

Abstract
The combination of four proteins and their paralogues including MBD2/3, GATAD2A/B, CDK2AP1 and CHD3/4/5, which we refer to as the MGCC module, form the chromatin remodelling module of the nucleosome remodelling and deacetylase (NuRD) complex. To date, mechanisms by which the MGCC module acquires paralogue‐specific function and specificity have not been addressed. Understanding the protein–protein interaction (PPI) network of the MGCC subunits is essential for defining underlying mechanisms of gene regulation. Therefore, using pulldown followed by mass spectrometry analysis (PD‐MS), we report a proteome‐wide interaction network of the MGCC module in a paralogue‐specific manner. Our data also demonstrate that the disordered C‐terminal region of CHD3/4/5 is a gateway to incorporate remodelling activity into both ChAHP (CHD4, ADNP, HP1γ) and NuRD complexes in a mutually exclusive manner. We define a short aggregation‐prone region (APR) within the C‐terminal segment of GATAD2B that is essential for the interaction of CHD4 and CDK2AP1 with the NuRD complex. Finally, we also report an association of CDK2AP1 with the nuclear receptor co‐repressor (NCOR) complex. Overall, this study provides insight into the possible mechanisms through which the MGCC module can achieve specificity and diverse biological functions.
Keywords: aggregation prone region, ChAHP complex, CHD4, chromatin remodelling, NCOR complex, NuRD complex, protein–protein interactions
The protein interaction network of the remodelling module of the NuRD complex reveals paralogue‐specific interaction signatures. Paralogue‐specific interactions occur via CHD3/4/5, GATAD2A/B and MBD2/3 subunits, whilst CDK2AP1 and CHD3/4/5 are also individually found in the NCoR and ChAHP complexes, respectively.
Abbreviations
- APR
aggregation prone region
- CHD
chromodomain helicase DNA binding
- FLAG
epitope DYKDDDDK
- HA
human influenza haemagglutinin, epitope YPYDVPDYA
- iBAQ
intensity‐based absolute quantification
- IVT
in vitro translation
- LC‐MS/MS
liquid chromatography‐tandem mass spectrometry
- LFQ
label‐free quantification
- MGCC
MBD/GATAD2/CDK2AP1/CHD module
- MHR
MTA/HDAC/RBBP module
- MS
mass spectrometry
- NCOR
nuclear receptor co‐repressor complex
- NuRD
nucleosome remodelling and deacetylase complex
- PD‐MS
pulldown followed by mass spectrometry analysis
Introduction
The nucleosome remodelling and deacetylase (NuRD) complex plays a crucial function in embryonic stem cell biology and development, and cancer [1, 2, 3]. The NuRD complex consists of seven core subunits, namely MTA, HDAC, RBBP, MBD, GATAD2, CDK2AP1 and chromodomain helicase DNA binding (CHD), with each subunit interchangeable with its paralogous family members: MTA1, MTA2 and MTA3; HDAC1 and HDAC2; RBBP4 and RBBP7; MBD3 and MBD2; GATAD2A and GATAD2B; CHD3, CHD4 and CHD5 [4, 5, 6]. This ~1 MDa nuclear protein complex harbours both chromatin remodelling and lysine deacetylase activities [7, 9]. A symmetric module encompassing MTA, HDAC and RBBP proteins (MHR) conveys the lysine deacetylation activity via the HDAC catalytic subunit [9, 10, 11, 12], whereas MBD, GATAD2, CDK2AP1 and CHD proteins (MGCC) collaborate to translocate nucleosomal DNA via the CHD subunit [13, 14]. The canonical NuRD complex comprises a stable asymmetric composition of two MHR modules and a single MGCC module [12]. Among other functions, MGCC components regulate the expression of adult globin genes and are linked to β‐haemoglobinopathies [15, 16, 17]. Therefore, subunits of MGCC are considered as potential molecular targets for therapeutic activation of fetal gamma globin genes [18]. Understanding the auxiliary binding partners of the MGCC module will facilitate understanding of the molecular basis of MGCC‐mediated gene silencing.
The domains responsible for mediating intra‐NuRD architecture are being uncovered through careful biochemical analysis. Previous structural studies have documented that two HDAC molecules are activated by homo‐ or heterodimerisation of two molecules of MTA through ELM2‐SANT domains [19]. MTA1/2 and MTA3 provide two and one binding regions, respectively, within their C terminus for RBBP proteins to facilitate their access to histone tails to possibly remove acetyl marks from lysine residues [9, 10]. The bromo‐adjacent homology (BAH) domain of MTA proteins is well conserved but is structurally and functionally poorly characterised [20]. MBD connects to the N‐terminal region of the GATAD2 protein through a coiled‐coil interface [21]. GATAD2 uses the GATA zinc finger domain in the C‐terminal region to facilitate CHD binding [7, 16].
Focusing on the MGCC module, MBD2 and MBD3, which are highly homologous and mutually exclusive within the NuRD complex [22], structurally bridge GATAD2/CDK2AP1/CHD subunits with the MHR module to form the intact NuRD complex [11, 12]. Although MBD2 and MBD3 are highly similar, their affinity and selectivity towards DNA is markedly different [23, 24]. MBD3 has been extensively studied, but the function of MBD2 isoforms MBD2a and MBD2b are poorly understood. MBD2a contains an extra 148 amino acid Gly‐ and Arg‐rich domain at the N‐terminal end as compared to MBD2b [25, 26]. Within GATAD2, the C‐terminal portion is responsible for the interaction with CHD proteins [7, 16]. However, the minimal region mediating this interaction has remained elusive. To date, CDK2AP1 is the least studied subunit of NuRD biochemically and possibly overlooked in some studies because of its small size. CHD4 contains several functional domains essential for ATP‐dependent nucleosome remodelling. The N‐terminal region of CHD4 binds DNA via an HMG‐box‐like domain [27]. CHD4 can bind to histone tails (such as H3K4 and trimethylated H3K9) via two plant homeodomain (PHD) zinc fingers and remodels nucleosomes via its ATPase motor domain assisted by tandem chromodomains (CHD) [28]. The C‐terminal region of CHD4, however, is functionally and structurally the least characterised part of CHD4.
Recently, the NuRD subunit CHD4 was identified as a component of the ChAHP complex, which also contains ADNP and HP1γ (otherwise known as CBX3) subunits [29]. ChAHP modulates chromatin organisation by neutralising loops formed by CTCF at specific genomic regions and plays an important role in embryonic neural development and heterochromatin organisation [30]. ADNP is the requisite subunit, without which the ChAHP complex cannot form [29]. How CHD4 can be independently involved in two different chromatin‐binding complexes and whether CHD3 and CHD5 can also form stable complexes with ADNP have not been resolved. The minimal domains for these interactions are also unknown.
Based on the above understanding, we set out to address the following: (a) what proteins interact with MBD2 isoforms? (b) What is the minimal region in GATAD2 proteins that mediates their interaction with CHD family members? (c) Is CDK2AP1 an exclusive subunit of NuRD or is it found in other complexes? And (d) what regions in CHD4 and ADNP mediate their interaction? We have sought to address these questions by focusing on the interaction network of the MGCC module of NuRD and core component of the ChAHP complex, ADNP. We define novel factors and complexes that can interact with paralogues of the NuRD subunits. We also refine minimal interacting domains between certain subunits and isoforms of the NuRD MGCC module. The importance of these results lies in providing clarification of how the MGCC module functions to co‐ordinate diverse cellular functions.
Results
The domain architecture of the NuRD and ChAHP complexes is depicted in Fig. 1A, which shows known domains and sites of interaction (Fig. 1A). Domains responsible for mediating intra‐NuRD subunit interactions that have not previously been described or characterised are highlighted with a question mark (Fig. 1A).
Fig. 1.
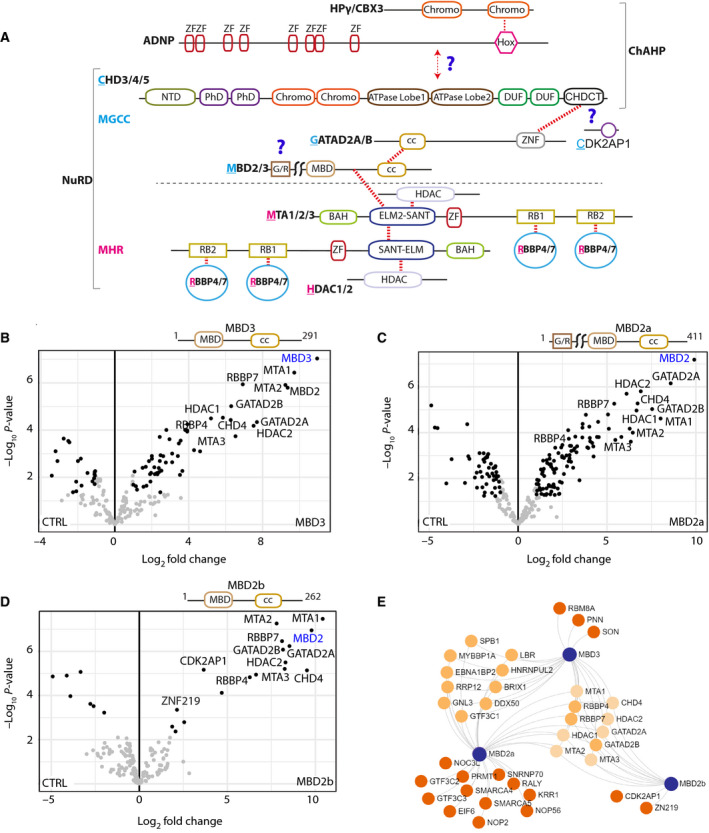
Mutual and distinct MBD binding partners revealed by PD‐MS. (A) Schematic diagram depicting the domain organisation of the NuRD and ChAHP complexes. Coloured regions indicate domains with known structures, or which are predicted to be ordered; and single lines specify regions predicted to be disordered. The total NuRD complex contains MHR and MGCC modules separated by a dashed line. CHD4 is a component of both ChAHP and NuRD complexes. Red dotted lines represent the intersubunit binding regions. Question marks indicate the regions of interaction that have not been well defined. (B–D) FLAG pulldowns were performed in triplicate followed by label‐free quantitative MS analysis, and LFQ intensities were used to generate the volcano plots. The canonical subunits of the NuRD complex are highlighted in black and noncanonical or potential new interactors in grey. Significantly enriched proteins in (B) MBD3 (C) MBD2a and (D) MBD2b pulldowns. Significantly enriched proteins are indicated with black dots; nonsignificant proteins are indicated with grey dots; the bait is indicated as blue text. (E) Network representation of shared and specific interactors of MBD proteins. Significantly enriched proteins with at least twofold change were used to generate the network.
MBD2 isoforms and MBD3 have both mutual and unique binding partners
Initially, we focused on the MBD subunits, which bridge the MGCC to the MHR module. Relative label‐free quantification (LFQ) analysis of MBD2a, MBD2b and MBD3 pulldown followed by mass spectrometry analysis (PD‐MS) data demonstrate that all three MBDs significantly copurify the canonical subunits of the NuRD complex (Fig. 1B–D). Both MBD2a and MBD3 mutually co‐immunoprecipitated a substantial number of possible new interactors involved in chromatin biology, transcriptional gene regulation and RNA processing (Fig. 1B,C). MBD2b had comparatively few interactors, but significantly enriched the canonical NuRD subunit CDK2AP1 (Fig. 1D). Interestingly, CDK2AP1 was significantly enriched only with the MBD2b isoform but not MBD2a, or MBD3. This shows the preference of CDK2AP1 for binding to the MBD2/NuRD complex but not MBD3/NuRD. We also identified other proteins specifically enriched with each MBD family member. For example, the MBD2a isoform, which contains an N‐terminal Gly‐ and Arg‐rich domain, captures numerous proteins, including the chromatin remodelling proteins SMARCA4 and SMARCA5 as well as the arginine N‐methyltransferase enzyme PRMT1 (Fig. 1E). Evidence for arginine methylation of MBD2a [31] suggests a possible mechanism by which the function of the MBD2/NuRD complex is regulated. Interestingly, ZNF219, a transcriptional repressor that is a known interactor of the NuRD complex, was only pulled down with MBD2b (Fig. 1D,E) [6]. Given that MBD2b is not the major isoform of MBD2, this supports the substoichiometric nature of ZNF219 noted previously [6] and possibly other noncanonical partners of the NuRD complex that have been observed. Our data reveal that the two MBD2 isoforms show distinct molecular interactions and could form NuRD complexes associating with distinct sets of partner proteins. Hence, these diverse NuRD subspecies might perform different gene regulatory activities at different loci.
A 40‐residue region in the GATAD2B C terminus is important for connecting CHD4 to the NuRD complex
Previous biochemical studies have demonstrated that the C‐terminal region (residues 276‐593) in GATAD2 proteins is necessary for binding to CHD4 [7, 16]. Using GATAD2B as an exemplar, we examined whether other proteins can compete with CHD4 for binding to GATAD2B and then defined the minimal region that mediates this interaction. LFQ analysis of proteins copurified with full‐length GATAD2B showed marked enrichment of the canonical NuRD subunits (Fig. 2A). We also observed significant enrichment of auxiliary proteins important for chromatin biology and gene repression, namely BEND3 and KCRM. PD‐MS of the N‐terminal half of GATAD2B (GATAD2B‐N, residues 1‐276) revealed that all canonical NuRD subunits except for CDK2AP1 and CHD4 were enriched (Fig. 2B).
Fig. 2.
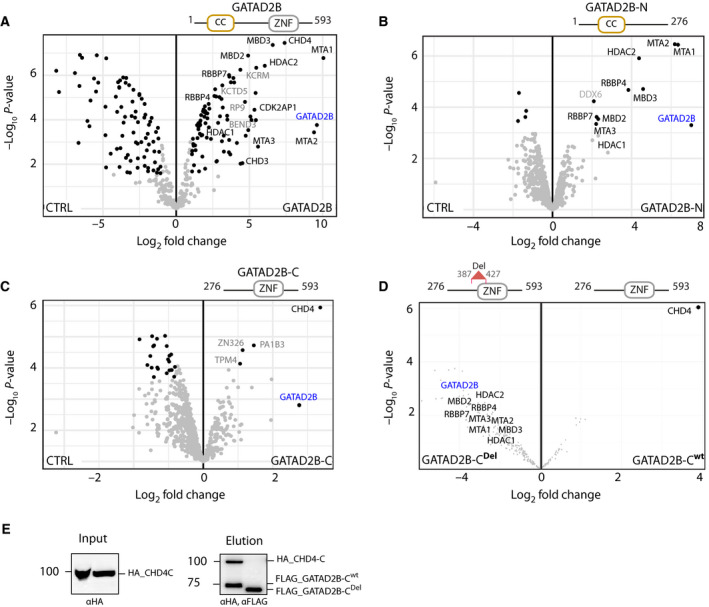
GATAD2B C terminus mediates interaction with CHD4. Volcano plots were generated using LFQ intensities of FLAG PD‐MS and the canonical subunits of the NuRD complex are highlighted in black and noncanonical or potential new interactors in grey. (A) Full‐length GATAD2B, (B) N terminus (GATAD2B‐N) and (C) C terminus (GATAD2B‐C). FLAG‐only was used as a control (CTRL) to account for background contamination. (D) FLAG PD‐MS of GATAD2B‐CDel versus WT GATAD2B‐C. Schematic of GATAD2B constructs used as baits are indicated on top of the volcano plots. (E) Western blots of GATAD2B proteins co‐expressed in the IVT system and purified on anti‐FLAG beads. Input (5% of total, left panel) blot was developed with anti‐HA and the elution (70% of total, right panel) was developed with both HA and FLAG antibodies.
On the other hand, using GATAD2B C‐terminal region (GATAD2B‐C) as bait, CHD4 was the only NuRD subunit that prefers the C‐terminal portion of GATAD2B for binding (Fig. 2C); it was also the most highly enriched protein in this experiment across the whole proteome. Examination of intensity‐based absolute quantification (iBAQ) values in all GATAD2B bait replicates revealed that all three replicates of GATAD2B (full‐length) and two replicates of GATAD2B‐C but not GATAD2B‐N had high‐intensity values for CDK2AP1 (Fig. S1), indicating the C‐terminal GATAD2B binding preference of CDK2AP1.
To narrow down the CHD4‐binding region in the C terminus of GATAD2B, residues 387‐427 of GATAD2B were deleted from GATAD2B‐C (herein named GATAD2B‐CDel) and PD‐MS was performed. This pulldown showed marked depletion of CHD4 (but not other NuRD subunits), compared to the wild‐type GATAD2B‐C (Fig. 2D). Notably, this deletion did not disrupt the interaction of other NuRD subunits. To corroborate this finding, we performed co‐immunoprecipitation using epitope DYKDDDDK (FLAG)‐tagged GATAD2B‐C or GATAD2B‐CDel versus an human influenza haemagglutinin, epitope YPYDVPDYA (HA)‐tagged C‐terminal CHD4 construct (HA‐CHD4‐C, residues 1230‐1912) using an in vitro translation (IVT) system. IVT confirmed that the GATAD2B‐CHD4 interaction was direct and that residues 387‐427 are necessary for the integrity of this interaction (Fig. 2E).
The GATAD2 interaction interface with CHD4 includes an aggregation prone region
Protein–protein interaction (PPI) interfaces typically contain high proportions of hydrophobic residues. These interfaces can display characteristics of aggregation‐prone regions (APRs) with fibril‐forming capacities. Analysis of GATAD2A and GATAD2B protein sequences using the TANGO aggregation prediction algorithm [32] revealed a number of mutually exclusive as well as overlapping APRs (Fig. 3A). The most prominent of these APRs was a small and highly similar stretch of seven hydrophobic and aliphatic amino acids in GATAD2A (residues 384‐390, FIYLVGL) and GATAD2B (residues 388‐394–FIYMVGL; Fig. 3A,B). We designed peptidomimetics containing the 7‐residue APRs from GATAD2A and GATAD2B, conjugated to a 11‐residue portion of HIV‐1 Tat protein for cell permeability, referred to herein as APRA and APRB, respectively. We also designed APRs flanked by gatekeeper glutamic acid residue and named them APRAGK and APRBGK. We first tested the impact of these APR and APRA/BGK peptides on K562 cell viability compared to a Tat‐conjugated control peptide comprising seven alanine residues (CTRL). After dose‐dependent addition of APRA, APRB, APRAGK, APRBGK and CTRL peptides, K562 cell proliferation was analysed after 48 h by MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide) assay. APRA and APRB induced cell death with IC50 values of ~ 30 μm, whereas the CTRL peptide had no effect (Fig. 3C). Interestingly, addition of the flanking gatekeeper glutamic acid residues (Fig. 3B) to both peptides lessened the impact on K562 cell viability, most likely as a result of disrupting β‐aggregation tendency in GATAD2 proteins (Fig. 3C).
Fig. 3.
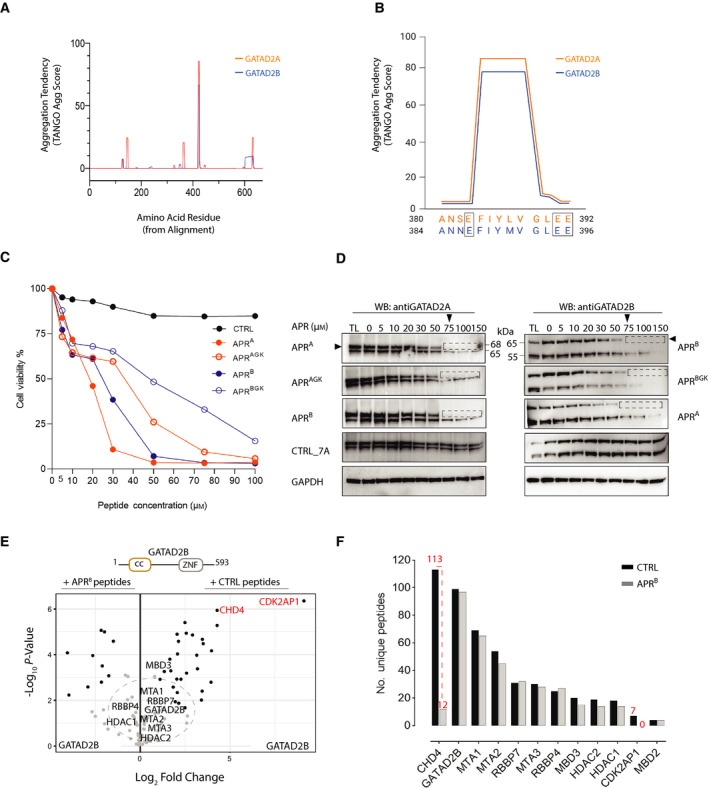
An APR within GATAD2 proteins co‐ordinates CDK2AP1 and CHD4 association with NuRD. (A) TANGO analysis shows β‐sheet aggregation tendency for the APR within GATAD2A and GATAD2B (B) A close‐up schematic of the β‐sheet aggregation tendency of the APRs in GATAD2A (384‐390) and GATAD2B (388‐394) and gatekeeper residues (boxed). (C) Graph representing the MTT assay performed in 2 biological and three technical replicates (n = 6). Cell viability of K562 cells treated with a range of APR peptide concentrations was measured after 48 h. Data are presented as a per cent of untreated cell viability measured using the MTT assay. (D) Western blots of endogenous GATAD2 proteins after treatment of K562 cell lysates with APR‐tat peptides. APR peptidomimetics (0–150 μm) were added to the lysates before sonication. Total lysates (TL, 5%) were taken as input before separation of the soluble and insoluble fractions by centrifugation. Cleared lysate (5% of soluble fraction) was run on SDS‐PAGE and probed for GATAD2 proteins. Black arrows indicate the canonical isoform of GATAD2A and GATAD2B proteins, and dotted rectangular box shows loss of soluble proteins post‐APR treatment. (E) Volcano plot comparing the interactors of overexpressed GATAD2B in the presence of 50 μm APRB (left panel) or CTRL (right panel) peptides. (F) The number of unique peptides of each subunit of the NuRD complex detected by MS after FLAG‐GATAD2B PD‐MS in the presence of APRB and CTRL peptides.
To confirm whether these peptidomimetics were inducing protein aggregation, we titrated APR peptidomimetics into K562 total cell lysates (after sonication and before centrifugation). Addition of APRA and APRB resulted in the aggregation and depletion of soluble endogenous GATAD2A and GATAD2B proteins, whereas the control peptidomimetic had no impact on GATAD2 protein solubility (Fig. 3D). Due to the similarity between the GATAD2A/B APR regions (Fig. 3B), APRA and APRB depleted GATAD2B and GATAD2A equally, indicating no specificity for a particular GATAD2 protein (Fig. 3D). In addition, we did not see significant differences between APR and APRGK peptides’ ability to deplete specific GATAD2 proteins as measured by western blots. We next performed FLAG‐GATAD2B PD‐MS in the presence of 50 μm APRB or CTRL to determine whether the APR mimetic peptides could directly interfere with any PPIs that require the APR (Fig. 3E). We observed a significant loss of NuRD subunits CDK2AP1 and CHD4 in the presence of APRB peptides compared to the CTRL, whilst no significant change was observed for GATAD2B and other NuRD subunits (Fig. 3E). We also examined the total number of peptides detected for all NuRD subunits to ensure that the loss of CHD4 and CDK2AP1 was not due to a reduction in GATAD2B or other subunits of the NuRD complex. Interestingly, we saw no significant difference in the total number of unique peptides (99 vs 97) of GATAD2B and NuRD subunits other than CHD4 and CDK2AP1 (Fig. 3F). In contrast, the number of unique peptides for CHD4 and CDK2AP1 decreased from 113 to 12 and 7 to 0, respectively (Fig. 3F). Based on these results, we conclude that APRB competes away CHD4 and CDK2AP1 from binding to the NuRD complex.
CDK2AP1 interacts with both NuRD and NCOR complexes
CDK2AP1 is a 12‐kDa protein involved in cell cycle regulation and is the least defined subunit of the NuRD complex. Pulldown of a CDK2AP1‐GFP fusion previously demonstrated that CDK2AP1 is a canonical component of the NuRD complex [33]. To corroborate these findings, we used the smaller (~ 1 kDa) FLAG tag to immunoprecipitate CDK2AP1, and our PD‐MS confirmed the enrichment of canonical NuRD subunits using medium stringency washes (Fig. 4A). Notably, we also observed enrichment of nuclear receptor co‐repressor (NCOR)1, NCOR2, TBL1X, TBL1R, GSP2 and HDAC3, which are all canonical subunits of the NCOR complex (Fig. 4A).
Fig. 4.
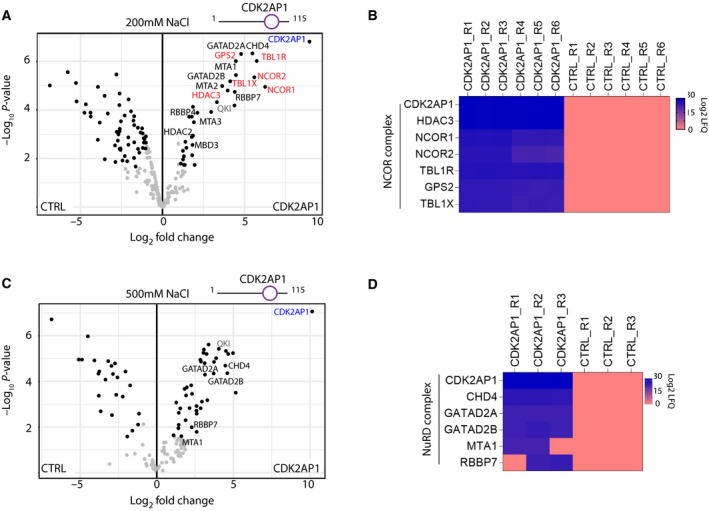
CDK2AP1 interacts with NuRD and NCOR complexes. (A and C) Volcano plots represent enrichment of the NuRD and NCOR subunits in FLAG‐CDK2AP1 and FLAG‐only CTRL pulldowns in the presence of (A) 200 mm NaCl (n = 6) and (C) 500 mm NaCl (n = 3). (B and D) LFQ intensity‐based heatmap of FLAG‐CDK2AP1 PD‐MS versus FLAG‐alone control (CTRL) showing: (B) NCOR complex subunits pulled down only with CDK2AP1 and not in any replicates of CTRL in presence of 200 mm NaCl; (D) Canonical NuRD subunits at 500 mm NaCl.
An LFQ intensity‐based heatmap of the NCOR subunits detected in FLAG‐CDK2AP1 versus CTRL samples confirmed that the interaction was not mediated by beads or FLAG tag (Fig. 4B). This result is supported by the number of unique NCOR complex peptides obtained as well as the low representation of these proteins in databases of common mass spectrometry (MS) contaminants (Table S1). With high stringency washes (500 mm NaCl), the interaction of CDK2AP1 with CHD4 and GATAD2 proteins in particular was not abrogated, suggesting this is a high affinity and direct interaction (Fig. 4C,D). However, at 500 mm NaCl, all interactions of CDK2AP1 with the NCOR complex subunits were lost, suggesting that this interaction might be of low‐to‐medium affinity (Fig. 4C). Interestingly, the RNA‐binding protein QKI, which is involved in mRNA stability, translation and splicing, was also significantly enriched in CDK2AP1 PD‐MS, even with stringent washing conditions (Fig. 4A,C). CDK2AP1‐QKI is a strong and possibly direct interaction but occurs independently of the MGCC module as it was not detected in other PD‐MS experiments in this study.
The C‐terminal end of CHD proteins facilitates engagement with the NuRD and ChAHP complexes
Our previous in vitro co‐IP studies of NuRD subunits revealed that the CHD4 C terminus (aa 1230‐1912) interacts with GATAD2 proteins [7]. In addition, a recent report showed that CHD4 also interacts with the ChAHP complex [16]. Given that the NuRD complex can also incorporate CHD3 or CHD5, we investigated the interactome networks mediated through the C termini of all NuRD‐associated CHD proteins. Accordingly, FLAG‐tagged CHD3‐C, CHD4‐C and CHD5‐C complexes were purified and analysed by MS. We observed strong enrichment of most NuRD canonical subunits (Fig. 5A–C). Interestingly, CDK2AP1 enrichment was seen only with CHD4‐C but not with CHD4‐N and CHD4‐M (Figs 5D and S2), indicating that CDK2AP1 only recognises the C‐terminal half of CHD4 (Fig. 5A).
Fig. 5.
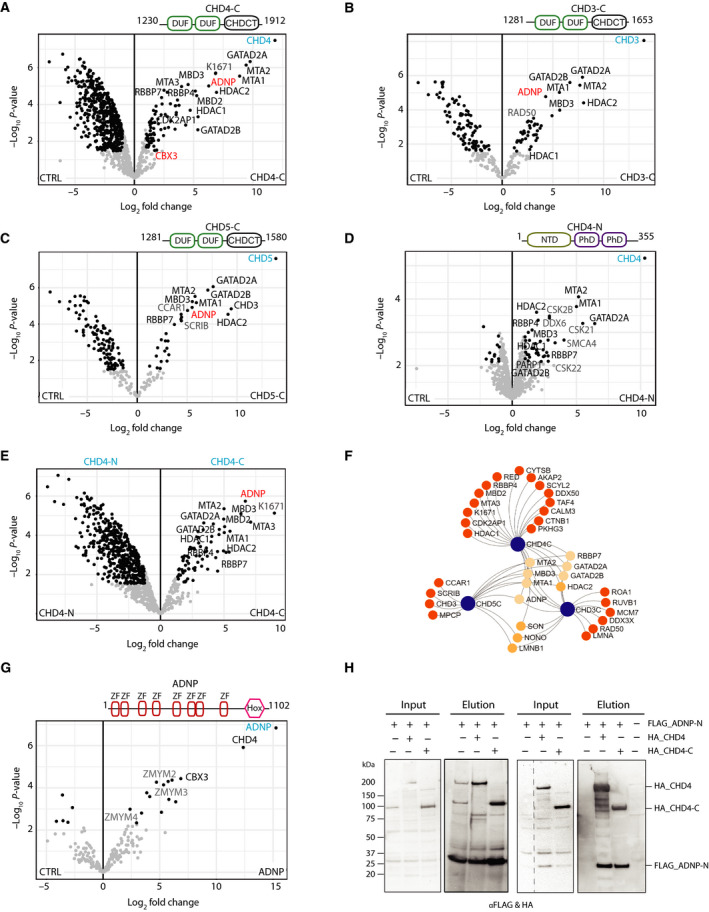
CHD3/4/5 C termini preferentially bind NuRD subunits and ADNP. The canonical subunits of the NuRD are highlighted in black, ADNP in red and potential new interactors in grey. Interaction partners of (A) CHD4‐N (B) CHD3‐C (C) CHD5‐C and (D) CHD4‐N compared to CTRL, and (E) CHD4‐N vs CHD4‐C (F) Network representation of shared and specific interactors of CHD proteins. Significantly enriched proteins with at least twofold change were used to generate the network. Bait proteins are indicated in blue circles, and unique interactors of each family member are indicated in dark orange and shared interactors in light orange. (G) ADNP copurifies with CHD4 and CBX3/HP1γ, known subunits of ChAHP complex. ZMYM family proteins are indicated in grey. (H) Western blots of input and elution samples from Flag‐ADNP‐N (1‐228 aa) co‐expressed with CHD4 protein. Left, pulldown showing that FLAG‐ADNP‐N purified on anti‐FLAG beads pulls down co‐expressed CHD4 full‐length (Lane 2) or CHD4‐C (Lane 3). Right, pulldown showing that CHD4 full‐length (Lane 2) or CHD4‐C (Lane 3) purified on anti‐HA beads pulls down co‐expressed FLAG‐ADNP‐N.
PD‐MS performed with the CHD4 N‐terminal region (CHD4‐N) also showed enrichment of NuRD subunits, suggesting that CHD4‐C is not the sole region responsible for engagement with NuRD (Fig. 5D). However, notable enrichment of NuRD subunits was seen with CHD4‐C when compared to CHD4‐N (Fig. 5E). Notably, we observed enrichment of CSK2B (CSNK2B), CSK21 (CSNK2A1) and CSK22 (CSNK2A2), well‐known subunits of the casein kinase II (CSK2) serine/threonine protein kinase complex, in the CHD4‐N pulldown (Fig. 5D). The CSK2 complex regulates the function of many regulators of chromatin organisation and function and has been shown to phosphorylate serine residues in the CHD4 N terminus [34].
We also observed specific enrichment of activity‐dependent neuroprotective protein (ADNP) with all CHD family members (Fig. 5A–C,F). Since the discovery of the ChAHP complex containing ADNP, CBX3 and CHD4 in 2017 [29], attention has focused on its molecular and cellular function in cancer and neurodegenerative diseases. It has been shown by PD‐MS of full‐length FLAG‐tagged ADNP in HEK293 cells that CHD4 and CBX3 are the top‐enriched proteins [29]. In addition, we saw marked enrichment of three members of the MYM‐type zinc finger family, namely ZMYM2, ZMYM3 and ZMYM4 (Fig. 5G). Significant enrichment of these proteins with ChAHP may suggest a role for MYM‐type proteins in genome organisation and architecture. To corroborate CHD4 interaction with ADNP, we performed co‐immunoprecipitation using FLAG‐tagged ADNP N terminus (ADNP‐N, aa 1‐228) and full‐length HA‐CHD4 or HA‐CHD4‐C (aa 1230‐1912) proteins in HEK293 cells. FLAG‐ADNP‐N immobilised on FLAG beads could pulldown both full‐length HA‐CHD4 and the HA‐CHD4‐C (Fig. 5 H, left panel). Similarly, HA‐CHD4 proteins immobilised on HA beads could also precipitate FLAG‐ADNP‐N (Fig. 5H, right panel). Notably, the C‐terminal half of ADNP failed to express (data not shown).
Cancer missense mutations may change the balance of CHD/NuRD and CHD/ChAHP complexes
The impact of somatic or germline mutations on large, multisubunit complexes such as NuRD is being recognised. We focused on CHD4 as a mutually exclusive partner of both NuRD and ChAHP complexes to determine whether any cancer‐specific missense mutations in the C‐terminal region of CHD4 disrupted its binding to either GATAD2 or ADNP proteins. Previous CRISPR/Cas9 screens in erythroid cells revealed that disruption of aa 1872‐1883 of CHD4 abrogated the CHD4‐GATAD2B interaction [16]. We therefore examined the six missense mutations in CHD4 within or adjacent to this region, which could potentially affect the interaction of CHD4 with either ADNP or GATAD2B (Fig. S3A). To understand the effect of the CHD4 C‐terminal missense mutations in the context of NuRD and ChAHP assembly, we performed pairwise interaction experiments to evaluate the binding of CHD4‐C to full‐length ADNP and GATAD2B proteins. Wild‐type (WT) or mutant HA‐tagged CHD4‐C as well as FLAG‐tagged ADNP and GATAD2B proteins were co‐expressed and their interaction examined in pulldowns followed by western blot experiments. Four of these mutations (D1867N, P1879S, R1890C and N1891D) had no impact on CHD4 interaction with either GATAD2B or ADNP (D1867N is exemplified in Fig. S3B, Lane 2). However, a reduction in CHD4‐CA1866D interaction with full‐length ADNP was observed, when compared to WT CHD4‐C (Fig. S3B). With regard to GATAD2B, only CHD4‐CE1889K showed a clear effect on the interaction when compared to WT (Fig. S3C). These mutations were not completely disruptive but reduced the affinity of CHD4 for either NuRD or ChAHP subunits. These results may suggest that the composition and proportion of CHD4‐NuRD and CHD4‐ChAHP complexes might be perturbed in cancer cells carrying these particular CHD4‐C mutations.
Fixed and altered stoichiometries were observed for MHR and MGCC modules, respectively
Understanding the stoichiometry of the NuRD complex subunits will help delineate its structure and function. To this aim, we used the iBAQ‐adjusted intensity values as described previously [6, 35]. Because of the high sequence similarity between paralogues (i.e. RBBP4 is 92% identical to RBBP7), we first considered each set of paralogues as a single group and averaged their iBAQ values to assess stoichiometry. The averaged values were divided by the averaged MTA value and multiplied by 2 because based on the published X‐ray crystal structure, in which two molecules of MTA are found in an intact NuRD complex. Of note, bait proteins were excluded because they are in excess and introduce a bias in stoichiometry calculations. MBD and GATAD2 have been considered as exclusive subunits of the NuRD complex. Thus, we used their iBAQ data for stoichiometry calculations. Using MBD PD‐MS data, we calculated stoichiometric ratios of ~ 1 : 0.3 : 0.1 : 2 : 2 : 4 for GATAD2:CHD:CDK2AP1:MTA:HDAC:RBBP4 (Fig. 6A). The 2 : 2 : 4 ratios for MHR exactly matches the previous crystal structures [19], as well as MS quantifications [6, 12, 35, 36]. Similarly, when GATAD2B was used as bait, the ratio for the MHR module remained constant (2 : 2 : 4) but we calculated a ratio of ~ 2 : 1 : 2 for MBD:CHD:CDK2AP1 (Fig. 6B).
Fig. 6.
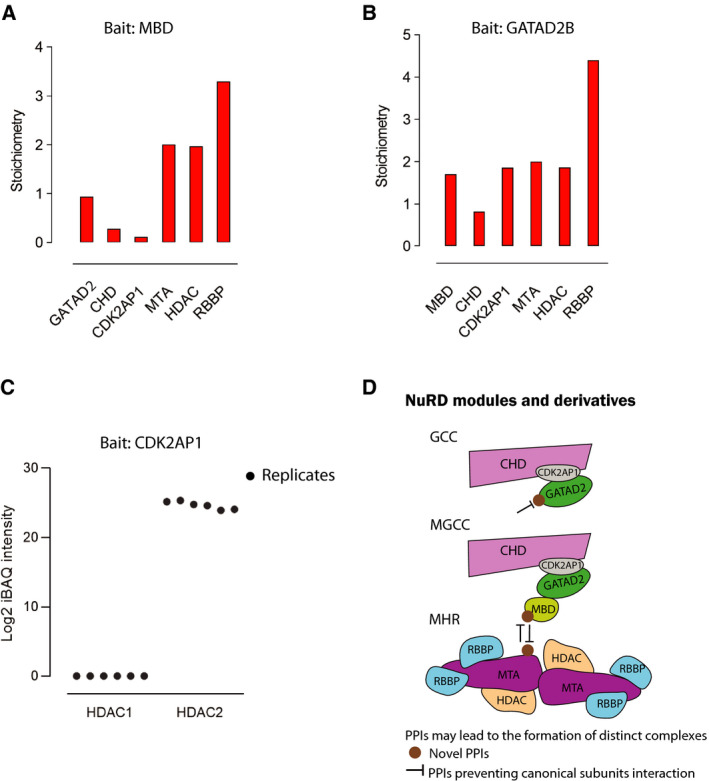
Stoichiometry of the NuRD subunits. MBD2/3 and GATAD2B were used as bait. Bait proteins were excluded from calculations, because they are in excess amount and introduce a bias in stoichiometry calculations. Stoichiometry of the subunits where (A) MBD2/3 and (B) GATAD2B were used as bait. (C) Scatter plot showing log2 of iBAQ intensities for HDAC1 and HDAC2 in all six replicates of CDK2AP1 PD‐MS. (D) Schematic representation of the possible new subcomplexes independent of the NuRD complex that can be formed via PPIs occurring with bridging subunits.
Approximately the same number of unique peptides were detected for HDAC1/2 (~ 20) in NuRD subunits PD‐MS, but no iBAQ intensity values were calculated for HDAC1 in CDK2AP1 PD‐MS suggesting that only HDAC2 but not HDAC1 was copurified with the CDK2AP1/NuRD complex (Fig. 6C). Further investigation of the number of unique peptides detected for HDAC2 vs HDAC1 in CDK2AP1 PD‐MS was 6 to 2, respectively (Fig. S4A). To ensure that this observation is not due to lack of HDAC1 expression , we further analysed the transcript and protein expression levels of both HDAC1/2 (RNA‐Seq dataset from Human Protein Atlas [37] and shotgun proteomics dataset from ProteomicsDB [38]) in HEK293 cells. These data show that HDAC1/2 express at the same level, and thus, absence of HDAC1 in CDK2AP1 pulldowns was not related to expression. We conclude that this observation is linked to the assembly and architecture and consequently interaction of the HDAC1 with the CDK2AP1/NuRD complex (Fig. S4B,C).
Discussion
The MGCC module of the NuRD complex plays a diverse role in almost all stages of development and in many disease states in metazoans [39, 40]. A central question concerning multisubunit assemblies such as MGCC is how specificity is achieved whilst the complex is recruited to specific genomic loci. It is generally acknowledged that subunit‐specific protein interactions provide some of this regulatory specificity to multiprotein complexes. For example, two groups recently demonstrated that PWWP2A, an H2A.Z‐binding protein, binds to MTA1 and separates MHR from MGCC, thus forming PWWP2A‐MTA1‐HDAC1/2‐RBBP4/7 complexes [41, 42]. It is therefore plausible that NuRD subunit‐specific binding partners that we report in this study could give rise to a combination of additional NuRD subcomplexes that would add to this functional diversity by creating other NuRD species with distinct compositions and stoichiometries. Our interactome data lay the foundation for future studies to investigate the functional readout of such complexes.
Recently, Sher et al. demonstrated that disruption of the MBD2/NuRD axis, but not MBD3/NuRD, leads to the derepression of fetal haemoglobin genes [16, 17]. MBD2 is a methyl‐CpG‐binding protein; however, the proximal promoter of γ‐globin and the entire β‐globin locus is depleted of CpG islands, suggesting that MBD2 may not be acting to directly bind methylated DNA at the β‐globin locus. Identification of a PPI network for MBD2a and MBD2b isoforms could potentially help to define a mechanism of their function and pave the way for more targeted molecular therapies. Here, we demonstrate that MBD2 isoforms copurify with dozens of proteins involved in gene regulation and genome structure organisation; these proteins could potentially contribute to γ‐globin repression. It is highly likely that other proteins – such as ZNF219 – might facilitate the binding of MBD2/NuRD to the β‐globin gene locus.
Notably, enrichment of ZNF219 and CDK2AP1 with MBD2 but not MBD3 does not necessarily indicate that they do not interact with MBD3/NuRD. Regarding ZNF219, we have previously reported the enrichment of ZNF219 with MBD3/NuRD in NTERA‐2 cells, where MBD2 is not expressed [6]. Together, these data might indicate that there might be a competition between MBD2/NuRD and MBD3/NuRD for binding to ZNF219. Thus, in the presence of MBD2, the interaction of ZNF219‐MBD3/NuRD could occur at very low stoichiometry and below the detection limit of our liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) method in this cell line (HEK293). Our data reveal that ZNF219/MBD2b‐NuRD and CDK2AP1/MBD2(a/b)‐NuRD are more abundant compared to the MBD3‐NuRD complex in HEK293 cells. Future functional studies of these complexes in relevant cell lines such as Human Umbilical Cord Derived Erythroid Progenitor‐2 (HUDEP) cells could help define their function.
MBD PD‐MS iBAQ data show substoichiometric ratio for CDK2AP1 and CHD4, which is consistent with previous studies. This may also indicate that the majority of MBD/NuRD complexes lack CHD and CDK2AP1 proteins. If true, the MBD/NuRD complex that carries both deacetylation and remodelling activities might be less abundant compared to MBD/NuRD with only deacetylase activity. The increase in ratios of MBD, CHD4 and CDK2AP1 when GATAD2B is used as bait might also imply that the MBD‐GATAD2‐CDK2AP1‐CHD assembly is present as an independent complex with remodelling activity.
In conclusion, we report several specific protein interactions for the subunits of the MGCC module and further report the presence of new subcomplexes independent of the NuRD complex such as GATAD2‐CDK2AP1‐CHD and CDK2AP1/NCOR complexes (Fig. 6D). Future studies may shed more light on the biological function of these interactors and subcomplexes in gene regulation in normal and disease states. The generation of recombinant protein complexes or tandem purification of the complexes followed by biophysical analysis might reveal the precise stoichiometry and structure and molecular function of these derivative complexes.
Materials and methods
Plasmid constructs
All genes used in this study were cloned into pcDNA3.1(+) vector using Gibson Assembly and were either N‐terminally FLAG‐ or HA‐tagged. Except for ADNP (obtained as a cDNA from Horizon Discovery (GenBank #BC075794)) and GATAD2B‐CDel constructs, the rest were a kind gift from Professor Joel Mackay, The University of Sydney. A list of primers used for Gibson Assembly of all cDNAs into pcDNA3.1(+) are available on request.
Design of APR peptides
The GATAD2A/B protein sequences were analysed by TANGO to detect APRs [32]. Default physicochemical parameters were selected as below: temperature, 298 K; pH 7.5; ionic strength, 0.02 m; and concentration, 1 m. An aggregation score of 5 was set as a cut‐off per residue. The residues spanning each APR were combined with an 11 aa portion of HIV‐1 Tat protein (YGRKKRRQRRR) to enhance cell permeability. Peptides were synthesised to at least 80% purity by HPLC at Mimotopes, Australia.
Cell culture and transfection
K562 cells were grown in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum, penicillin (100 U·mL−1) and streptomycin (100 μg·mL−1). Expi293F™ cells were grown to a density of 1.5 × 106 cells·mL−1 in Expi293™ Expression Medium (Thermo Fisher Scientific, Waltham, MA, USA). Combinations of equimolar quantities of constructs were cotransfected into cells using linear polyethylenimine (PEI) (Polysciences, Warrington, PA, USA). DNA (4 μg) was first diluted in 200 μL of PBS and vortexed briefly. PEI (8 μL, 1 mg·mL−1) was then added, and the mixture was vortexed again, incubated for 20 min at room temperature and then added to 1.9 mL of cells in a 12‐well plate. The cells were incubated for 65–72 h at 37 °C, 5% CO2 in a humidified incubator on a horizontal orbital shaker (130 rpm). Aliquots of cells (1 mL) were then harvested, washed twice with PBS, centrifuged (300 g , 5 min), snap‐frozen in liquid nitrogen and stored at −80 °C.
Cell lysate and APR treatment for aggregation analysis
K562 cells were lysed using 500 μL lysis buffer containing 50 mm Tris‐HCl, 150 mm NaCl, 0.5% IGEPAL, pH 7.5, 1x protease inhibitor cocktail (Sigma‐Aldrich, St. Louis, MO, USA), 1 mm DTT and 1 μL Pierce™ Universal Nuclease (Thermo Fisher Scientific). Then, cells were sonicated for 5 cycles, 1 min ON/30 s OFF. After sonication, total lysate was collected and the rest of the lysate was aliquoted into new 1.5 mL Eppendorf tubes, and then, known concentration of APR peptides was added to each tube and incubated for 30 min at 4 °C. Next, tubes were spun at 20 000 g for 30 min to separate the soluble and insoluble fractions.
In vitro protein expression and Co‐IP
DNA constructs were transcribed and translated in vitro in pairs using equimolar plasmids in 70 μL of TNT® Quick Coupled Transcription/Translation System (Promega). RNaseOUT (0.5 μL, Thermo Fisher Scientific), methionine (2 μL, 1 mm) and 1x protease inhibitor (Sigma‐Aldrich) were added to each reaction. The reactions were incubated at 30 °C for 3 h. Prior to immunoprecipitation, 500 μL lysis buffer (50 mm Tris‐HCl, 150 mm NaCl, 0.1% (v/v) Triton X‐100, 1 mm DTT, pH 7.5) was added to the reactions. Input (5% of total) was collected, and the remainder was mixed with 20 μL of anti‐FLAG Sepharose 4B beads (Sigma‐Aldrich) at 4 °C for 2 h on a rotator. Beads then were washed with 500 μL of wash buffer (50 mm Tris‐HCl, 200 or 500 mm NaCl, 0.5% (v/v) IGEPAL® CA630, 0.2 mm DTT, pH 7.5) for 5 times. Finally, elution was performed three times each using 20 μL of a 150 μg·μL−1 stock of 3xFLAG peptides (Sigma‐Aldrich). Western blot analysis was done as previously described [7]. Antibodies used in this study are as follows: GATAD2A (#A302‐358A, Bethyl Laboratories, Montgomery, TX, USA), GATAD2B (#A301‐281A, Bethyl Laboratories), FLAG (#A8592‐1, Sigma‐Aldrich), HA (#2999, Cell Signalling Technology, Beverly, MA, USA) and GAPDH (#ab8245, Abcam, Inc., Cambridge, MA, USA).
Sample preparation and tandem MS
Label‐free FLAG pulldowns were performed in at least triplicate. Nuclei of transiently‐transfected Expi293F cells were lysed in the same lysis buffer as mentioned above and after sonication and spinning incubated with 20 μL anti‐FLAG beads (Sigma‐Aldrich). After incubation for 2 h, five washes were performed: 3 washes with a buffer containing [200 mm NaCl, 50 mm Tris‐HCl, 0.5% (v/v) IGEPAL, PH 7.5], and two washes with the same buffer lacking IGEPAL. Affinity‐purified proteins were subject to on‐bead trypsin digestion, where 20 μL of digestion buffer (2 m urea freshly dissolved in 50 mm Tris‐HCl, 1 mm DTT, 100 ng trypsin, 20 ng LysC (Promega)) was added and then vortexed in 30 °C for 2 h. Next, the beads were collected, and supernatant was transferred into LoBind tubes. Beads were resuspended in 20 μL 2 m urea containing 10 mm IAA in the dark for 20 min. The supernatant was transferred to the previous tube and incubated at 30 °C for 16 h. The following day, tryptic peptides were acidified to a final concentration of 2% (v/v) with formic acid (Sigma‐Aldrich) and desalted using StageTips (Thermo Fisher Scientific). Peptides were dried in a SpeediVac and dissolved in 10 μL 0.1% (v/v) formic acid. LC‐MS/MS analysis was performed on an UltiMate™ 3000 RSLCnano System (Thermo Fisher Scientific) system connected to a Thermo Scientific Q‐Exactive HF‐X hybrid quadrupole‐Orbitrap mass spectrometer equipped with a standard nanoelectrospray source (Thermo Fisher Scientific). Peptides (3 μL) were injected onto a C18 column (35 cm x 75 μm inner diameter column packed in‐house with 1.9 μm C18AQ particles). Peptides were separated at a flow rate of 200 nL·min−1 using a linear gradient of 5%–30% buffer B over 30 min. Solvent A consisted of 0.1% (v/v) formic acid, and solvent B consisted of 80% (v/v) acetonitrile and 0.1% (v/v) formic acid. The end‐to‐end run time was 45 min, including sample loading and column equilibration times. The mass spectrometer was set to a data‐dependent acquisition mode (DDA). In the DDA run, each full‐scan MS1 was operated as follows: mass scan range was between 300 and 1600 m·z−1 at resolution of 60 000. The top 15 most intense precursor ions were selected to be fragmented in the Orbitrap via high‐energy collision dissociation activation. MS2 scan was operated as follows: mass scan range was between 200 and 2000 m·z−1 at resolution of 15 000, 1 x 105 AGC target and 1.4 m·z−1 isolation window.
Raw data analysis
Raw data were analysed by MaxQuant (version 1.6.6.0) using standard settings. Additional parameters including carbamidomethyl cysteine (C) and methionine oxidation (M) were selected as fixed and variable modifications, respectively, and LysC and trypsin were selected as proteolytic enzymes. The human proteome (Proteome ID UP000005640) was used as the reference proteome. The generated proteingroups.txt table in conjunction with an experimental design text file was used to perform all statistical analyses using LFQ values in R studio as described elsewhere [43, 44]. Perseus algorithm was used to impute the missing values. Proteins that had two missing values were discarded from the analysis . For better data visualisation, the output files were further processed. First, all proteins with fold change > 2 and statistically significant were kept. Then, proteins including heat shock, ribosomal, keratin or proteins with mitochondrial and cytoplasmic localisation were excluded from the list. A full list of unfiltered interactors is listed in the data file. To determine the stoichiometry of the subunits, the iBAQ‐adjusted intensity values (iBAQ; is an approximate calculation of protein copy numbers by dividing the sum of intensities of all experimentally detected peptides by the number of theoretically observable peptides for each protein) for MTAs were averaged and then intensity values for known NuRD components were divided by the MTA value and multiplied by 2 because based on published X‐ray crystal structure two molecules of HDAC1 and two molecules of MTA1 make the core of the NuRD complex [19]. Due to high sequence similarity between paralogues (i.e. RBBP4 is 92% identical to RBBP7), we first considered each set of paralogues as a single group. NetworkAnalyst tool was used for network visualisation of the unique and shared interactors LFQ and iBAQ are two different quantification algorithms: iBAQ is an approximate calculation of protein copy numbers and is the best at determining ratio changes within samples not across samples, whereas LFQ is the best representative of ratio changes between samples. Of note, the presence of false positives is inevitable in immunoprecipitation studies.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
MST and CGB conceived the study, designed MS experiments, analysed data and wrote the manuscript. CG, YF and HF prepared samples for MS experiments and performed western blots and MTT assays. JKKL designed and cloned constructs and reviewed the manuscript. HM provided additional data. JPM reviewed the manuscript and provided intellectual input on the studies and manuscript. JEJR reviewed the manuscript and provided intellectual input, research governance and scientific discussion.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.16112.
Supporting information
Fig. S1. CDK2AP1 interacts with the C‐terminal of GATAD2B.
Fig. S2. Enriched proteins with CHD4‐M.
Fig. S3. Cancer‐associated missense mutation in the C‐terminal region of CHD4 may change the balance of CHD4/NuRD vs CHD4/ChAHP.
Fig. S4. HDAC2‐NuRD is more abundant as compared to HDAC1‐NuRD.
Table. S1. The NCOR complex.
Acknowledgements
We thank Cynthia Metierre for assisting with MTT assays. We also appreciate the technical assistance we received from the Mass Spectrometry core facility at The University of Sydney. Financial support was provided by Tour de Cure grants to CGB and JEJR; National Health & Medical Research Council funding (Investigator Grant #1177305 and Project Grant and #1128748 to J.E.J.R); support grants from Cure The Future Foundation and an Anonymous Foundation.
Data accessibility
All raw MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD025263.
References
- 1. Mor N, Rais Y, Sheban D, Peles S, Aguilera‐Castrejon A, Zviran A, Elinger D, Viukov S, Geula S, Krupalnik V et al. (2018) Neutralizing Gatad2a‐Chd4‐Mbd3/NuRD complex facilitates deterministic induction of naive pluripotency. Cell Stem Cell 23, 412–425.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lai AY & Wade PA (2011) Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 11, 588–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaji K, Caballero IM, MacLeod R, Nichols J, Wilson VA & Hendrich B (2006) The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat Cell Biol 8, 285–292. [DOI] [PubMed] [Google Scholar]
- 4. Zhang Y, Ng HH, Erdjument‐Bromage H, Tempst P, Bird A & Reinberg D (1999) Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev 13, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xue Y, Wong J, Moreno GT, Young MK, Cote J & Wang W (1998) NURD, a novel complex with both ATP‐dependent chromatin‐remodeling and histone deacetylase activities. Mol Cell 2, 851–861. [DOI] [PubMed] [Google Scholar]
- 6. Sharifi Tabar M, Mackay JP & Low JKK (2019) The stoichiometry and interactome of the Nucleosome Remodeling and Deacetylase (NuRD) complex are conserved across multiple cell lines. FEBS J 286, 2043–2061. [DOI] [PubMed] [Google Scholar]
- 7. Torrado M, Low JKK, Silva APG, Schmidberger JW, Sana M, Sharifi Tabar M, Isilak ME, Winning CS, Kwong C, Bedward MJ et al. (2017) Refinement of the subunit interaction network within the nucleosome remodelling and deacetylase (NuRD) complex. FEBS J 284, 4216–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cox J & Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.‐range mass accuracies and proteome‐wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- 9. Schmidberger JW, Sharifi Tabar M, Torrado M, Silva AP, Landsberg MJ, Brillault L, AlQarni S, Zeng YC, Parker BL, Low JK et al. (2016) The MTA1 subunit of the nucleosome remodeling and deacetylase complex can recruit two copies of RBBP4/7. Protein Sci 25, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Millard CJ, Varma N, Saleh A, Morris K, Watson PJ, Bottrill AR, Fairall L, Smith CJ & Schwabe JW (2016) The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife 5, e13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Millard CJ, Fairall L, Ragan TJ, Savva CG & Schwabe JWR (2020) The topology of chromatin‐binding domains in the NuRD deacetylase complex. Nucleic Acids Res 48, 12972–12982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Low JKK, Silva APG, Sharifi Tabar M, Torrado M, Webb SR, Parker BL, Sana M, Smits C, Schmidberger JW, Brillault L et al. (2020) The nucleosome remodeling and deacetylase complex has an asymmetric, dynamic, and modular architecture. Cell Rep 33, 108450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhong Y, Paudel BP, Ryan DP, Low JKK, Franck C, Patel K, Bedward MJ, Torrado M, Payne RJ, van Oijen AM et al. (2020) CHD4 slides nucleosomes by decoupling entry‐ and exit‐side DNA translocation. Nat Commun 11, 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Farnung L, Ochmann M & Cramer P (2020) Nucleosome‐CHD4 chromatin remodeler structure maps human disease mutations. Elife 9, e56178. 10.7554/eLife.56178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amaya M, Desai M, Gnanapragasam MN, Wang SZ, Zu Zhu S, Williams DC Jr & Ginder GD (2013) Mi2β‐mediated silencing of the fetal γ‐globin gene in adult erythroid cells. Blood 121, 3493–3501. 10.1182/blood-2012-11-466227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sher F, Hossain M, Seruggia D, Schoonenberg VAC, Yao Q, Cifani P, Dassama LMK, Cole MA, Ren C, Vinjamur DS et al. (2019) Rational targeting of a NuRD subcomplex guided by comprehensive in situ mutagenesis. Nat Genet 51, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu X, Azzo A, Bilinovich SM, Li X, Dozmorov M, Kurita R, Nakamura Y, Williams DC Jr & Ginder GD (2019) Disruption of the MBD2‐NuRD complex but not MBD3‐NuRD induces high level HbF expression in human adult erythroid cells. Haematologica 104, 2361–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kim MY, Na I, Kim JS, Son SH, Choi S, Lee SE, Kim JH, Jang K, Alterovitz G, Chen Y et al. (2019) Rational discovery of antimetastatic agents targeting the intrinsically disordered region of MBD2. Sci Adv 5, eaav9810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L & Schwabe JW (2013) Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell 51, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Millard CJ, Fairall L & Schwabe JW (2014) Towards an understanding of the structure and function of MTA1. Cancer Metastasis Rev 33, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gnanapragasam MN, Scarsdale JN, Amaya ML, Webb HD, Desai MA, Walavalkar NM, Wang SZ, Zu Zhu S, Ginder GD & Williams DC Jr (2011) p66Alpha‐MBD2 coiled‐coil interaction and recruitment of Mi‐2 are critical for globin gene silencing by the MBD2‐NuRD complex. Proc Natl Acad Sci USA 108, 7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E & Stunnenberg HG (2006) MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol 26, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cramer JM, Scarsdale JN, Walavalkar NM, Buchwald WA, Ginder GD & Williams DC Jr (2014) Probing the dynamic distribution of bound states for methylcytosine‐binding domains on DNA. J Biol Chem 289, 1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Desai MA, Webb HD, Sinanan LM, Scarsdale JN, Walavalkar NM, Ginder GD & Williams DC Jr (2015) An intrinsically disordered region of methyl‐CpG binding domain protein 2 (MBD2) recruits the histone deacetylase core of the NuRD complex. Nucleic Acids Res 43, 3100–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fujita H, Fujii R, Aratani S, Amano T, Fukamizu A & Nakajima T (2003) Antithetic effects of MBD2a on gene regulation. Mol Cell Biol 23, 2645–2657. 10.1128/mcb.23.8.2645-2657.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wood KH & Zhou Z (2016) Emerging molecular and biological functions of MBD2, a reader of DNA methylation. Front Genet 7, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Silva AP, Ryan DP, Galanty Y, Low JK, Vandevenne M, Jackson SP & Mackay JP (2016) The N‐terminal Region of Chromodomain Helicase DNA‐binding Protein 4 (CHD4) is essential for activity and contains a high mobility group (HMG) box‐like‐domain that can bind poly(ADP‐ribose). J Biol Chem 291, 924–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mansfield RE, Musselman CA, Kwan AH, Oliver SS, Garske AL, Davrazou F, Denu JM, Kutateladze TG & Mackay JP (2011) Plant homeodomain (PHD) fingers of CHD4 are histone H3‐binding modules with preference for unmodified H3K4 and methylated H3K9. J Biol Chem 286, 11779–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ostapcuk V, Mohn F, Carl SH, Basters A, Hess D, Iesmantavicius V, Lampersberger L, Flemr M, Pandey A, Thoma NH et al. (2018) Activity‐dependent neuroprotective protein recruits HP1 and CHD4 to control lineage‐specifying genes. Nature 557, 739–743. [DOI] [PubMed] [Google Scholar]
- 30. Kaaij LJT, Mohn F, van der Weide RH, de Wit E & Buhler M (2019) The ChAHP complex counteracts chromatin looping at CTCF sites that emerged from SINE expansions in mouse. Cell 178, 1437–1451.e14. [DOI] [PubMed] [Google Scholar]
- 31. Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V & Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res 43, D512–D520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fernandez‐Escamilla AM, Rousseau F, Schymkowitz J & Serrano L (2004) Prediction of sequence‐dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22, 1302–1306. [DOI] [PubMed] [Google Scholar]
- 33. Spruijt CG, Bartels SJ, Brinkman AB, Tjeertes JV, Poser I, Stunnenberg HG & Vermeulen M (2010) CDK2AP1/DOC‐1 is a bona fide subunit of the Mi‐2/NuRD complex. Mol Biosyst 6, 1700–1706. [DOI] [PubMed] [Google Scholar]
- 34. Zhao Z, Senturk N, Song C & Grummt I (2018) lncRNA PAPAS tethered to the rDNA enhancer recruits hypophosphorylated CHD4/NuRD to repress rRNA synthesis at elevated temperatures. Genes Dev 32, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smits AH, Jansen PW, Poser I, Hyman AA & Vermeulen M (2013) Stoichiometry of chromatin‐associated protein complexes revealed by label‐free quantitative mass spectrometry‐based proteomics. Nucleic Acids Res 41, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kloet SL, Baymaz HI, Makowski M, Groenewold V, Jansen PW, Berendsen M, Niazi H, Kops GJ & Vermeulen M (2015) Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J 282, 1774–1785. [DOI] [PubMed] [Google Scholar]
- 37. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A et al. (2015) Tissue‐based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 38. Samaras P, Schmidt T, Frejno M, Gessulat S, Reinecke M, Jarzab A, Zecha J, Mergner J, Giansanti P, Ehrlich HC et al. (2020) ProteomicsDB: a multi‐omics and multi‐organism resource for life science research. Nucleic Acids Res 48, D1153–D1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O'Shaughnessy‐Kirwan A, Signolet J, Costello I, Gharbi S & Hendrich B (2015) Constraint of gene expression by the chromatin remodelling protein CHD4 facilitates lineage specification. Development 142, 2586–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reynolds N, Latos P, Hynes‐Allen A, Loos R, Leaford D, O'Shaughnessy A, Mosaku O, Signolet J, Brennecke P, Kalkan T et al. (2012) NuRD suppresses pluripotency gene expression to promote transcriptional heterogeneity and lineage commitment. Cell Stem Cell 10, 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Link S, Spitzer RMM, Sana M, Torrado M, Völker‐Albert MC, Keilhauer EC, Burgold T, Pünzeler S, Low JKK, Lindström I et al. (2018) PWWP2A binds distinct chromatin moieties and interacts with an MTA1‐specific core NuRD complex. Nat Commun 9. 10.1038/s41467-018-06665-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang T, Wei G, Millard CJ, Fischer R, Konietzny R, Kessler BM, Schwabe JWR & Brockdorff N (2018) A variant NuRD complex containing PWWP2A/B excludes MBD2/3 to regulate transcription at active genes. Nat Commun 9, 3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shah AD, Goode RJA, Huang C, Powell DR & Schittenhelm RB (2020) LFQ‐analyst: an easy‐to‐use interactive web platform to analyze and visualize label‐free proteomics data preprocessed with MaxQuant. J Proteome Res 19, 204–211. [DOI] [PubMed] [Google Scholar]
- 44. Tyanova S & Cox J (2018) Perseus: a bioinformatics platform for integrative analysis of proteomics data in cancer research. Methods Mol Biol 1711, 133–148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. CDK2AP1 interacts with the C‐terminal of GATAD2B.
Fig. S2. Enriched proteins with CHD4‐M.
Fig. S3. Cancer‐associated missense mutation in the C‐terminal region of CHD4 may change the balance of CHD4/NuRD vs CHD4/ChAHP.
Fig. S4. HDAC2‐NuRD is more abundant as compared to HDAC1‐NuRD.
Table. S1. The NCOR complex.
Data Availability Statement
All raw MS data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD025263.