

Abstract
Background
Treatment choices for individual patients with an inborn bleeding disorder are increasingly challenging due to increasing options and rising costs for society. We have initiated an integrated interdisciplinary national research program.
Objectives
The SYMPHONY consortium strives to orchestrate personalized treatment in patients with an inborn bleeding disorder, by unraveling the mechanisms behind interindividual variations of bleeding phenotype.
Patients
The SYMPHONY consortium will investigate patients with an inborn bleeding disorder, both diagnosed and not yet diagnosed.
Results
Research questions are categorized under the themes: (1) diagnosis, (2) treatment, and (3) fundamental research, and consist of work packages addressing specific domains. Importantly, collaborations between patients and talented researchers from different areas of expertise promise to augment the impact of the SYMPHONY consortium, leading to unique interactions and intellectual property.
Conclusions
SYMPHONY will perform research on all aspects of care, treatment individualization in patients with inborn bleeding disorders, as well as diagnostic innovations and results of molecular genetics and cellular model technology with regard to the hemostatic process. We believe that these research investments will lead to health‐care innovations with long‐term clinical and societal impact.
This consortium has been made possible by a governmental, competitive grant from the Netherlands Organization for Scientific Research (NWO) within the framework of the NWA‐ORC Call grant agreement NWA.1160.18.038.
Keywords: bleeding disorders, cellular models, personalized treatment, pharmacokinetics, proteomics, value‐based health care
Essentials.
Treatment choices for individual patients with an inborn bleeding disorder are increasingly challenging due to increasing options and rising costs for society.
The SYMPHONY consortium strives to orchestrate personalized treatment in patients with an inborn bleeding disorder, diagnosed and not yet diagnosed, by unraveling the mechanisms behind interindividual variations of bleeding phenotype.
Greater insight into these mechanisms will support individualization of treatment, potentially leading to regimens based more on individual bleeding tendency than on diagnosis.
This will support personalization of health‐care innovations and shared decision making with regard to treatment choice, taking societal costs into account.
1. INTRODUCTION
Care for patients with an inborn bleeding disorder has improved drastically in the last decades and has progressed from high morbidity and mortality due to debilitating arthropathy and intracranial or gastrointestinal bleeds, to a normal life expectancy and high quality of life. 1 , 2 The introduction of factor replacement therapy for hemophilia in the 1970s has proved to be life changing and led to the concept of prophylaxis in severely affected patients. 3 , 4 However, long‐term effects of intravenously administered prophylaxis on joint bleeds and joint damage was only shown in a randomized controlled trial in 2007. 5 The capacity to produce recombinant factor concentrates in the 1990s, instead of deriving them from human plasma, further increased safety and quality of care by avoiding viral infections. Recombinant products in vials has made it possible to implement home treatment worldwide, attaining prophylactic trough levels >0.01 IU/ml. Meanwhile, hemophilia treatment has progressed further, being one of the first genetic diseases to develop and implement effective gene therapy. 6 , 7 , 8 , 9 Moreover, extended half‐life factor concentrates and novel subcutaneously administered monoclonal antibodies with a long half‐life and therefore infrequent infusions, have further advanced care and health outcomes for this patient group. 10 , 11
These developments have concomitantly influenced research and treatment modalities in other inborn bleeding disorders concerning both primary and secondary hemostasis, and/or fibrinolysis, such as von Willebrand disease, platelet function disorders, isolated factor deficiencies, fibrinolytic disorders, and bleeding of unknown cause. Historically, the Netherlands has taken a leading position in advancing care for patients with bleeding disorders, initiating multicenter (inter)national clinical studies into symptoms, complications, pharmacokinetic‐guided dosing of medication, and quality of life, 1 , 12 , 13 , 14 , 15 , 16 , 17 , 18 concomitantly also improving diagnostic tests and performing research into the etiology of hemostatic disorders and the development of inhibiting antibodies. 19
Notwithstanding this progress, which has mainly benefited individuals with hemophilia, there are still unmet needs. As the unmet needs described in inborn bleeding disorders may be recognized by other rare diseases, the described approach in this design article may provide a valuable research framework for other fields as well.
Knowledge gaps and unmet needs within the field of hemostasis can be structured according to the following four categories.
First, presently, various novel expensive therapeutic approaches are emerging and becoming standard of care such as gene therapy and subcutaneously administered bispecific monoclonal antibodies. 6 , 11 , 20 Potentially these will further improve patient outcomes but cost and benefit for patients and society are yet unclear. The effectiveness, (long term) side effects, and therefore positioning and optimal use of these new treatments are still to be established. 21 In addition, we are obligated to identify which patients will benefit from which therapeutic approach. These new treatment options make personalized treatment for bleeding disorders not only possible, but mandatory.
Second, treatment strategies with intravenously administered factor concentrates and desmopressin, currently increasingly available in lower resource countries, are suboptimal and lead to both under‐ and overtreatment, as shown by the multicenter OPTI‐CLOT studies, with subsequently either risk of bleeding, thrombosis, and/or excessive costs. 12 , 22 , 23
Third, patient and treatment outcomes are currently not monitored adequately. Systematic documentation of both patient‐reported and patient‐related outcomes and experience measures as well as treatment costs can determine which (future) treatment is best for each individual, taking costs for society into account. 24 , 25 , 26 This is needed to inform decision making by all involved stakeholders, including governmental health‐care institutions and insurance companies.
Finally, despite the growing knowledge over the years there are still significant gaps in our knowledge of the hemostatic process. Knowledge on determinants of interindividual variation in bleeding phenotype is lacking, 27 but is crucial for personalization of treatment according to severity and defining necessity of more innovative, expensive therapies. 28 The limitations in health‐care management of inborn bleeding disorders warrants development of both novel diagnostic tests and investments in fundamental research to identify disease modifiers and more precisely establish therapeutic requirements. Techniques such as proteomic profiling, flow models, and cellular disease models are instrumental to gain more insight.
1.1. An integrated interdisciplinary research program
The SYMPHONY consortium aims to personalize treatment in patients with inborn bleeding disorders, either due to defects in the known pathways (primary hemostasis, secondary hemostasis, and fibrinolysis), or due to not yet identified pathways. To achieve this aim, an integrated interdisciplinary national research program in close collaboration with patients, was set up to gain greater insight into the mechanisms behind interindividual variations in bleeding tendency. We believe that elucidation of these mechanisms will further personalize treatment, potentially leading to regimens that are increasingly based on individual bleeding tendency and needs rather than on diagnosis per se, as is now the case. Moreover, it will support development of algorithms for shared decision making with regard to best treatment choice for each individual patient, taking societal costs into account and further personalize health‐care innovations and clinical management.
Research questions have been categorized according to a three‐way approach to achieve precision diagnosis, and to install safe, innovative, and cost‐effective treatment strategies by implementation of the results of enterprising fundamental research. The themes (1) diagnosis, (2) treatment, and (3) fundamental research and respective work packages (WPs), are depicted in Figure 1.
FIGURE 1.
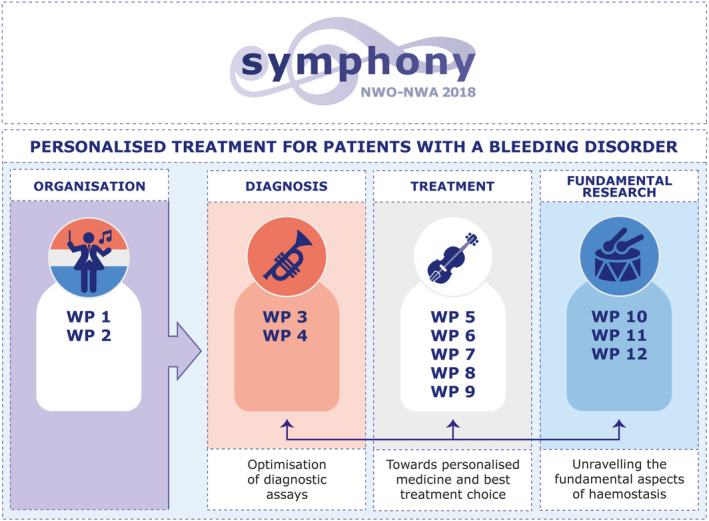
Three theme approach to achieve personalized treatment in patient with bleeding disorders. WP, work package.
This interdisciplinary consortium provides a unique framework to implement health‐care innovations, and to harmonize and amplify results from earlier multicenter (inter)national studies in which clinical data and blood samples have been collected, 1 , 12 , 13 , 14 , 15 , 16 , 17 , 29 expanding and linking these to fundamental research on the biochemistry of hemostasis, molecular genetics, proteomics, and cellular disease models. The patient perspective is well integrated in all SYMPHONY themes, especially those considering the realization and implementation of treatment and/or health‐care innovations.
We believe that these investments will lead to health‐care innovations and personalization of management and treatment with clinical and societal impact for patients with inborn bleeding disorders, and provide an example for other patient groups with a rare disease.
1.2. Objective
The general research question we aim to answer in SYMPHONY is: How can we improve and personalize diagnosis and the treatment of inborn bleeding disorders at acceptable costs for society, by identifying, diagnosing, and modifying the factors that define interindividual variation in bleeding phenotype and treatment response?
1.3. Participants
Beneficiaries of the SYMPHONY consortium are Erasmus University Medical Center and Erasmus MC Sophia Children’s Hospital, University Medical Center Rotterdam (Erasmus MC) (project leadership and coordination); Erasmus University Rotterdam (EUR); Sanquin Diagnostics; Sanquin Research; Amsterdam University Medical Centers (Amsterdam UMC); University Medical Center Groningen (UMCG); University Medical Center Utrecht (UMCU); Leiden University Medical Center (LUMC); Radboud University Medical Center (Radboud UMC); Netherlands Society of Hemophilia Patients (NVHP); Netherlands Society for Thrombosis and Hemostasis (NVTH); Bayer B.V., CSL Behring B.V., Swedish Orphan Biovitrum (Belgium) BVBA/SPRL.
Patient involvement in SYMPHONY is key and represented by intense involvement of the NVHP patient society especially in the treatment WPs. Clinical care and research units involved in SYMPHONY include all Hemophilia Treatment Centers in the Netherlands as well as scientists from various Sanquin departments. In addition, three pharmaceutical companies participate in the consortium, sharing their expertise with regard to therapeutics for bleeding disorders as well as knowledge on data sharing, marketing, and intellectual property. All partners and disciplines involved fulfill a specific need within the project for which they are valuable to the consortium.
The following disciplines and specialties are united within the SYMPHONY consortium: (pediatric) hematology, vascular medicine, clinical genetics, clinical pharmacology, (medical) psychology, ethics, epidemiology and methodology, mathematics, health care economics, laboratory medicine, information and data technology, biochemistry, molecular biology, molecular genetics, cell biology, (clinical) proteomics, expertise with regard to cellular (induced pluripotent stem cell [iPSC] technology) and vascular endothelial cell models, business economics, marketing, sales, patient advocacy, and industrial design.
As a consortium, we have consciously focused on complementarity and diversity within the consortium, both in choices of participating co‐applicants as well as cooperating partners, but also with regard to positions within the management structure. We have taken characteristics into account with regard to: professional experience (senior and junior), area of expertise, educational background, gender, diversity, and type of institution, as well as representatives from small groups, large businesses and institutions, and start‐ups.
2. PATIENTS AND METHODS
2.1. Study population(s)
Within SYMPHONY all patients with a diagnosed and undiagnosed bleeding disorder can be included. SYMPHONY is a result of the expertise and experience of Dutch researchers, the Dutch hemophilia community, patients, and caregivers. It builds on several (inter)national, multicenter initiatives, of which the most important are named.
Existing national initiatives/expertise areas and their importance to SYMPHONY:
HemoNed
National registry for hemophilia patients, developed in close collaboration with the patient organization NVHP and patients. SYMPHONY will use HemoNed, and the App‐based registration of prophylaxis, bleeding episodes, and treatment (Vasteprik®) as the digital platform to extend registration to other bleeding disorders and to implement proposed e‐health modules for value‐based health‐care outcome registration and pharmacokinetic‐pharmacodynamic (PK‐PD)–guided dosing. 30
OPTI‐CLOT
Multicenter studies that aim to optimize treatment by PK‐ and ultimately PK‐PD‐‐guided dosing of factor concentrates and desmopressin and emicizumab in hemophilia by construction of population PK‐PD models. Subsequent To WiN and DAVID studies specifically aim to improve treatment in von Willebrand and non‐severe hemophilia A with a comparable approach. 16 , 31 SYMPHONY provides the opportunity to expand to prospective clinical trials 12 to achieve population PK‐PD models that will finally associate coagulation factor levels with bleeding events, which is urgently needed, and additionally to develop a user‐friendly PK‐PD tool. 32
Other clinical multicenter studies in bleeding disorders initiated in the Netherlands:
Hemophilia in the Netherlands (HiN); von Willebrand in the Netherlands (WiN); Thrombocytopathy in the Netherlands (TiN); Rare Bleeding Disorders in the Netherlands (RBiN); studies into bleeding of unknown cause (CRESCENDO); international studies initiated in the Netherlands, including INSIGHT/RISE/DYNAMO. 1 , 13 , 14 , 15 , 17 , 18 , 29 All studies provide interesting patient material to deepen our understanding of interindividual differences. SYMPHONY provides the framework to maximize these collaborations and data sharing to its full potential.
Value‐based health‐care methodology/patient‐reported outcome measures
SYMPHONY will build on the expertise of several co‐applicants, cooperating partners, and external advisors with long‐term follow‐up of patient‐important outcome measures. This will proceed in collaboration with International Consortium for Health Outcomes Measurements (ICHOM) and KLIK (a Dutch initiative enabling long‐term follow‐up of quality of life parameters in children and parents/caregivers) and in cooperation with other groups specialized in value‐based health‐care development and implementation. 33 , 34
All Dutch Hemophilia Treatment Centers, EAHAD‐certified, and also accredited European Reference Network (ERN)/EuroBloodNet members have joined forces in SYMPHONY. Therefore, cross‐border health‐care potential and knowledge utilization and dissemination both nationally and in Europe is able to take place. This is further supported by the important leading (inter)national clinical, scientific positions fulfilled by many co‐applicants, cooperating partners in the field of hemostasis.
Participation in phase II, III, and IV drug trials of all clinical SYMPHONY members in, for instance, gene therapy for hemophilia A and B, long‐acting extended half‐life products, antibody‐based therapy, novel coagulation factor bypassing therapies and previously untreated patients (PUP) studies, ensures knowledge of advantages and side effects of novel therapies and dilemmas described.
Although previously collected cohorts are valuable, there is still a need to generate additional well‐characterized clinical cohorts with associated (longitudinally) collected biological materials (plasma, platelets, endothelial cells). Within the consortium a protocol for biomaterial will be created to ensure optimal efficient use of patient samples.
2.2. Study design
Within the three themes (1) diagnosis, (2) treatment, and (3) fundamental research questions are addressed within defined work packages (WPs 1–12) and if applicable answered interactively. Titles of WPs are depicted in Figure 2. Overall objectives, methods, and techniques per WP are described below.
FIGURE 2.
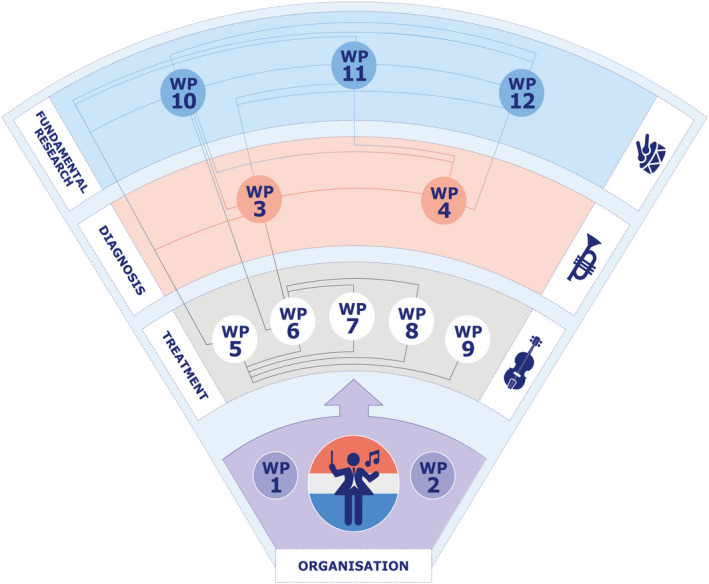
Orchestrated interactions between work packages (WPs) in SYMPHONY consortium.
2.2.1. Theme 0: management, organization, and dissemination of results
WP01 Objective: Manage SYMPHONY professionally, ensuring optimal collaboration, communication, and overall results. WP02 Objective: Assure good utilization of knowledge and entrepreneurship. In WP01 and 02, we will manage the consortium accordingly and allocate sufficient funds for overall management and knowledge transfer.
2.2.2. Theme 1: diagnosis
WP03 Objective: Develop reliable tests and flow models which more optimally quantify hemostatic potential and identify modifiers of hemostasis. In WP03, we will ensure results by the combined expertise of biochemists on determinants of coagulation, fibrin, and clot formation, and on development and validation of diagnostic tests and alternative hemostatic models. This will include detailed analyses of clot formation using the explicit expertise of thrombodynamics (Hemacore), modifications of thrombin generation, and plasmin generation assays. 35 , 36 Experiments will optimize type and concentration of phospholipids, factor‐deficient plasma, and other specific alterations of procedures and develop a first flow model in collaboration with vascular endothelial cell model experts in WP12.
WP04 Objective: Develop precision tests that characterize platelet function disorders and compare results with current standard diagnostic and genetic techniques.
In WP04, we will apply novel diagnostic tests for analysis of platelet function to functionally characterize platelets from both adult and pediatric patients with inherited platelet disorders in the nationwide Thrombocytopathy in the Netherlands (TiN) study. 37 This will include measurement of platelet activation markers with flow cytometry, which has increased diagnostic range compared to the current gold standard for platelet function analysis, light transmission aggregometry (LTA). 38 We will expand the current panel of platelet activation markers to improve sensitivity for platelet function disorders. Within the TiN study, whole exome sequencing (WES) data are collected on all patients with inherited platelet disorders. By combining WES data with extensive platelet phenotyping and the platelet proteome profiles determined in WP10, we aim to identify new genetic variants for inherited platelet disorders. Causality will subsequently be confirmed with iPSC models in WP11.
2.2.3. Theme 2: treatment
WP05 Objective: Improve quality of care by systematic measurement of outcomes that matter to patients. Implementation of value‐based health care into the field of bleeding disorders is vital to balance both patient‐ and doctor‐reported health‐care outcomes with costs of treatment, especially in the light of emerging even more expensive therapeutic agents for bleeding disorders, especially hemophilia. 24 Value‐based health care comprises several advantages: (1) patients may achieve a higher state of health at similar or lower costs, (2) health‐care providers are more efficient and succeed in achieving greater patient satisfaction, (3) payers (e.g., health insurerd) are better able to control costs with reduction of risks, (4) suppliers can align prices with patient outcomes, and (5) society as a whole becomes healthier while reducing overall health‐care spending.
WP06 Objective: Devise a dosing tool based on PK and PD principles instead of body weight, applicable in all patients with bleeding disorders to optimize quality of care and cost‐effectiveness. In WP06, we will perform PK‐PD modeling using non‐linear mixed effects modeling (NONMEM). NONMEM is a statistical technique and recommended in US Food and Drug Administration and European Medicines Agency guidelines for the evaluation of population PK‐PD of new drugs. Uniquely, NONMEM allows the analysis of population data obtained during clinical routine with often sparse and heterogeneous sampling from different treatment centers. The OPTI‐CLOT research group has extensive experience with this technique and has applied NONMEM for the development of population PK models for factor VIII (FVIII), factor IX (FIX), von Willebrand factor (VWF)/FVIII, desmopressin, and most recently also emicizumab. 12 , 39 They will apply these models in unique prospective multicenter trials in which doses are individualized on basis of individual patient PK using Bayesian statistical techniques.
WP07 Objective: Prioritize implementation of personalized care strategies and value‐based health‐care principles by installing a personal communication platform for all patients with an inborn bleeding disorder. Persons with inborn bleeding disorders in the Netherlands receive comprehensive care at Hemophilia Treatment Centers, including psychosocial support with a strong emphasis on self‐management and home treatment. 40 Data on patient‐reported health outcomes is currently collected to enable personalized treatment. However, individuals’ medical information is fragmented across health‐care information systems, digital treatment diaries, and questionnaire portals, hampering integrated care. We intend to develop a nationwide digital personal health record for patients to manage and share relevant medical information in collaboration with existing initiatives such as the national hemophilia patient registry HemoNed and the digital PROM portal KLIK.
WP08 Objective: Calculate costs and cost effectiveness of treatment for standard and new treatment modalities. In WP08, we will use innovative quasi‐experimental designs to study the effectiveness of the novel treatment options available for patients with bleeding disorders. In medicine, effectiveness of interventions is usually studied within randomized controlled trials with good internal validity but less feasibility or in observational data, the complete other end of the spectrum, with poor validity. Quasi‐experimental designs form the optimal balance. These quasi‐experimental designs will be combined with cost‐effectiveness analysis to obtain cost‐effectiveness estimates of the newer treatment options. Empirical data collection will be combined with a decision analytic modeling approach to obtain model‐based estimates of the cost effectiveness of the joint interventions.
WP09 Objective: Evaluate the patient’s perspective on ethical dilemmas with regard to medical innovations. In WP09, we will take an empirical‐‐ethical approach to bioethics. Such an approach starts from the assumption that ethical analysis and evaluation of a given practice does not occur in isolation of a certain context and that there is a certain wisdom in practices that needs to be taken into account for a thorough ethical analysis or evaluation. Several approaches exist within empirical‐ethics. In this project, we will take a broad, pluralistic perspective on empirical‐ethics by integrating an ethical clinical and patient‐participatory approach.
2.2.4. Theme 3: fundamental research
WP10 Objective: Determine which modifying hemostatic plasma proteins are associated with the bleeding phenotype in hemophilia A, von Willebrand disease, and bleedings of unknown cause, as regularly performed in platelets. In WP10, we will apply nanoscale liquid chromatography mass spectrometry approaches to identify hemostatic plasma and cellular protein signatures as determinants of bleeding. To this end, we assess protein expression and activation profiles of plasma and cellular systems of healthy individuals and patients with explained and unexplained bleedings. The full catalogue of profiles will assist in the identification as well as the understanding of the mechanisms behind the poorly understood bleedings.
WP11 Objective: Investigate if the new platelet‐function‐disorder mutations found in WP4 and previous studies are causative and if so, how do they affect megakaryopoiesis, platelet counts, and/or platelet function? In WP11, we will generate iPSCs from patients or will introduce platelet‐disorder–specific mutations using CRISPR/Cas9 into iPSC lines. Using these iPSC lines as model systems, ideal conditions to study causality and mechanisms behind novel identified mutations in platelet function disorders can be obtained. 41 In addition, identified putative causative mutations can be investigated in both patient genomic and patient independent genetic backgrounds. In general, iPSC lines have been used as successful disease model systems in an array of pathologies both hematopoietic and non‐hematopoietic. 42 , 43 For instance, at Sanquin, differentiation of GFI1B mutated gray platelet syndrome–specific iPSCs recapitulated disease characteristics and uncovered mechanistic insights. Numerous patient‐specific and control iPSC lines have been generated at the Sanquin iPSC facility using integrative and non‐integrative methodology. 43 , 44 , 45 The combined expertise to generate, maintain, differentiate, and study megakaryopoiesis using both patient specific and de novo generated (CRISPR/CAS9) iPSC lines is only sparsely available, internationally. This allows for quick and adaptive responses to specific research questions.
WP12 Objective: Investigate if interindividual variation in bleeding phenotype can be explained by cellular vascular endothelial mechanisms. In WP12, we will apply several state‐of‐the‐art approaches to unravel the cellular mechanisms that are at the basis of (unexplained) bleeding abnormalities in patients. This will be done by isolating endothelial colony forming cells (ECFCs), in which patient‐derived endothelial cell models of patients with various bleeding disorders are cultured. 46 This approach is particularly suited as it is capable of studying the effects of causative disease mutations in the endothelial context. We will construct a model system that reflects the patient’s diseased‐affected endothelium more accurately than has been done with general endothelial cell lines. In order to validate candidate disease modifiers we will use CRISPR/Cas9 gene editing in cord blood ECFCs. This research group was the first to establish a protocol to generate clonal CRISPR‐engineered endothelial lines and the first to recapitulate a bleeding disorder (HPS‐2) in endothelial cells using CRISPR/Cas9 gene editing of its causative gene, AP3B1, in cord blood ECFCs. 47
2.3. Governance
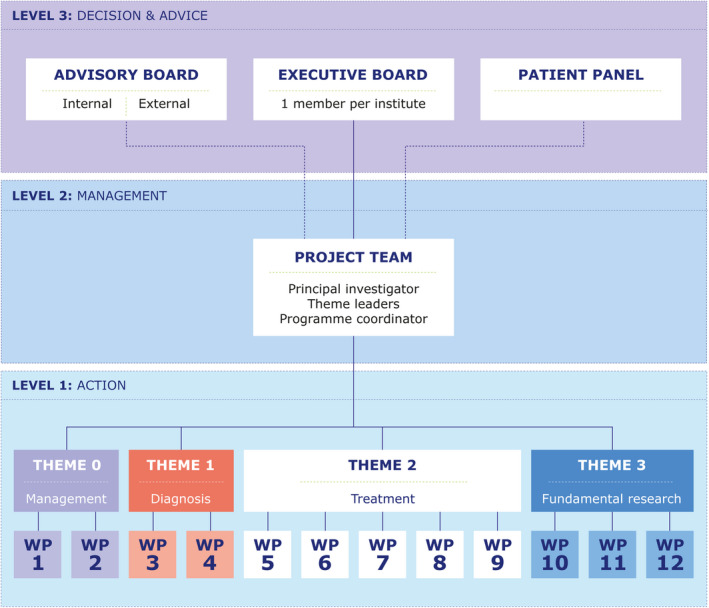
The SYMPHONY consortium is managed at three levels as illustrated in Figure 3 and described below.
FIGURE 3.

SYMPHONY consortium: Titles of work packages (WPs) per theme. iPSC, induced pluripotent stem cell.
2.3.1. Management level 1: action
The execution of each WP is managed at WP level under supervision of the respective WP leaders. The WP leaders communicate monthly with the project team on highlights and issues. The research WPs are split into two groups of five WPs (theme 1 and 3: WPs 03–04 and 10–12; Theme 2: WPs 05–09). These two groups report to the project team alternatively at 3 monthly theme meetings.
2.3.2. Management level 2: management
The project team is responsible for day‐to‐day management of the overall SYMPHONY research program including monitoring of milestones and deliverables, intellectual property and ethics monitoring, dissemination strategy, publication policy, and execution of the data management plan. It also manages contractual, legal, financial, and administrative affairs as well as safeguards SYMPHONY governance, knowledge transfer, and utilization, within and beyond the consortium.
2.3.3. Management level 3: decision and advice
The executive committee is the ultimate decision‐making entity. It consists of a representative from each institute involved in SYMPHONY, and is authorized to make binding decisions on behalf of his/her party. The advisory board consists of experts in the field. In addition, a patient panel assists the NVHP Working Group Care & Research to evaluate patient‐related topics. Both the advisory board and the patient panel meet annually during the general assembly. Both advise the project team as stated in level 2. Independent advisors can be invited for specific topics.
2.4. Endpoints and reporting
Project endpoints are defined per WP as milestones and deliverables. Monthly reports are made by WP leaders and summarized in a yearly overview. Milestones and deliverables are reported to Netherlands Organization for Scientific Research (now) by way of an impact plan as described by the theory of change, 48 which defines output, outcome, and societal impact and addresses assumptions made between problem areas according to spheres of influence. Consortium output is regulated according to a communication, publication, and authorship plan that defines how output is registered and safeguarded as well as how branding of NWO and SYMPHONY is organized.
2.5. Registration
This research received funding from the NWO in the framework of the NWA‐ORC Call grant agreement NWA.1160.18.038. Principal investigator: Dr. M.H. Cnossen. Project manager: Dr. S.H. Reitsma.
3. CONCLUSION
In the coming years, SYMPHONY will produce cutting‐edge papers on all aspects of care, treatment individualization in patients with inborn bleeding disorders, as well as diagnostic innovations and results of molecular genetic and cellular model technology with regard to the hemostatic process. We believe that these research investments will lead to health‐care innovations with long‐term clinical and societal impact. Moreover, we are convinced that rare diseases in general may benefit from SYMPHONY’s pioneering example to innovate by integration of interdisciplinary efforts to improve health care and outcomes.
INFORMED CONSENT
All authors have agreed to the publication of the article.
AUTHOR CONTRIBUTIONS
M.H. Cnossen and I. van Moort are the main authors of the manuscript. All authors substantially contributed to the writing, critically revised the manuscript, and approved the final draft. A complete overview of all collaborators is stated in the acknowledgments section.
CONFLICTS OF INTEREST
M.H. Cnossen has received grants from governmental and societal research institutes, for example, NWO‐ZonMW, NWO‐NWA, Innovation fund; from private funds, institutional grants, and unrestricted investigator research grants/educational and travel funding from the following companies over the years: Pfizer, Baxter/Baxalta/Shire, Bayer Schering Pharma, CSL Behring, Sobi Biogen, Novo Nordisk, Novartis, Nordic Pharma and Roche; and has served as a member on steering boards of Roche and Bayer. All grants, awards, and fees go to the Erasmus MC as an institution. I. van Moort has received grants from Sobi‐Biogen (Dutch junior researcher award), and CSL Behring (Prof. Heimburger Award) of which the fees go to the Erasmus MC as an institution. R.E.G. Schutgens reports grants from Bayer, Baxalta, Pfizer, and Novo Nordisk outside the submitted work. R.A.A. Mathôt has received grants from governmental and societal research institutes such as NWO, ZonMW, Dutch Kidney Foundation, and Innovation Fund and unrestricted investigator research grants from Baxter/Baxalta/Shire/Takeda, Bayer, CSL Behring, Sobi‐Biogen, and CelltrionHC. He has served as advisor for Bayer, CSL Behring, Merck Sharp & Dohme, Baxter/Baxalta/Shire/Takeda. All grants and fees paid to the institution. S.C. Gouw received an unrestricted medical research grant from CSL Behring (Prof. Heimburger Award), Sobi, of which the fees go to the Amsterdam UMC as an institution. K. Meijer received speaker fees from Alexion, Bayer, and CSL Behring; participation in trial steering committee for Bayer; consulting fees from UniQure; participation in data monitoring and endpoint adjudication committee for Octapharma; all payments are made to her institution. A. Bredenoord is a member of the Senate of the Dutch Parliament. The institution of K. Fijnvandraat has received unrestricted research grants from CSL Behring, Sobi‐Biogen, and Novo Nordisk and her institution received consultancy fees from Grifols, Takeda, Novo Nordisk, and Roche. H.C.J. Eikenboom has received grants from CSL Behring of which the fees go to the Leiden University Medical Center as an institution. M. de Haas has received grants from governmental institutes (RIVM), from the Landsteiner Foundation for Blood Transfusion Research. All grants and fees go to Sanquin Research as an institution. F.W.G. Leebeek received unrestricted research grants from CSL Behring, Shire/Takeda, SOBI, and UniQure. He is a consultant for CSL Behring, Takeda, Biomarin, and UniQure, of which the fees go to the Erasmus MC as an institution. He served as DSMB member of a study sponsored by Roche. The remaining authors declare no competing financial interests.
ACKNOWLEDGMENTS
Symphony is an interdisciplinary research program performed by all Dutch Hemophilia Centers, academic hospitals, and research institutes, in collaboration with the patient society (NVHP), and both hematologists (NVHB) and hemostasis specialists (NVTH) as well as three pharmaceutical companies (Figures 4 and 5).
FIGURE 4.
Governance structure SYMPHONY consortium. WP, work package.
FIGURE 5.
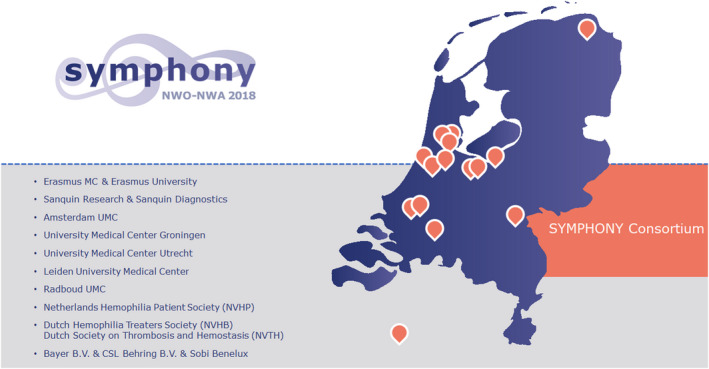
Interdisciplinary research within the SYMPHONY consortium is performed by all Dutch Hemophilia Centers, academic hospitals, research institutes, in collaboration with the patient society (NVHP), and both treaters (NVHB) and hemostasis specialists (NVTH) as well as three pharmaceutical companies. MC, medical center; UMC, university medical center.
Project team: Principal investigator: Dr. M.H. Cnossen. Project manager: Dr. S.H. Reitsma.
Theme leaders: Prof. Dr. M. de Haas, Dr. M. van den Biggelaar (Diagnosis), Prof. Dr. F.W.G. Leebeek (Treatment), Prof. Dr. J. Voorberg (Fundamental research).
WP‐leaders (according to WP): WP3: Prof. dr. M.P.M. de Maat; WP4: Prof. dr. R.E.G. Schutgens, Dr. R.T. Urbanus; WP5: Dr. H.F. Lingsma; WP6: Prof. dr. R.A.A. Mathot; WP7: Dr. S.C. Gouw; WP8: Dr. H.F. Lingsma; WP9: Prof. dr. K. Meijer, Prof. dr. A.L. Bredenoord, Dr. R. van der Graaf; WP10: Prof. Dr. K. Fijnvandraat, Prof. dr. A.B. Meijer; WP11: Dr. E. van den Akker; WP12: Dr. R. Bierings, Prof.dr. H.C.J. Eikenboom.
Post‐doc & PhD students (according to WP): Dr. I. van Moort (WP12; post‐doc); WP3: R.A. Arisz; WP4: M. Zivkovic; WP5: E.S. van Hoorn; WP6: L.H. Bukkems, M.C.H.J. Goedhart, L.G.R. Romano, W. Al Arashi, M.E. Cloesmeijer, A. Janssen; WP7: M.R. Brands; WP9: L. Baas; WP10: J. del Castillo Alferez; WP11: H. Zhang, WP12: S.N.J. Laan.
Supporting and co‐investigators (alphabetical order):
J. Boender (Sobi‐Biogen), Prof. dr. J.G. van der Bom, M.H.A. Bos (NVTH), Prof. dr. L. Burdorf.
Dr. M. Coppens, Dr. M. Driessens (NVHP), Dr. K.F. Fischer, Dr. R. van der Graaf, Dr. L. Haverman, Prof. dr. J.A. Hazelzet, Drs. E.J. Huisman, N. Jansen (CSL Behring), S. de Jong (Bayer), Dr. M. Kruip, Dr. N. van Leeuwen, Dr. F. van der Meer, S. Meijer (NVHP), Prof. dr. J.K. Ploos van Amstel, Dr. S. Polinder, Dr. S.E.M. Schols, G. Wijfjes (NVHP), K. Kluft, W.L. van Heerde, G. Goedhart, Prof.dr. C. Uyl, J. Timp, A. Stekelenburg, Dr. F. Moenen, Dr. P. Ypma, Dr. L. Nieuwenhuizen, A. Plat (NVHP).
Cnossen MH, van Moort I, Reitsma SH, et al.. SYMPHONY consortium: Orchestrating personalized treatment for patients with bleeding disorders. J Thromb Haemost. 2022;20:2001‐2011. doi: 10.1111/jth.15778
Manuscript handled by: Riitta Lassila
Final decision: Riitta Lassila, 27 May 2022
This manuscript has been read and approved for submission to Journal of Thrombosis and Haemostasis by all authors.
Contributor Information
Marjon H. Cnossen, Email: m.cnossen@erasmusmc.nl, @CnossenM.
Iris van Moort, @IrisMoort.
Annelien L. Bredenoord, @ALBredenoord.
Ruben Bierings, @Rbierings.
Jan Voorberg, @VoorbergJ.
Frank W. G. Leebeek, @FLeebeek.
REFERENCES
- 1. Hassan S, van Balen EC, Smit C, et al. Health and treatment outcomes of patients with hemophilia in The Netherlands, 1972‐2019. J Thromb Haemost. 2021;19(10):2394‐2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Plug I, van der Bom JG, Peters M, et al. Thirty years of hemophilia treatment in The Netherlands, 1972‐2001. Blood. 2004;104(12):3494‐3500. [DOI] [PubMed] [Google Scholar]
- 3. Ahlberg A. Haemophilia in Sweden. VII. Incidence, treatment and prophylaxis of arthropathy and other musculo‐skeletal manifestations of haemophilia A and B. Acta Orthop Scand Suppl. 1965;Suppl 77:3‐132. [DOI] [PubMed] [Google Scholar]
- 4. Fijnvandraat K, Cnossen MH, Leebeek FW, Peters M. Diagnosis and management of haemophilia. Bmj. 2012;344:e2707. [DOI] [PubMed] [Google Scholar]
- 5. Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535‐544. [DOI] [PubMed] [Google Scholar]
- 6. Nathwani AC, Reiss UM, Tuddenham EG, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow‐up of AAV5‐hFVIII‐SQ gene therapy for hemophilia A. N Engl J Med. 2020;382(1):29‐40. [DOI] [PubMed] [Google Scholar]
- 8. Miesbach W, Meijer K, Coppens M, et al. Gene therapy with adeno‐associated virus vector 5‐human factor IX in adults with hemophilia B. Blood. 2018;131(9):1022‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leebeek FWG, Miesbach W. Gene therapy for hemophilia: a review on clinical benefit, limitations, and remaining issues. Blood. 2021;138(11):923‐931. [DOI] [PubMed] [Google Scholar]
- 10. Collins P, Chalmers E, Chowdary P, et al. The use of enhanced half‐life coagulation factor concentrates in routine clinical practice: guidance from UKHCDO. Haemophilia. 2016;22:487‐498. [DOI] [PubMed] [Google Scholar]
- 11. Shima M, Hanabusa H, Taki M, et al. Factor VIII‐mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374(21):2044‐2053. [DOI] [PubMed] [Google Scholar]
- 12. van Moort I, Preijers T, Bukkems LH, et al. Perioperative pharmacokinetic‐guided factor VIII concentrate dosing in haemophilia (OPTI‐CLOT trial): an open‐label, multicentre, randomised, controlled trial. Lancet Haematol. 2021;8(7):e492‐e502. [DOI] [PubMed] [Google Scholar]
- 13. de Wee EM, Sanders YV, Mauser‐Bunschoten EP, et al. Determinants of bleeding phenotype in adult patients with moderate or severe von Willebrand disease. Thromb Haemost. 2012;108(4):683‐692. [DOI] [PubMed] [Google Scholar]
- 14. Blaauwgeers MW, Kruip MJHA, Beckers EAM, et al. Bleeding phenotype and diagnostic characterization of patients with congenital platelet defects. Am J Hematol. 2020;95(10):1142‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Saes JL, Verhagen MJA, Meijer K, et al. Bleeding severity in patients with rare bleeding disorders: real‐life data from the RBiN study. Blood Adv. 2020;4(20):5025‐5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schütte LM, Cnossen MH, van Hest RM, et al. Desmopressin treatment combined with clotting factor VIII concentrates in patients with non‐severe haemophilia A: protocol for a multicentre single‐armed trial, the DAVID study. BMJ Open. 2019;9(4):e022719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Veen CSB, Huisman EJ, Cnossen MH, et al. Evaluation of thromboelastometry, thrombin generation and plasma clot lysis time in patients with bleeding of unknown cause: a prospective cohort study. Haemophilia. 2020;26(3):e106‐e115. [DOI] [PubMed] [Google Scholar]
- 18. Eckhardt CL, van Velzen AS, Peters M, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122(11):1954‐1962. [DOI] [PubMed] [Google Scholar]
- 19. Eckhardt CL, Astermark J, Nagelkerke SQ, et al. The Fc gamma receptor IIa R131H polymorphism is associated with inhibitor development in severe hemophilia A. J Thromb Haemost. 2014;12(8):1294‐1301. [DOI] [PubMed] [Google Scholar]
- 20. Lambert T, Benson G, Dolan G, et al. Practical aspects of extended half‐life products for the treatment of haemophilia. Ther Adv Hematol. 2018;9(9):295‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ten Ham RMT, Walker SM, Soares MO, et al. Modeling benefits, costs, and affordability of a novel gene therapy in hemophilia A. Hema. 2022;6(2):e679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hazendonk HC, Lock J, Mathot RA, et al. Perioperative treatment of hemophilia A patients: blood group O patients are at risk of bleeding complications. J Thromb Haemost. 2016;14(3):468‐478. [DOI] [PubMed] [Google Scholar]
- 23. Zwagemaker AF, Gouw SC, Jansen JS, et al. Incidence and mortality rates of intracranial hemorrhage in hemophilia: a systematic review and meta‐analysis. Blood. 2021;138(26):2853‐2873. [DOI] [PubMed] [Google Scholar]
- 24. van Balen EC, O'Mahony B, Cnossen MH, et al. Patient‐relevant health outcomes for hemophilia care: development of an international standard outcomes set. Res Pract Thromb Haemost. 2021;5(4):e12488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aiyegbusi OL, Isa F, Kyte D, et al. Patient and clinician opinions of patient reported outcome measures (PROMs) in the management of patients with rare diseases: a qualitative study. Health Qual Life Outcomes. 2020;18(1):177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kuijlaars IAR, Teela L, van Vulpen LFD, et al. Generic PROMIS item banks in adults with hemophilia for patient‐reported outcome assessment: feasibility, measurement properties, and relevance. Res Pract Thromb Haemost. 2021;5(8):e12621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abrantes JA, Solms A, Garmann D, Nielsen EI, Jonsson S, Karlsson MO. Relationship between factor VIII activity, bleeds and individual characteristics in severe hemophilia A patients. Haematologica. 2019;105:1443‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Denis CV, Susen S, Lenting PJ. von Willebrand disease: what does the future hold? Blood. 2021;137(17):2299‐2306. [DOI] [PubMed] [Google Scholar]
- 29. Abdi A, Kloosterman FR, Eckhardt CL, et al. The factor VIII treatment history of non‐severe hemophilia A. J Thromb Haemost. 2020;18(12):3203‐3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.M.Driessens H.E., Goedhart G., Taal E.M., et al. Abstract WFH: the Dutch haemophilia registry HemoNED – Building an ecosystem. Haemophilia 2020; 26(S4): 3–140. [Google Scholar]
- 31. Hazendonk H, Heijdra JM, de Jager NCB, et al. Analysis of current perioperative management with Haemate([R]) P/Humate P([R]) in von Willebrand disease: Identifying the need for personalized treatment. Haemophilia. 2018;24:460‐470. [DOI] [PubMed] [Google Scholar]
- 32. Hazendonk H, van Moort I, Mathot RAA, et al. Setting the stage for individualized therapy in hemophilia: what role can pharmacokinetics play? Blood Rev. 2018;32(4):265‐271. [DOI] [PubMed] [Google Scholar]
- 33. Haverman L, van Oers HA, van Muilekom MM, Grootenhuis MA. Options for the interpretation of and recommendations for acting on different PROMs in daily clinical practice using KLIK. Med Care. 2019;57 Suppl(5 Suppl 1):S52‐S58. [DOI] [PubMed] [Google Scholar]
- 34. Terwee CB, Zuidgeest M, Vonkeman HE, Cella D, Haverman L, Roorda LD. Common patient‐reported outcomes across ICHOM standard sets: the potential contribution of PROMIS®. BMC Med Inform Decis Mak. 2021;21(1):259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. van Geffen M, Loof A, Lap P, et al. A novel hemostasis assay for the simultaneous measurement of coagulation and fibrinolysis. Hematology. 2011;16(6):327‐336. [DOI] [PubMed] [Google Scholar]
- 36. Tarandovskiy ID, Balandina AN, Kopylov KG, et al. Investigation of the phenotype heterogeneity in severe hemophilia A using thromboelastography, thrombin generation, and thrombodynamics. Thromb Res. 2013;131(6):e274‐e280. [DOI] [PubMed] [Google Scholar]
- 37. Blaauwgeers MW, van Asten I, Kruip M, et al. The limitation of genetic testing in diagnosing patients suspected for congenital platelet defects. Am J Hematol. 2020;95(1):E26‐E28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. van Asten I, Schutgens REG, Baaij M, et al. Validation of flow cytometric analysis of platelet function in patients with a suspected platelet function defect. J Thromb Haemost. 2018;16(4):689‐698. [DOI] [PubMed] [Google Scholar]
- 39. Bukkems LH, Heijdra JM, de Jager NCB, et al. Population pharmacokinetics of the von Willebrand factor‐factor VIII interaction in patients with von Willebrand disease. Blood Adv. 2021;5(5):1513‐1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Leebeek FW, Fischer K. Quality of haemophilia care in The Netherlands: new standards for optimal care. Blood Transfus. 2014;12 Suppl 3(Suppl 3):s501‐s504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hansen M, von Lindern M, van den Akker E, Varga E. Human‐induced pluripotent stem cell‐derived blood products: state of the art and future directions. FEBS Lett. 2019;593(23):3288‐3303. [DOI] [PubMed] [Google Scholar]
- 42. Yi G, Mandoli A, Jussen L, et al. CBFβ‐MYH11 interferes with megakaryocyte differentiation via modulating a gene program that includes GATA2 and KLF1. Blood Cancer J. 2019;9(3):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hansen M, Varga E, Wust T, et al. Generation and characterization of a human iPSC line SANi005‐A containing the gray platelet associated heterozygous mutation p.Q287* in GFI1B. Stem Cell Res. 2017;25:34‐37. [DOI] [PubMed] [Google Scholar]
- 44. Aarts CEM, Karampini E, Wüst T, et al. Generation and characterization of a control and patient‐derived human iPSC line containing the Hermansky Pudlak type 2 (HPS2) associated heterozygous compound mutation in AP3B1. Stem Cell Res. 2021;54:102444. [DOI] [PubMed] [Google Scholar]
- 45. Aarts CEM, Varga E, Webbers S, et al. Generation and characterization of a human iPSC line SANi006‐A from a gray platelet syndrome patient. Stem Cell Res. 2021;55:102443. [DOI] [PubMed] [Google Scholar]
- 46. Martin‐Ramirez J, Hofman M, van den Biggelaar M, Hebbel RP, Voorberg J. Establishment of outgrowth endothelial cells from peripheral blood. Nat Protoc. 2012;7(9):1709‐1715. [DOI] [PubMed] [Google Scholar]
- 47. Karampini E, Schillemans M, Hofman M, et al. Defective AP‐3‐dependent VAMP8 trafficking impairs Weibel‐Palade body exocytosis in Hermansky‐Pudlak Syndrome type 2 blood outgrowth endothelial cells. Haematologica. 2019;104(10):2091‐2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weiss CH. Nothing as Practical as Good Theory : Exploring Theory‐Based Evaluation for Comprehensive Community Initiatives for Children and Families 2011 2011.