5 mM EDTA, and 1 mM DTT for 30 min at 37°C. After repeated vortexing, the IEL fraction was released from the tissue and isolated by filtering over a 70 μm cell strainer. Subsequently, IEL‐depleted pieces of the intestine were washed in HBSS supplemented with 2% FCS and enzymatically digested for 30 min at 37°C with 375 U/mL Collagenase Type I (Worthington) and 0.15 mg/mL DNase I (Roche, from bovine pancreas, grade II) in RPMI 1640 (supplemented with 10% FCS) to isolate the LPL fraction. Similarly, kidneys, which were cut into pieces of 1 mm3, were enzymatically digested for 30 min at 37°C with 750 U/mL Collagenase Type I (Worthington) and 0.31 mg/mL DNase I (Roche, from bovine pancreas, grade II) in RPMI 1640 (supplemented with 10% FCS). Single‐cell suspensions from the LPL fraction of SI, kidneys, spleen, LNs, and liver were prepared by mechanical disruption via passing over a 70 μm cell strainer. The isolated lymphocytes from liver, kidneys, SI IEL, and LPL were purified by density centrifugation on a 60%/40% Percoll gradient (GE Healthcare). BM was isolated from tibia and femur by crushing the bones in PBS or flushing them with PBS. Single‐cell suspensions of BM were obtained by passing through a 70 μm cell strainer. Contaminating erythrocytes were removed using RBC lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA).
Flow cytometry
Cells were incubated with antibodies and tetramers for 30 min at 4°C and washed with PBS (supplemented with 0.5 % [v/v] FCS) to remove unbound reagents. Antibodies were purchased from BioLegend, eBioscience, BD Biosciences, or Thermo Fisher Scientific as listed in Table 1. H‐2 Db KAVYNFATC (GP33) tetramers (kindly provided by R. Arens, Leiden University Medical Center, Leiden) were used to detect LCMV‐specific CD8+ T cells. These cells are referred to as virus‐specific or antigen‐experienced CD8+ T cells in the context of LCMV infection. Exclusion of dead cells was performed with the live or dead fixable near‐IR dead cell stain kit (Thermo Fisher Scientific). Samples were acquired on LSR Fortessa or FACSymphony flow cytometers (BD Biosciences), and data were analyzed using FlowJo V10 software (Tree Star). Cell sorting was performed using an Aria III (BD Biosciences). The analyses were performed in accordance with the guidelines for the use of flow cytometry and cell sorting in immunological studies [38].
Table 1.
List of antibodies
| Antibody | Clone | Supplier | Catalogue No. |
|---|---|---|---|
| CD103 | 2E7 | BioLegend | 121408 |
| CD103 | M290 | BD Biosciences | 557495 |
| CD127 | A7R34 | BioLegend | 135027 |
| CD3 | 17A2 | eBioscience | 56‐0032‐82 |
| CD4 | GK1.5 | eBioscience | 11‐0041‐85 |
| CD4 | RM4‐5 | Thermo Fisher Scientific | Q10092 |
| CD44 | IM7 | BD Biosciences | 564392 |
| CD44 | IM7 | BioLegend | 103030 |
| CD45.1 | A20 | BioLegend | 110714, 110706 |
| CD45.2 | 104 | eBioscience | 56‐0454 |
| CD62L | MEL‐14 | BioLegend | 104441 |
| CD69 | H1.2F3 | BD Biosciences | 564684 |
| CD69 | H1.2F3 | BioLegend | 104506 |
| CD69 | H1.2F3 | eBioscience | 25‐0691 |
| CD8α | 53‐6.7 | BD Biosciences | 563786 |
| CD8α | 53‐6.7 | BioLegend | 100734 |
| CD8α | 53‐6.7 | eBioscience | 56‐0081‐82 |
| KLRG1 | 2F1 | BD Biosciences | 740279 |
| KLRG1 | 2F1 | BioLegend | 138416 |
| TCRβ | H57‐597 | BioLegend | 109224 |
DNA isolation
Memory CD8+ T‐cell populations were FACS‐sorted from the liver and SI IEL of LCMV‐infected HobitWT/CRE and HobitKO/CRE × Blimpflox/flox mice based on TCRβ+ CD8+ expression and separated into Hobit+ and Hobit‐ fractions using tdTomato expression. Control naïve CD8+ T cells (CD44‐ CD62L+) were isolated from the spleen of HobitWT/CRE mice. Sorted cells were lysed overnight in lysis buffer (100 mM Tris‐HCl; 5 mM EDTA pH 8.0; 0.2% SDS; 200 mM NaCl, 200 μg/mL proteinase K) at 56°C in a rotator set at 400 rpm. The lysates were centrifuged (13 000 rpm, 30 min, RT) and the supernatants were mixed with an equal volume of isopropanol to precipitate the genomic DNA. After centrifugation (13 000 rpm, 15 min, RT), the DNA pellet was washed in 70% (v/v) ethanol (13 000 rpm, 15 min, RT). After air drying, the DNA pellet was dissolved in ddH2O (37°C, 400 rpm, ≥30 min) for analysis by PCR.
PCR analysis
A double set of primers, Blimp‐1 deleted (Blimp1DEL) and Blimp nondeleted (Blimp1ND), was used to assess the efficiency of the Hobit‐driven Cre recombinase to delete Blimp‐1 (Table 2) [8]. The expected amplicon sizes of the PCR for Blimp1DEL and Blimp1ND are provided in Table 2. The amplified material was loaded on a 2% agarose gel, separated by gel electrophoresis (100 V, Mupid‐One, BioRad Sub‐Cell GT) and imaged using the InGenius LHR Gel Imagining System (Syngene) to examine CRE recombinase‐driven deletion of the Blimp‐1 locus.
Table 2.
List of primers for identification of Blimp‐1 gene deletion
| Gene | Primer | Sequence | Product | Amplicon size (bp) |
|---|---|---|---|---|
| Blimp1ND | Floxed non deleted Fw | GGCAAGATCAAGTATGAGTGC | Nondeleted | 765 |
| Floxed common Rv | TGAGTAGTCACAGAGTACCCA | WT | 611 | |
| Blimp1DEL | Floxed deleted Fw | AGGTGTCTAGCCTTTGTATTTG | Deleted | 646 |
| Floxed common Rv | TGAGTAGTCACAGAGTACCCA |
RNA‐seq analysis
Previously published and normalized RNA‐seq data of sorted murine CD8+ T‐cell subsets after LCMV infection [17] and after Listeria monocytogenes OVA [23] was analyzed for the expression of Prdm1, and the indicated panels of S1P receptors and chemokine receptors at day 8 p.i. and/or day >30.
Quantitative PCR analysis
To perform qPCR analysis, RNA was isolated using Trizol according to the manufacturer's instructions (Life Technologies). RNA was synthesized into cDNA using iScript cDNA Synthesis Kit (BioRad). qPCR was run on a StepOne Plus (Applied Biosystems) using FAST SYBR Green Master Mix (Applied Biosystems). The following primer pairs were used: S1pr1 (forward: 5′‐GTGTAGACCCAGAGTCCTGCG‐3′, reverse: 5′‐AGCTTTTCCTTGGCTGGAGAG‐3′), Ccr7 (forward: 5′‐ CAGCCTTCCTGTGTGATTTCTACA‐3′, reverse: 5′‐ACCACCAGCACGTTTTTCCT‐3′), and Hprt (forward: 5′‐TGAAGAGCTACTGTAATGATCAGTCAAC‐3′, reverse: 5′‐AGCAAGCTTGCAACCTTAACCA‐3′). Expression was normalized using Hprt and the expression was quantified relative to expression in naïve WT CD8 T cells set to 1.
Statistical analysis
Statistical analysis was performed using Prism 8 (GraphPad). Statistical significance was calculated using the unpaired Student's t test for groups that were normally distributed, and using the Mann‐Whitney U test for groups that were not normally distributed. For paired samples, paired two‐tailed Student's t test was employed. For comparison of more than two groups, one‐way ANOVA was used. Unless otherwise indicated, differences were not statistically significant. p values of <0.05 were considered statistically significant (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Author contributions
L.P.V., R.L.R.E.T., A.B.C., H.A., R.A.W.v.L., R.S., and K.P.J.M.v.G. designed the experiments. L.P.V., R.L.R.E.T., A.B.C, H.A., N.A.M.K., A.A.B., and F.M.B. performed the experiments. L.P.V., R.L.R.E.T., A.B.C., and H.A. analyzed the data. L.P.V. and K.P.J.M.v.G. wrote the manuscript. All of the authors critically revised the manuscript for important intellectual content.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202149665.
Abbreviations
- IEL
intraepithelial lymphocytes
- LPL
lamina propria lymphocytes
- MPECs
memory precursor effector cells
- pLN
peripheral LNs
- p.i.
postinfection
- SI
the small intestine
- SLECs
short‐lived effector cells
- Tcm
central memory CD8+ T cells
- Tem
effector memory CD8+ T cells
- Trm
tissue‐resident memory CD8+ T cells
Supporting information
Supporting Information
Acknowledgements
We would like to thank the members of the van Gisbergen laboratory and the Department of Hematopoiesis for fruitful discussions. We thank Dr. Ramon Arens (Leiden University Medical Center) for providing LCMV Db GP33 tetramers. L.P.V. and K.P.J.M.v.G. were supported by Vidi grant 917.13.338 from ZonMw and a fellowship of the Landsteiner Foundation of Blood Transfusion Research. R.S. was supported by a Fellowship from the Alexander von Humboldt Foundation and by Veni grant 016.186.116 from ZonMw.
Renske L.R.E. Taggenbrock and Ammarina Beumer‐Chuwonpad contributed equally to this work.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Butz, E. A. and Bevan, M. J. , Massive expansion of antigen‐specific CD8+ T cells during an acute virus infection. Immunity 1998. 8: 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Williams, M. A. and Bevan, M. J. , Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007. 25: 171–192. [DOI] [PubMed] [Google Scholar]
- 3. Joshi, N. S. , Cui, W. , Chandele, A. , Lee, H. K. , Urso, D. R. , Hagman, J. , Gapin, L. et al., Inflammation directs memory precursor and short‐lived effector CD8(+) T cell fates via the graded expression of T‐bet transcription factor. Immunity 2007. 27: 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaech, S. M. , Tan, J. T. , Wherry, E. J. , Konieczny, B. T. , Surh, C. D. and Ahmed, R. , Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long‐lived memory cells. Nat. Immunol. 2003. 4: 1191–1198. [DOI] [PubMed] [Google Scholar]
- 5. Sallusto, F. , Lenig, D. , Forster, R. , Lipp, M. and Lanzavecchia, A. , Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999. 401: 708–712. [DOI] [PubMed] [Google Scholar]
- 6. Gebhardt, T. , Wakim, L. M. , Eidsmo, L. , Reading, P. C. , Heath, W. R. and Carbone, F. R. , Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009. 10: 524–530. [DOI] [PubMed] [Google Scholar]
- 7. Schenkel, J. M. , Fraser, K. A. , Vezys, V. and Masopust, D. , Sensing and alarm function of resident memory CD8(+) T cells. Nat. Immunol. 2013. 14: 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kallies, A. , Xin, A. , Belz, G. T. and Nutt, S. L. , Blimp‐1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity 2009. 31: 283–295. [DOI] [PubMed] [Google Scholar]
- 9. Rutishauser, R. L. , Martins, G. A. , Kalachikov, S. , Chandele, A. , Parish, I. A. , Meffre, E. , Jacob, J. et al., Transcriptional repressor Blimp‐1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity 2009. 31: 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Backer, R. A. , Helbig, C. , Gentek, R. , Kent, A. , Laidlaw, B. J. , Dominguez, C. X. , de Souza, Y. S. et al., A central role for Notch in effector CD8(+) T cell differentiation. Nat. Immunol. 2014. 15: 1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cruz‐Guilloty, F. , Pipkin, M. E. , Djuretic, I. M. , Levanon, D. , Lotem, J. , Lichtenheld, M. G. , Groner, Y. et al., Runx3 and T‐box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 2009. 206: 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang, C. Y. , Best, J. A. , Knell, J. , Yang, E. , Sheridan, A. D. , Jesionek, A. K. , Li, H. S. et al., The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat. Immunol. 2011. 12: 1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Banerjee, A. , Gordon, S. M. , Intlekofer, A. M. , Paley, M. A. , Mooney, E. C. , Lindsten, T. , Wherry, E. J. et al., Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol. 2010. 185: 4988–4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Intlekofer, A. M. , Takemoto, N. , Wherry, E. J. , Longworth, S. A. , Northrup, J. T. , Palanivel, V. R. , Mullen, A. C. et al., Effector and memory CD8+ T cell fate coupled by T‐bet and eomesodermin. Nat. Immunol. 2005. 6: 1236–1244. [DOI] [PubMed] [Google Scholar]
- 15. Ichii, H. , Sakamoto, A. , Kuroda, Y. and Tokuhisa, T. , Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J. Immunol. 2004. 173: 883–891. [DOI] [PubMed] [Google Scholar]
- 16. Ji, Y. , Pos, Z. , Rao, M. , Klebanoff, C. A. , Yu, Z. , Sukumar, M. , Reger, R. N. et al., Repression of the DNA‐binding inhibitor Id3 by Blimp‐1 limits the formation of memory CD8+ T cells. Nat. Immunol. 2011. 12: 1230–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parga‐Vidal, L. , Behr, F. M. , Kragten, N. A. M. , Nota, B. , Wesselink, T. H. , Kavazović, I. , Covill, L. E. et al., Hobit identifies tissue‐resident memory T cell precursors that are regulated by Eomes. Sci. Immunology 2021. 6: eabg3533. [DOI] [PubMed] [Google Scholar]
- 18. Iborra, S. , Martinez‐Lopez, M. , Khouili, S. C. , Enamorado, M. , Cueto, F. J. , Conde‐Garrosa, R. , Del Fresno, C. et al., Optimal generation of tissue‐resident but not circulating memory T cells during viral infection requires crosspriming by DNGR‐1(+) dendritic cells. Immunity 2016. 45: 847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kok, L. , Dijkgraaf, F. E. , Urbanus, J. , Bresser, K. , Vredevoogd, D. W. , Cardoso, R. F. , Perié, L. et al., A committed tissue‐resident memory T cell precursor within the circulating CD8+ effector T cell pool. J. Exp. Med. 2020. 217: e20191711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Milner, J. J. , Toma, C. , He, Z. , Kurd, N. S. , Nguyen, Q. P. , McDonald, B. , Quezada, L. et al., Heterogenous populations of tissue‐resident CD8(+) T cells are generated in response to infection and malignancy. Immunity 2020. 52: 808–824.e807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kurd, N. S. , He, Z. , Louis, T. L. , Milner, J. J. , Omilusik, K. D. , Jin, W. , Tsai, M. S. et al., Early precursors and molecular determinants of tissue‐resident memory CD8+T lymphocytes revealed by single‐cell RNA sequencing. Science Immunol. 2020. 5: eaaz6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mackay, L. K. , Minnich, M. , Kragten, N. A. , Liao, Y. , Nota, B. , Seillet, C. , Zaid, A. et al., Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 2016. 352: 459–463. [DOI] [PubMed] [Google Scholar]
- 23. Behr, F. M. , Parga‐Vidal, L. , Kragten, N. A. M. , van Dam, T. J. P. , Wesselink, T. H. , Sheridan, B. S. , Arens, R. et al., Tissue‐resident memory CD8(+) T cells shape local and systemic secondary T cell responses. Nat. Immunol. 2020. 21: 1070–1081. [DOI] [PubMed] [Google Scholar]
- 24. Kragten, N. A. M. , Behr, F. M. , Vieira Braga, F. A. , Remmerswaal, E. B. M. , Wesselink, T. H. , Oja, A. E. , Hombrink, P. et al., Blimp‐1 induces and Hobit maintains the cytotoxic mediator granzyme B in CD8 T cells. Eur. J. Immunol. 2018. 48: 1644–1662. [DOI] [PubMed] [Google Scholar]
- 25. Shin, H. , Blackburn, S. D. , Intlekofer, A. M. , Kao, C. , Angelosanto, J. M. , Reiner, S. L. and Wherry, E. J. , A role for the transcriptional repressor Blimp‐1 in CD8(+) T cell exhaustion during chronic viral infection. Immunity 2009. 31: 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matloubian, M. , Lo, C. G. , Cinamon, G. , Lesneski, M. J. , Xu, Y. , Brinkmann, V. , Allende, M. L. et al., Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004. 427: 355–360. [DOI] [PubMed] [Google Scholar]
- 27. Mackay, L. K. , Rahimpour, A. , Ma, J. Z. , Collins, N. , Stock, A. T. , Hafon, M. L. , Vega‐Ramos, J. et al., The developmental pathway for CD103(+)CD8+ tissue‐resident memory T cells of skin. Nat. Immunol. 2013. 14: 1294–1301. [DOI] [PubMed] [Google Scholar]
- 28. Shiow, L. R. , Rosen, D. B. , Brdickova, N. , Xu, Y. , An, J. , Lanier, L. L. , Cyster, J. G. et al., CD69 acts downstream of interferon‐alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 2006. 440: 540–544. [DOI] [PubMed] [Google Scholar]
- 29. Zhou, X. , Yu, S. , Zhao, D. M. , Harty, J. T. , Badovinac, V. P. and Xue, H. H. , Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 2010. 33: 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Gisbergen, K. P. , Kragten, N. A. , Hertoghs, K. M. , Wensveen, F. M. , Jonjic, S. , Hamann, J. , Nolte, M. A. et al., Mouse Hobit is a homolog of the transcriptional repressor Blimp‐1 that regulates NKT cell effector differentiation. Nat. Immunol. 2012. 13: 864–871. [DOI] [PubMed] [Google Scholar]
- 31. Vieira Braga, F. A. , Hertoghs, K. M. , Kragten, N. A. , Doody, G. M. , Barnes, N. A. , Remmerswaal, E. B. , Hsiao, C. C. et al., Blimp‐1 homolog Hobit identifies effector‐type lymphocytes in humans. Eur. J. Immunol. 2015. 45: 2945–2958. [DOI] [PubMed] [Google Scholar]
- 32. Martins, G. A. , Cimmino, L. , Shapiro‐Shelef, M. , Szabolcs, M. , Herron, A. , Magnusdottir, E. and Calame, K. , Transcriptional repressor Blimp‐1 regulates T cell homeostasis and function. Nat. Immunol. 2006. 7: 457–465. [DOI] [PubMed] [Google Scholar]
- 33. Kallies, A. , Hawkins, E. D. , Belz, G. T. , Metcalf, D. , Hommel, M. , Corcoran, L. M. , Hodgkin, P. D. et al., Transcriptional repressor Blimp‐1 is essential for T cell homeostasis and self‐tolerance. Nat. Immunol. 2006. 7: 466–474. [DOI] [PubMed] [Google Scholar]
- 34. Mackay, L. K. , Wynne‐Jones, E. , Freestone, D. , Pellicci, D. G. , Mielke, L. A. , Newman, D. M. , Braun, A. et al., T‐box transcription factors combine with the cytokines TGF‐beta and IL‐15 to control tissue‐resident memory T cell fate. Immunity 2015. 43: 1101–1111. [DOI] [PubMed] [Google Scholar]
- 35. Milner, J. J. , Toma, C. , Yu, B. , Zhang, K. , Omilusik, K. , Phan, A. T. , Wang, D. et al., Runx3 programs CD8+ T cell residency in non‐lymphoid tissues and tumours. Nature 2017. 552: 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hombrink, P. , Helbig, C. , Backer, R. A. , Piet, B. , Oja, A. E. , Stark, R. , Brasser, G. et al., Programs for the persistence, vigilance and control of human CD8(+) lung‐resident memory T cells. Nat. Immunol. 2016. 17: 1467–1478. [DOI] [PubMed] [Google Scholar]
- 37. Kallies, A. , Hasbold, J. , Tarlinton, D. M. , Dietrich, W. , Corcoran, L. M. , Hodgkin, P. D. and Nutt, S. L. , Plasma cell ontogeny defined by quantitative changes in blimp‐1 expression. J. Exp. Med. 2004. 200: 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cossarizza, A. , Chang, H. D. , Radbruch, A. , Abrignani, S. , Addo, R. , Akdis, M. , Andra, I. et al., Guidelines for the use of flow cytometry and cell sorting in immunological studies (third edition). Eur. J. Immunol. 2021. 51: 2708–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.