

Abstract
Cycloaddition‐dehydration involving a BNBN‐butadiene analogue at the bay region of a dibenzoperylene and a non‐enolizable aldehyde provides a novel strategy for incorporation of the oxadiazadiborinane (B2N2CO) ring into the scaffold of a polycyclic aromatic hydrocarbon resulting in highly emissive compounds.
Keywords: boron, cycloaddition, dibenzoperylene, nitrogen, polycycles
We report the first acid‐catalyzed heteroatom cycloaddition at the (BN)2 bay region of a dibenzoperylene with different aldehydes. This new method allows the synthesis of heteroatom‐doped polycyclic aromatic hydrocarbons (PAHs) and the formation of 1,3,5,2,4‐oxadiazadiborinane (B2N2CO) rings.
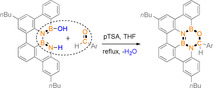
The introduction of heteroatoms is a very popular strategy to modify the electronic properties of polycyclic aromatic hydrocarbons (PAHs) for application as organic electronic materials. [1] In particular, the isoelectronic and isosteric exchange of carbon‐carbon (CC) by boron and nitrogen pairs (BN) opened a huge field of novel organic materials since the report of the first BN doped PAH in 1958. [2] In addition to BN substitution, the incorporation of oxygen atoms is a useful approach to develop new compounds for material applications and catalysis.[ 1 , 3 ]
Among the available methods for synthesizing 1,2‐azaborine containing PAHs, the electrophilic borylation, introduced by Dewar [2a] and optimized further, is attractive as both B−N and B−C bonds are constructed in one synthetic step (Scheme 1 a).[ 2h , 3a , 4 ] Alternatively, the coupling of carbon‐carbon (CC) bonds around existing BN units has occasionally been employed, e.g., by Piers, [5] Bonifazi,[ 3b , 6 ] and our group (Scheme 1 b). [7] The Wagner group [8] described the Au catalyzed NC bond formation as the final step of the synthesis of a doubly BN substituted perylene (Scheme 1 c).[ 8 , 9 ]
Scheme 1.
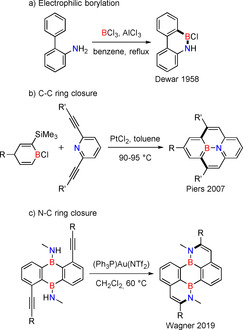
Selected representative examples of available methods for synthesis of 1,2‐azaborine containing PAHs.
Perylenes and dibenzoperylenes with BN doping have been synthesized by Wagner, Bonifazi and our group.[ 3b , 4j , 8 , 10 ] We have developed a multistep synthesis of a dibenzoperylene 1 that features a BNBN unit at its bay region. [4j] It would be interesting to use this heterodiene as functional group for widening the chemical space in heteroatom doped PAH synthesis by cycloaddition reactions. Cycloaddition reactions at the bay region of perylene are well known (Scheme 2), [11] and they were even suggested for growth of carbon nanotubes by reaction of armchair rims with acetylene. [12] However, the analogous cycloaddition reaction of heteroatom bay region substituted PAHs are unknown. The closest to this are cycloaddition reactions of diazadiboretidines, which are the quite reactive BN analogs of antiaromatic cyclobutadiene. Paetzold postulated hetero‐Diels–Alder reactions of diazadiboretidines with ketones and aldehydes forming 1,3,5,2,4‐oxadiazadiborinan (B2N2CO) rings under ring extension (Scheme 2). [13]
Scheme 2.
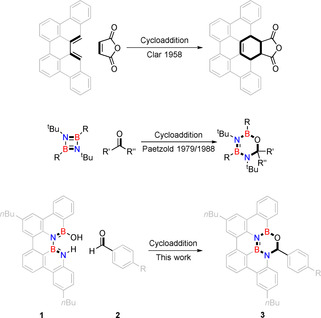
Cycloaddition reactions of dibenzoperylene (top), diazadiboretidines (center) and BN bay region doped dibenzoperylene (bottom).
The cycloaddition of A (a model for 1 without the n‐Bu groups) with typical electron rich (TCNE, TME) or electron poor dienophiles (methyl vinyl ether, see Figure 1) is unfavorable as revealed by quantum chemical calculations (M062X/6–311+G**), even after dehydration. However, the reaction A → C + H2O becomes mildly exergonic if the polar carbonyl group of benzaldehyde is employed as dienophile. For reactions with other hetero dienophiles, see Supporting Information (SI).
Figure 1.
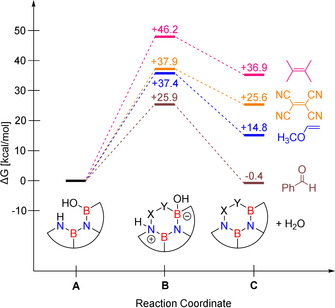
Free energy differences for different dienophiles calculated at the M062X/6–311+G** level of theory (T=298.15 K); (BN)2 dibenzoperylene scaffolds A, B and C are abbreviated for clarity.
Heating 1 and benzaldehyde in THF even for a long period of time does not result in any reaction. To facilitate the dehydration that is necessary to have an exergonic reaction, we added an excess of para‐toluenesulfonic acid (pTSA) in order to induce an acid catalyzed dehydration. To remove emerging water from equilibrium, we heated the mixture in the presence of molecular sieve in a Soxhlet extractor. Then, a slow reaction proceeds and after 5 days of reaction time, the product 3 a, which is stable against oxygen and moisture, can be isolated in a good yield of 67 % by column chromatography.
Compound 3 a (Scheme 3) can be characterized by multinuclear (1H, 13C, 11B) and correlated NMR spectroscopy (2D spectra) as well as by high resolution mass spectrometry. Notably, the 1H NMR shows one singlet for the benzylic proton in the ON2B2C‐ring that experiences significant deshielding and resonates at 7.02 ppm (see SI, Figure S1). The 11B spectrum of 3 a shows a broad signal at 28.3 ppm for both boron atoms (see SI, Figure S3).
Scheme 3.
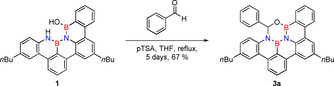
Heteroatom cycloaddition reaction with benzaldehydes; 1 equiv 1, 10 equiv benzaldehyde, 10 equiv pTSA, THF, Soxhlet extractor with 3 Å molecular sieve, reflux, 5 days.
Single crystals for X‐ray crystallography were obtained by recrystallization from THF (for crystallographic data see SI). Compound 3 a crystallizes in the triclinic space group with two enantiomeric molecules due to the asymmetric sp3 carbon in the B2N2CO ring (see Figure 2). The dibenzoperylene motif is responsible for intermolecular π‐π stacking with a distance of 3.5 Å between molecular planes, while the single phenyl groups as well as one butyl group feature towards the next molecular layer. The BN bond lengths average 1.43 Å, slightly shorter than analogous bond lengths in typical BN doped PAHs (1.45–1.47 Å).[ 5a , 14 ] The CO, BO and NC bond length are in good agreement with the literature. [15] The newly formed heterocycle adopts a puckered geometry with a tilt angle of 18.15° involving the BNB and NCO planes. This ring has a sp3 hybridized carbon center and is expected to be non‐aromatic as confirmed by NICS(1) computations (see SI, Figure S40). [16] While the four all‐carbon rings of the PAH backbone have large negative NICS(1), the heteroatom containing rings have small NICS(1) values, reminiscent of the related boroxazine derivative. [3c]
Figure 2.
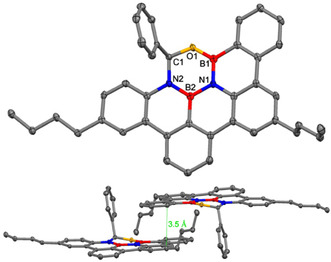
Molecular structure of 3 a (hydrogen atoms omitted for clarity (top), ellipsoids set at 50 % probability; selected bond length of 3 a in Å: O1‐B1 1.376, B1‐N1 1.420, N1‐B2 1.444, B2‐N2 1.422, N2‐C1 1.468, C1‐O1 1.430; packing of 3 a (bottom). [20]
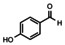
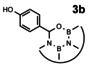
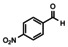
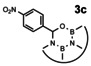
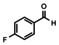
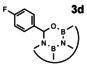
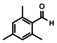
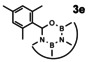
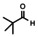
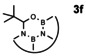
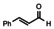
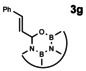
In order to investigate substituent effects, a number of aryl aldehydes were employed in the reaction (Table 1). The reaction time and yield are not affected significantly by the electronic nature of the substituent (Table 1, entries 1–4) as revealed by TLC monitoring of the disappearance of 1. Mesitaldehyde can also be used as a dienophile, but provides lower yields due to steric hindrance (Table 1, entry 5). The product with the sterically more encumbered pivaldehyde can only be detected in low yields (Table 1, entry 6) and with some impurities (see SI Figure S32). The α,β‐unsaturated cinnamaldehyde undergoes this cycloaddition reaction in good yields (Table 1, entry 7) and selectively at the aldehyde function as no product resulting from cycloaddition to the C=C double bond is observed. All dehydrated cycloaddition products are stable against oxygen and moisture and can be purified by column chromatography.
Table 1.
Screening of different dienophiles in cycloaddition reaction with (BN)2‐dibenzoperylene and yield values. Conditions as in Scheme 3.
|
Entry |
Dienophile |
Product |
Yield [%] |
|---|---|---|---|
|
1 |
|
|
67 |
|
2 |
|
|
74 |
|
3 |
|
|
73 |
|
4 |
|
|
80 |
|
5 |
|
|
40 |
|
6 |
|
|
15 |
|
7 |
|
|
77 |
The influence of pTSA on the reagents was investigated by NMR spectroscopy in [D8]THF. A mixture of pTSA and benzaldehyde gives unchanged 1H NMR spectra compared to individual compounds. This observation is in line with the pK a values of pTSA (−2.8) [17] and of protonated benzaldehyde (−7.1). [18] On the other hand, the signal of the OH group of 1 at 9.06 ppm (assignment based on 2D NMR) turns broad at low concentrations of pTSA and becomes weaker and broader with increasing the pTSA concentration, indicating rapid exchange between the OH proton and the acidic proton of pTSA. The NH proton is not affected by the presence of pTSA and its signal remains sharp at 9.78 ppm. These results indicate that the OH group is involved in dynamic proton exchange with pTSA in THF solution. The action of pTSA could result in acid catalyzed dehydration of 1 to give the diazadiboretidine intermediate 4 (Scheme 4). Recall that these species were previously reported to undergo cycloaddition reactions with aldehydes (see Scheme 2).[ 13a , 13b ] Computations on the model system A without the n‐Bu groups at the M062X/6–311+G** level of theory with a continuum model to mimic the THF solvent effect show that formation of 4 is a very high energy process, ΔG(THF)=+81.1 kcal mol−1 (Scheme 4). Therefore, 4 cannot be relevant for the mechanism of the dehydrative cycloaddition reaction. However, neutral A can undergo the concerted cycloaddition with benzaldehyde with a barrier of 30.1 kcal mol−1, and this barrier is changed by at most 0.4 kcal mol−1 in the presence of para substituents (p‐OH, p‐NO2, p‐F) on the aryl group of the aldehyde (Scheme 4). The cycloaddition product B is a very shallow minimum on the potential energy surface as the barrier for the back reaction to the reagents is merely 1–3 kcal mol−1 depending on p‐X (Scheme 4). The pTSA could then cause acid catalyzed dehydration of B to give intermediate D (Scheme 4) that is higher in energy than B by roughly 8 kcal mol−1. The energy barriers of the cycloaddition step and its insensitivity to electronic substituent effects of the aryl aldehydes are in qualitative agreement with the experimental observations that the reaction rate is independent of the benzaldehyde concentration but that addition of acid is essential. Alternatively, the OH protonated form of 1, a borenium ion, could undergo the cycloaddition presumably with a decreased activation barrier, but the Gibbs free energy (T = 339 K) of this borenium ion is higher than that of D by roughly 12 kcal mol−1.
Scheme 4.
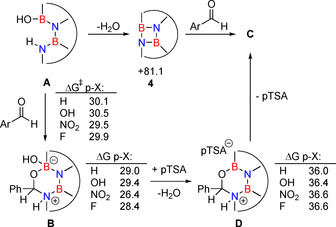
Possible mechanisms as computed at the M062X/6–311+G** level of theory in THF solution (T=339 K). Energy data is given in kcal mol−1.
The absorption and fluorescence spectra of compounds 3 a–e and 3 g were recorded in dichloromethane solutions (see Figure 3). The spectra resemble each other and are quite similar to those of 1. The substituents in 3 b–e or the presence of the C=C bridge in 3 g turned out to result in shifts of peak maxima of 1–2 nm. The Stokes shifts are small throughout (600 cm−1–755 cm−1, see SI for details). The fluorescence quantum yields of all products except for 3 c is 78 % and higher using 9,10‐diphenylanthracene in ethanol as reference (see SI). The cycloaddition product with p‐nitrobenzaldehyde 3 c only shows a low fluorescence quantum yield of 7 %. The decrease of the fluorescence quantum yield of aromatic compounds upon introduction of a nitro groups is well known. [19]
Figure 3.
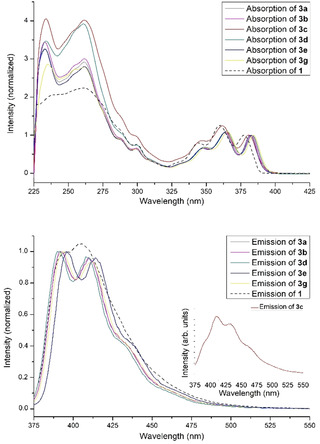
Absorption (normalized on maxima with longest wavelength, top) and fluorescence spectra (λ ex=347 nm, normalized on maxima with shortest wavelength, bottom) of the cycloaddition products in dichloromethane including absorption and fluorescence spectra of 1 (dashed line) for comparison. Inset: fluorescence spectrum of 3 c (Φ fl=0.07).
The electrochemical properties of 3 a were investigated by cyclic voltammetry. At scan rates greater than 2 V s−1, we find a quasi‐reversible reduction signal at E 0=−2.724 V vs. Fc/Fc+. At slow scan rates, the associated oxidation peak of this reduction signal is less pronounced, suggesting a slow chemical follow‐up reaction of the reduced species of 3 a (EC mechanism). Two further signals with peak potentials at approx. −3.0 V and −3.1 V vs. Fc/Fc+ were found in the forward scan at 50 mV s−1 within our electrochemical window (see Supporting Information for details).
In summary, we described the first cycloaddition‐dehydration reaction of a BNBN bay region doped dibenzoperylene with different non‐enolizable aldehydes forming new BN doped PAHs with an 1,3,5,2,4‐oxadiazadiborinan (B2N2CO) ring. The reaction is robust towards the electronic structure of the aldehydes, but is affected by steric effects. The novel reaction products, PAH with an embedded oxadiazadiborinane ring, show very high fluorescence quantum yields (>78 %), which make this conjugation of BN‐doped PAH with aldehydes a promising strategy towards enlarging the chemical scope in heteroatom doped PAH chemistry.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank Dr. P. Wagner for helpful discussions. This work was supported by the Vector Foundation. The authors acknowledge support by the state of Baden‐Württemberg through bwHPC and the German Research Foundation (DFG) through grant no INST 40/575‐1 FUGG (JUSTUS 2 cluster). Open access funding enabled and organized by Projekt DEAL.
M. Fingerle, J. Dingerkus, H. Schubert, K. M. Wurst, M. Scheele, H. F. Bettinger, Angew. Chem. Int. Ed. 2021, 60, 15798.
References
- 1. Stępień M., Gońka E., Żyła M., Sprutta N., Chem. Rev. 2017, 117, 3479–3716. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Dewar M. J. S., Kubba V. P., Pettit R., J. Chem. Soc. 1958, 3073–3076; [Google Scholar]
- 2b. Bosdet M. J. D., Piers W. E., Can. J. Chem. 2009, 87, 8–29; [Google Scholar]
- 2c. Campbell P. G., Marwitz A. J. V., Liu S.-Y., Angew. Chem. Int. Ed. 2012, 51, 6074–6092; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6178–6197; [Google Scholar]
- 2d. Wang X.-Y., Wang J.-Y., Pei J., Chem. Eur. J. 2015, 21, 3528–3539; [DOI] [PubMed] [Google Scholar]
- 2e. Helten H., Chem. Eur. J. 2016, 22, 12972–12982; [DOI] [PubMed] [Google Scholar]
- 2f. Morgan M. M., Piers W. E., Dalton Trans. 2016, 45, 5920–5924; [DOI] [PubMed] [Google Scholar]
- 2g. Giustra Z. X., Liu S.-Y., J. Am. Chem. Soc. 2018, 140, 1184–1194; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2h. Pati P. B., Jin E., Kim Y., Kim Y., Mun J., Kim S. J., Kang S. J., Choe W., Lee G., Shin H.-J., Park Y. S., Angew. Chem. Int. Ed. 2020, 59, 14891–14895; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 15001–15005; [Google Scholar]
- 2i. Scholz A. S., Massoth J. G., Bursch M., Mewes J.-M., Hetzke T., Wolf B., Bolte M., Lerner H.-W., Grimme S., Wagner M., J. Am. Chem. Soc. 2020, 142, 11072–11083. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Noda H., Furutachi M., Asada Y., Shibasaki M., Kumagai N., Nat. Chem. 2017, 9, 571–577; [DOI] [PubMed] [Google Scholar]
- 3b. Dosso J., Battisti T., Ward B. D., Demitri N., Hughes C. E., Williams P. A., Harris K. D. M., Bonifazi D., Chem. Eur. J. 2020, 26, 6608–6621; [DOI] [PubMed] [Google Scholar]
- 3c. Fingerle M., Bettinger H. F., Chem. Commun. 2020, 56, 3847–3850. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Dewar M. J. S., Poesche W. H., J. Am. Chem. Soc. 1963, 85, 2253–2256; [Google Scholar]
- 4b. Dewar M. J. S., Poesche W. H., J. Org. Chem. 1964, 29, 1757–1762; [Google Scholar]
- 4c. Hatakeyama T., Hashimoto S., Seki S., Nakamura M., J. Am. Chem. Soc. 2011, 133, 18614–18617; [DOI] [PubMed] [Google Scholar]
- 4d. Hatakeyama T., Hashimoto S., Oba T., Nakamura M., J. Am. Chem. Soc. 2012, 134, 19600–19603; [DOI] [PubMed] [Google Scholar]
- 4e. Wang X., Zhang F., Schellhammer K. S., Machata P., Ortmann F., Cuniberti G., Fu Y., Hunger J., Tang R., Popov A. A., Berger R., Müllen K., Feng X., J. Am. Chem. Soc. 2016, 138, 11606–11615; [DOI] [PubMed] [Google Scholar]
- 4f. Fingerle M., Maichle-Mössmer C., Schundelmeier S., Speiser B., Bettinger H. F., Org. Lett. 2017, 19, 4428–4431; [DOI] [PubMed] [Google Scholar]
- 4g. Yang D.-T., Nakamura T., He Z., Wang X., Wakamiya A., Peng T., Wang S., Org. Lett. 2018, 20, 6741–6745; [DOI] [PubMed] [Google Scholar]
- 4h. Fu Y., Zhang K., Dmitrieva E., Liu F., Ma J., Weigand J. J., Popov A. A., Berger R., Pisula W., Liu J., Feng X., Org. Lett. 2019, 21, 1354–1358; [DOI] [PubMed] [Google Scholar]
- 4i. Numano M., Nagami N., Nakatsuka S., Katayama T., Nakajima K., Tatsumi S., Yasuda N., Hatakeyama T., Chem. Eur. J. 2016, 22, 11574–11577; [DOI] [PubMed] [Google Scholar]
- 4j. Fingerle M., Stocker S., Bettinger H. F., Synthesis 2019, 51, 4147–4152; [Google Scholar]
- 4k. Sun Z., Yi C., Liang Q., Bingi C., Zhu W., Qiang P., Wu D., Zhang F., Org. Lett. 2020, 22, 209–213; [DOI] [PubMed] [Google Scholar]
- 4l. Wang X.-Y., Zhuang F.-D., Wang X.-C., Cao X.-Y., Wang J.-Y., Pei J., Chem. Commun. 2015, 51, 4368–4371; [DOI] [PubMed] [Google Scholar]
- 4m. Li G., Zhao Y., Li J., Cao J., Zhu J., Sun X. W., Zhang Q., J. Org. Chem. 2015, 80, 196–203. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Bosdet M. J. D., Piers W. E., Sorensen T. S., Parvez M., Angew. Chem. Int. Ed. 2007, 46, 4940–4943; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5028–5031; [Google Scholar]
- 5b. Neue B., Araneda J. F., Piers W. E., Parvez M., Angew. Chem. Int. Ed. 2013, 52, 9966–9969; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 10150–10153; [Google Scholar]
- 5c. Bosdet M. J. D., Jaska C. A., Piers W. E., Sorensen T. S., Parvez M., Org. Lett. 2007, 9, 1395–1398; [DOI] [PubMed] [Google Scholar]
- 5d. Jaska C. A., Piers W. E., McDonald R., Parvez M., J. Org. Chem. 2007, 72, 5234–5243. [DOI] [PubMed] [Google Scholar]
- 6. Dosso J., Tasseroul J., Fasano F., Marinelli D., Biot N., Fermi A., Bonifazi D., Angew. Chem. Int. Ed. 2017, 56, 4483–4487; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4554–4558. [Google Scholar]
- 7. Krieg M., Reicherter F., Haiss P., Ströbele M., Eichele K., Treanor M.-J., Schaub R., Bettinger H. F., Angew. Chem. Int. Ed. 2015, 54, 8284–8286; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8402–8404. [Google Scholar]
- 8. Kaehler T., Bolte M., Lerner H.-W., Wagner M., Angew. Chem. Int. Ed. 2019, 58, 11379–11384; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 11501–11506. [Google Scholar]
- 9. Alonso F., Beletskaya I. P., Yus M., Chem. Rev. 2004, 104, 3079–3160. [DOI] [PubMed] [Google Scholar]
- 10. Müller M., Behnle S., Maichle-Mössmer C., Bettinger H. F., Chem. Commun. 2014, 50, 7821–7823. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Clar E., Zander M., J. Chem. Soc. 1957, 4616–4619; [Google Scholar]
- 11b. Clar E., Zander M., J. Chem. Soc. 1958, 1861–1865; [Google Scholar]
- 11c. Lang K. F., Buffleb H., Kalowy J., Chem. Ber. 1960, 93, 303–309; [Google Scholar]
- 11d. Tokita S., Hiruta K., Ishikawa S., Kitahara K., Nishi H., Synthesis 1982, 854–855; [Google Scholar]
- 11e. Tokita S., Hiruta K., Kitahara K., Nishi H., Synthesis 1982, 229–231; [Google Scholar]
- 11f. Tokita S., Hiruta K., Yaginuma Y., Ishikawa S., Nishi H., Synthesis 1984, 270–271; [Google Scholar]
- 11g. Głodek M., Makal A., Plażuk D., J. Org. Chem. 2018, 83, 14165–14174; [DOI] [PubMed] [Google Scholar]
- 11h. Kurpanik A., Matussek M., Szafraniec-Gorol G., Filapek M., Lodowski P., Marcol-Szumilas B., Ignasiak W., Małecki J. G., Machura B., Małecka M., Danikiewicz W., Pawlus S., Krompiec S., Chem. Eur. J. 2020, 26, 12150–12157. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Fort E. H., Donovan P. M., Scott L. T., J. Am. Chem. Soc. 2009, 131, 16006–16007; [DOI] [PubMed] [Google Scholar]
- 12b. Fort E. H., Scott L. T., J. Mater. Chem. 2011, 21, 1373–1381. [Google Scholar]
- 13.
- 13a. Paetzold P., Richter A., Thijssen T., Wuertenberg S., Chem. Ber. 1979, 112, 3811–3827; [Google Scholar]
- 13b. Schreyer P., Paetzold P., Boese R., Chem. Ber. 1988, 121, 195–205; [Google Scholar]
- 13c. Paetzold P., Kiesgen J., Krahé K., Meier H.-U., Boese R., Z. Naturforsch. B 1991, 46, 853–860. [Google Scholar]
- 14. Liu Z., Marder T. B., Angew. Chem. Int. Ed. 2008, 47, 242–244; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 248–250. [Google Scholar]
- 15. Allen F. H., Watson D. G., Brammer L., Orpen A. G., Taylor R., International Tables for Crystallography, IUCr, Chester, 2006, pp. 790–811. [Google Scholar]
- 16.
- 16a. Schleyer P. v. R., Maerker C., Dransfeld A., Jiao H., van Eikema Hommes N. J. R., J. Am. Chem. Soc. 1996, 118, 6317–6318; [DOI] [PubMed] [Google Scholar]
- 16b. Schleyer P. v. R., Jiao H., van Eikema Hommes N. J. R., Malkin V. G., Malkina O. L., J. Am. Chem. Soc. 1997, 119, 12669–12670. [Google Scholar]
- 17. Guthrie J. P., Can. J. Chem. 1978, 56, 2342–2354. [Google Scholar]
- 18. de Lijser H. J. P., Rangel N. A., J. Org. Chem. 2004, 69, 8315–8322. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Catalfo A., Serrentino M. E., Librando V., Perrini G., de Guidi G., Appl. Spectrosc. 2008, 62, 1233–1237; [DOI] [PubMed] [Google Scholar]
- 19b. Reichardt C., Vogt R. A., Crespo-Hernández C. E., J. Chem. Phys. 2009, 131, 224518; [DOI] [PubMed] [Google Scholar]
- 19c. Mohammed O. F., Vauthey E., J. Phys. Chem. A 2008, 112, 3823–3830; [DOI] [PubMed] [Google Scholar]
- 19d. Vogt R. A., Reichardt C., Crespo-Hernández C. E., J. Phys. Chem. A 2013, 117, 6580–6588. [DOI] [PubMed] [Google Scholar]
- 20. Deposition Number 2050368 contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service www.ccdc.cam.ac.uk/structures.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary