

Abstract
Hemizygous missense variants in the RPL10 gene encoding a ribosomal unit are responsible for an X‐linked syndrome presenting with intellectual disability (ID), autism spectrum disorder, epilepsy, dysmorphic features, and multiple congenital anomalies. Among 15 individuals with RPL10‐related disorder reported so far, only one patient had retinitis pigmentosa and microcephaly was observed in approximately half of the cases. By exome sequencing, three Italian and one Spanish male children, from three independent families, were found to carry the same hemizygous novel missense variant p.(Arg32Leu) in RPL10, inherited by their unaffected mother in all cases. The variant, not reported in gnomAD, is located in the 28S rRNA binding region, affecting an evolutionary conserved residue and predicted to disrupt the salt‐bridge between Arg32 and Asp28. In addition to features consistent with RPL10‐related disorder, all four boys had retinal degeneration and postnatal microcephaly. Pathogenic variants in genes responsible for inherited retinal degenerations were ruled out in all the probands. A novel missense RPL10 variant was detected in four probands with a recurrent phenotype including ID, dysmorphic features, progressive postnatal microcephaly, and retinal anomalies. The presented individuals suggest that retinopathy and postnatal microcephaly are clinical clues of RPL10‐related disorder, and at least the retinal defect might be more specific for the p.(Arg32Leu) RPL10 variant, suggesting a specific genotype/phenotype correlation.
Keywords: intellectual disability, microcephaly, ocular, retinal dystrophy, RPL10
1. INTRODUCTION
More than 100 genes have been implicated in X‐linked intellectual disability (ID), and among them, the Ribosomal Protein L10 gene (RPL10) on the Xq28 region has been recently found to be responsible for a neurodevelopmental disorder with a wide spectrum of clinical presentations (Brooks et al., 2014; Chiocchetti et al., 2011; Klauck et al., 2006; Lubs et al., 2012; Striano & Zara, 2017). RPL10 encodes a multifunctional component of the ribosomal 60S subunit. 35 rRNA and 46 proteins are involved in the biogenesis of the 60S ribosomal subunit (Konikkat & Woolford, 2017). Pathogenic variants in several of the proteins forming the 60S ribosomal unit have been found to be responsible for an heterogeneous group of disorders named ribosomopathies (Farley‐Barnes et al., 2019; Kampen et al., 2020). Among these disorders, Diamond‐Blackfan anemia (DBA), Shwachman‐Diamond syndrome (SD‐S), and Treacher Collins syndrome (TCS) are the most studied. Despite ribosomes being ubiquitous, ribosomopathies might result in tissue‐specific impairments, since cell‐ and tissue‐type specific ribosomal proteins expresssion.
Hemizygous pathogenic variants in RPL10 have been described in only 15 patients with a wide spectrum of manifestations ranging from isolated autism spectrum disorder (ASD) and/or ID, to a complex disease including growth retardation, microcephaly, seizures, exotropia, laryngomalacia, genitourinary anomalies (cryptorchidism, hypospadias, and vesicoureteral reflux), spondyloepiphyseal dysplasia, and other skeletal anomalies (Bourque et al., 2018; Brooks et al., 2014; Chiocchetti et al., 2011; Klauck et al., 2006; Nijman et al., 2014; Thevenon et al., 2015; Vissers et al., 2016; Zanni et al., 2015).
We report four further boys, from three independent families, who all carry the same novel missense RPL10 variant. All patients presented with neurodevelopmental disorders, postnatal microcephaly, dysmorphic features, and retinal dysfunction. Moreover, we reviewed the clinical and genetic features of previously reported patients, to delineate the phenotypic spectrum of RPL10‐related disorder and establishing genotype–phenotype correlations.
2. PATIENTS AND METHODS
All patients were referred for clinical genetic evaluations because of ID and/or behavioral problems associated with dysmorphic features and congenital anomalies. After a normal diagnostic work‐up including array‐comparative genomic hybridization (CGH) and targeted next generation sequencing, the patients underwent trio exome sequencing (ES). Three of the four patients were enrolled in a research study approved by the Ethics Committee of Federico II University Hospital in Naples, Italy (48/16) whereas the fourth case underwent ES as part of his diagnostic evaluation. Genomic DNA from the patients and their parents underwent ES, enriched using the SureSelect Clinical Research Exome (Agilent, Technologies, Santa Clara, CA, USA) and NextSeq 500 sequencing system was used (Illumina, San Diego, CA, USA). A custom pipeline based on Burrows‐Wheeler Alignment tool, Genome Analysis Toolkit, and ANNOVAR (Wang et al., 2010) were used to call, annotate, filter, and prioritize variants (Musacchia et al., 2018). PyMOL (www.pymol.org) was used for in‐silico studies and molecular protein rendering.
2.1. Literature review
We reviewed publications (case reports and reviews) from 2006 to 2022 through PubMed (https://www.ncbi.nlm.nih.gov/pubmed), Decipher (https://www.deciphergenomics.org/), Online Mendelian Inheritance of Man, Human Gene Mutation Database, and Google Scholar using the following key words: “RPL10” and “ribosomopathy.” We retrieved 15 cases carrying pathogenic variants in RPL10. For each individual, we collected gender, sequencing data (including inheritance), clinical findings (perinatal issues, developmental milestones, organ and system involvement, and brain MRI abnormalities), and growth parameters.
3. CLINICAL REPORTS
3.1. Individual 1.1
The proband is the only child of a nonconsanguineous couple. Both the father and the paternal grandfather of the proband had isolated bilateral palpebral ptosis. Prenatal ultrasounds were unremarkable. He was born by cesarean section at 37 weeks of gestation complicated by maternal hypertension. Apgar score at 1' was 8 and at 5' was 9. The birth weight was 2900 g (7th centile). In the first days of life, he had an episode of symptomatic hypocalcemia with tremors that required calcium supplementation. He held his head at 6 months of age, he stood without support at 12 months, and at 13 months he started walking independently. He pronounced the first words at 7 months of age, but language skills were delayed. At 7 years and 7 months, he could pronounce 200 words and could make sentences of two to three words.
He suffered from constipation and underwent surgery for cryptorchidism. At a clinical evaluation, performed at the age of 8 years, his weight was 32.8 kg (94th centile), his height was 123 cm (20th centile) and his occipito‐frontal circumference (OFC) was 48.5 cm (standard deviation score [SDS] = −3). He had brachycephaly, bilateral palpebral ptosis, epicanthus, bilateral crumpled helix, depressed nasal root, long and flat philtrum, prognathism, bifid uvula, and small and spaced teeth (Figure 1a). He also was noted to have brachydactyly and bilateral clinodactyly of the fifth toe. X‐ray of the left hand showed hypoplasia of the middle phalanx of the fifth finger. Brain magnetic resonance imaging (MRI) showed mild hypoplasia of the lower portion of the cerebellar vermis. Array‐CGH and testing for Fragile X syndrome were both normal. For the suspicion of Kabuki syndrome Sanger sequencing of KMT2D and KDM6A prior of ES was performed but failed to reveal pathogenic variants.
FIGURE 1.
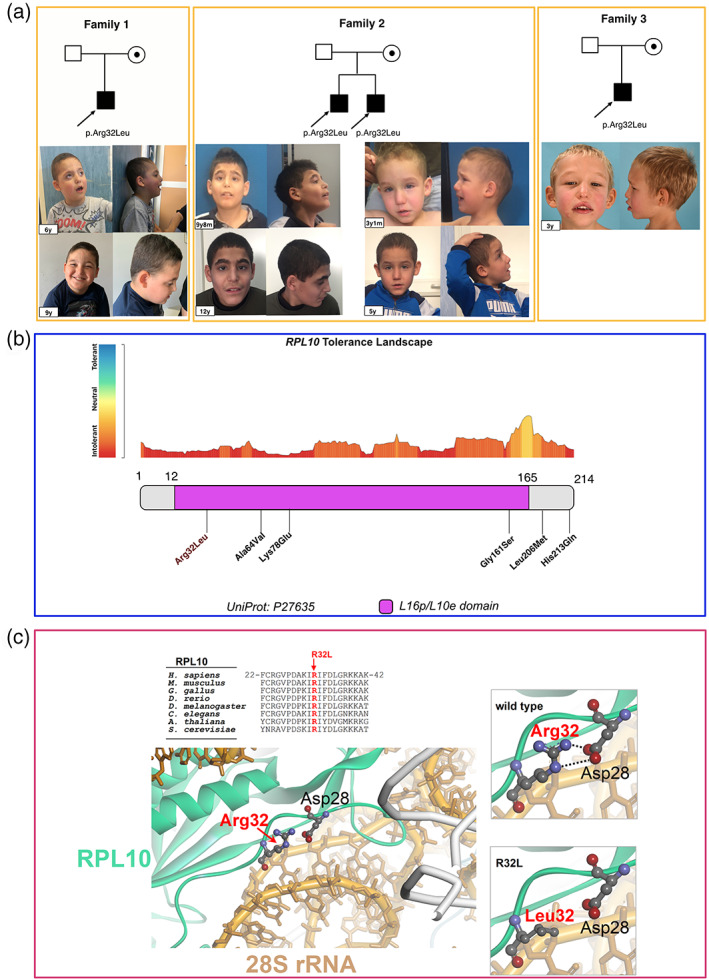
(a) Facial features and pedigree of individuals 1.1, 2.1, 2.2, and 3.1 at different ages. (b) RPL10 tolerance landscape showing the variant p.(Arg32Leu) in high intolerance domain. (c) The variant p.Arg32Leu affects the neutral charge of leucine disrupting the interaction with Asp28 residue, and RPL10 conformation and electrostatic interactions with the RNA molecules (PDB 5AJ0)
3.2. Individual 2.1
The proband is the first child of healthy nonconsanguineous parents. He was born by cesarean section after 37 weeks of gestation complicated by maternal hypertension. A fetal karyotype was normal. At birth, amniotic fluid was stained with meconium. At birth, his weight was 2950 g (10th centile) and his length was 51 cm (51st centile).
He walked independently at 3.5 years of age and he said his first words at 6 years of age, and he was able to pronounce 30 words at 9 years. He was noted to have poor eye contact and social interactions, stereotypic movements, and tip‐toe walking at 4 years of age. Because of hyperactivity and self‐injurious behaviors, he started risperidone. He had bilateral cryptorchidism that was surgically corrected.
At the age of 12 years, his weight was 44 kg (75th centile), his height was 163.5 cm (97th centile) with an arm span of 164 cm (arm span‐to‐height ratio: 1) and his OFC was 51.5 cm (SDS = −2.26). The systemic score for Marfan syndrome was 4 with positive wrist sign, pectus carinatum, and suggestive facial features. He had a long face with narrow and down‐slanting palpebral fissures, epicanthus, and esotropia of the left eye, long and flat philtrum, thin upper lip (Figure 1a), and enamel hypoplasia. He had shawl scrotum and bilateral syndactyly of second and third toe.
Cardiac ultrasound showed mild mitral prolapse and tricuspid prolapse with regurgitation without aortic root dilatation. Brain MRI was normal and the electroencephalogram showed mild electrical anomalies. Plasmatic homocysteine and 7‐dehydrocholesterol concentrations were both normal. The array‐CGH failed to detect pathogenic chromosomal microdeletions or microduplications.
3.3. Individual 2.2
The proband 2.2 is the youngest brother of 2.1 individual (Figure 1a) and he was born after 37 weeks of uncomplicated gestation. At birth, his weight was 3680 g (61st centile), his length was 49 cm (18th centile), and his OFC was 34 cm (22nd centile). Perinatal events were normal. He started walking independently at 2.5 years, and said his first words at 3 years of age when he was also noted to have poor eye contact, tendency to isolation, and hyperactivity. He underwent surgeries for vescicoureteral reflux and bilateral cryptorchidism.
On clinical evaluation, at 5 years of age, his weight was 20.5 kg (59th centile), his height was 107.7 cm (11st centile), and his OFC was 48.4 cm (SDS = −3). He has long face with narrow and down‐slanting palpebral fissures, long and flat philtrum, and thin upper lip (Figure 1a). He had bilateral syndactyly of second and third toe. Mild mitral prolapse and tricuspid regurgitation were noted by echocardiogram. Maternal serum phenylalanine was normal. The array‐CGH failed to detect pathogenic copy number variants.
3.4. Individual 3.1
The proband is the only child of healthy nonconsanguineous parents. Prenatal ultrasounds were unremarkable. He was born by emergency cesarean section because of abnormal cardiotocography after 39 weeks of gestation. At birth, his weight was 3700 g (63rd centile), his length was 49.5 cm (40th centile), and his OFC was 34 cm (20th centile). In the first months of life, he showed generalized hypotonia with severe orofacial hypotonia but normal deep tendon reflexes. He held his head at 5 months of age, he gained the standing position at 8 months, at 29 months he started walking and at 4 years of age he walked independently, often with tiptoeing. He started babbling at 12 months, and presently at 3 years of age he is not verbal. He had short attention span and frequent tantrums. He had intense hand and feet diaphoresis. He suffered from recurrent ear infections and obstructive sleep disorder. Hypertrophic adenoids were surgically removed. He had submucosal cleft palate but no signs of dysphagia. At a clinical evaluation at 3 years of age, his weight was 12.4 kg (8th centile), his height was 92 cm (16th centile), and his OFC was 48.0 cm (SDS = −2). He was noted to have dysmorphic features including long face with flat profile, sparse eyebrows, internal epicantal folds, full eyelids, narrows palpebral fissures, Cupid's bow upper lip, brachydactyly, and bilateral clinodactyly of the fifth fingers of hands and toes. A brain MRI showed a small corpus callosum without other abnormalities. Chromosome analysis and array‐CGH failed to detect pathogenic chromosomal abnormalities.
3.5. Ophthalmologic evaluations
3.5.1. Individual 1.1
The patient showed right eye exotropia at the age of 17 months but at that time visual acuity and stereoacuity could not be performed because of lack of cooperation. Objective examination showed exotropia (Hirschberg test −40 prism diopters) and hypertropia of right eye without excursion limitation in the nine gaze positions. Adnexa, anterior segment, and fundus examination were all normal (Figure 2a). Cicloplegic refraction (1% cyclopentolate eye drops) was +2.50 spherical equivalent in right eye and +2.00 spherical equivalent in left eye. When he was 7‐year‐old, optical coherence tomography (OCT) of the macular region and nerve fiber layer was normal (Figure 2b), but the electroretinography (ERG) showed a mild reduction of cones function, probably due to a bilateral defect of ganglional cells. The visual evoked potentials (VEP) were normal.
FIGURE 2.
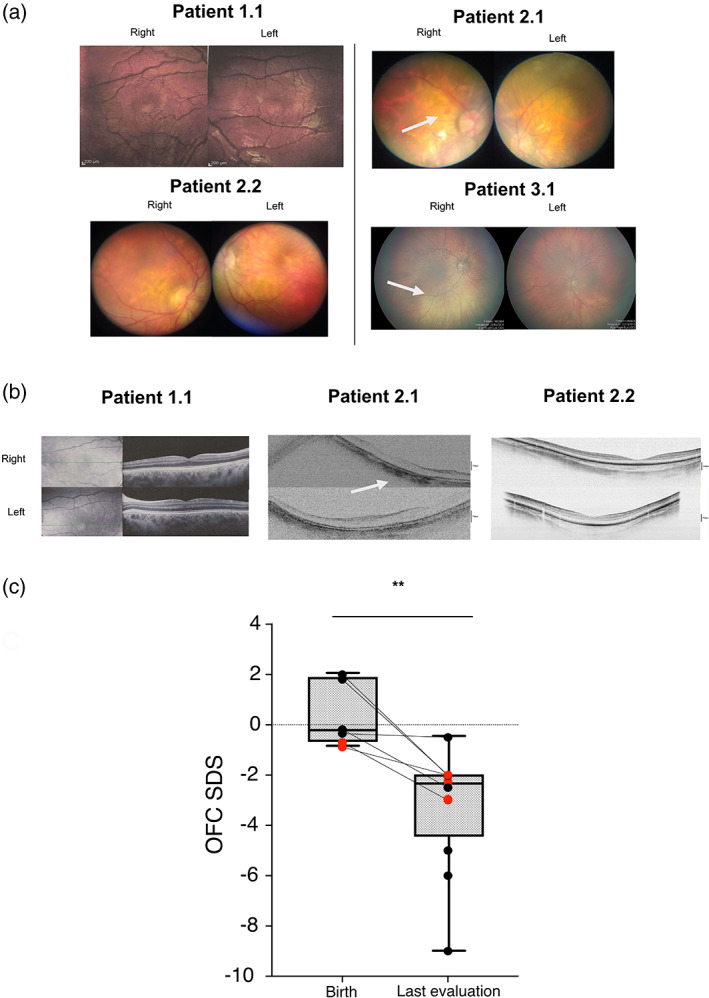
(a) Patients 1.1 and 2.2: Fundus image showed macular region within normal limits. Patient 2.1: Fundus image showed marked myopic retinopathy with a small retinal hemorrhage next to the optic disk of the right eye (white arrow). Patients 3.1: Fundus showed hypopigmented retinas (white arrow), while maculas are pigmented. (b) Patients 1.1 and 2.2: Optical choerence tomography (OCT) of the macular region and of the nerve fiber layer within normal limits. Patient 2.1: OCT of the macular region showing a diffuse reduction of neuroretinal rim thickness (white arrow). (c) SDS of OFC at birth (n = 6) and at last evaluation (n = 12) of all reported patients with RPL10‐related disorder so far. OFC SDS at the latest available evaluation is statistically significantly different from birth OFC SDS (p = 0.003). Red dots correspond to the four pateints reported herein
3.5.2. Individual 2.1
When the patient was 2‐year‐old difficulties in object fixation and following, along with squinting were noted. Visual acuity at 2 years of age was not evaluated because of lack of cooperation; stereoacuity was present (Lang test 200 arc second). Objective examination showed orthotropia and absence of excursion limitation in the nine gaze positions. A marked myopic retinopathy was detected on fundus exam along with a small retinal hemorrhage next to the optic disk of the right eye as incidental finding (Figure 2a). Cicloplegic refraction (1% cyclopentolate eye drops) was −22.00 spherical equivalent in both eyes. When he was 10‐year‐old, OCT of the macular region showed a diffuse reduction of neuroretinal rim thickness (Figure 2b) and the ERG showed rod dysfunction. The anterior segment exam showed bilateral superior subluxation of the lens.
3.5.3. Individual 2.2
The patient was noted to have difficulties with objects fixation by 18 months of age when he was first evaluated by ophthalmology but his visual acuity and stereoacuity could not be evaluated because of lack of cooperation. Objective examination showed esotropia (Hirschberg test +15 prism diopters) with mild limitation in the abduction of the left eye. Fundus examination were all normal (Figure 2a). Cicloplegic refraction (1% cyclopentolate eye drops) was +0.25 spherical equivalent in right eye and +0.75 spherical equivalent in left eye. When he was 3‐year‐old, OCT of the macular region was normal (Figure 2b) but the ERG showed a mild reduction of cone and rod function. Moreover, reduced VEP amplitude was detected in both eyes.
3.5.4. Individual 3.1
The patient was noted to have horizontal nystagmus at 12 months of age. Ophthalmological evaluation detected horizontal nystagmus with erratic ocular pursuit, photophobia, moderate myopia, and retinal hypopigmentation (Figure 2a). An ERG performed at 1 year of age by simultaneous stimulation of both eyes with a 3.0 flash elicited responses of sparse morphology and reduced amplitudes with preserved bilateral latency. Simultaneous stimulation of both eyes with flicker did not elicit a response on either side. The results were consistent with a macular alteration and achromatopsia and a suspicion of cone dystrophy was raised. At the age of 3 years, a repeated ERG showed normal flash and flicker stimulus responses.
4. RESULTS
The missense variant p.(Arg32Leu) in RPL10 (NM_006013:c.95G > T, chrX:153627840) in hemizygous state, inherited from their unaffected mother, was detected in the four individuals from the three unrelated pedigrees, by trio ES and was confirmed by Sanger sequencing. The p.(Arg32Leu) variant is absent in general population databases (gnomAD), is predicted to be damaging by multiple in silico tools (SIFT, Polyphen, Mutation taster), has a CADD score of 23.8, and affects a highly intolerant region of the protein (Figure 1b). According to American College of Medical Genetics classification, this variant was categorized as Likely Pathogenic based on PM1, PM2, PP1, PP3, and PP4 criteria in all four cases (Richards et al., 2015). The Arg32Leu mutation disrupts the salt‐brigde formed by Arg32 and Asp28, thus affecting RPL10 conformation and its electrostatic interactions with the RNA molecule (Figure 1c). In patient 2.1, a known pathogenic variant FBN1 was also detected, p.Arg1530Cys (c.4588C > T, NM_000138), explaining the lens subluxations (Loeys et al., 2001).
5. LITERATURE REVIEW
A total of 19 males with RPL10‐related disorder, including the four individuals in the present study, are known so far. Except for one case carrying a de novo variant, all patients inherited a RPL10 pathogenic missense variant from their mother. The clinical details of all 19 patients with RPL10‐related condition are shown in Table S1. We then compared the clinical findings of the patients with p.Arg32Leu variant in RPL10 (n = 4, p.Arg32Leu patients group) to the other 15 patients bearing different RPL10 variants (n = 15, literature patients group; Table 1). OFC was normal birth in all 19 patients, but microcephaly was observed in all the patients bearing p.Arg32Leu variant and in 75% of the other group (Table 1). OFC SDS measured in all the available patients at birth were significantly different from the last evaluation (SDS = +0.5128 at birth vs. SDS = −3.21 at last evaluations; p = 0.0032, t‐test) (Figure 2c), consistent with postnatal microcephaly. Short stature was absent in p.Arg32Leu patients while it was detected in 62% (5/8) of the literature group. While developmental delay (DD)/ID, as well as ASD, showed similar frequency in the p.Arg32Leu group compared to the literature patients, seizures, hearing loss, and hypotonia were more prevalent in literature patients group (Table 1). Considering all the patients with RPL10‐related disorder (n = 19), the average age for achieving autonomous ambulation was of 31.3 ± 14 months (range: 11 months–4 years), while the average age for the first words was 34.8 ± 27.5 months (range: 7–36 months). However, two individuals remained nonambulant and five individuals nonverbal (Table S1). Interestingly, all four individuals herein reported had eye anomalies with some degree of retinal involvement (4/4), while eye defects were identified in 50% of the literature group whose only one patient showed retinal involvement (Table 1). Genitourinary problems including cryptorchidism and vesicoureteral reflux were the most frequent anomalies, with similar frequency in p.Arg32Leu and the literature group, while constipation and gastroesophageal reflux disease were more frequent in latter group (Table 1). Dysmorphisms were present in all patients with the most recurrent features being long face, epicanthus, broad nasal ridge, long and flat philtrum, spaced teeth, prognathism, and prominent anteverted ears (Table S1).
TABLE 1.
Clinical features in individuals with RPL10‐related condition reported in literature (n=15) and in the four novel patients bearing the p.Arg32Leu variant in RPL10
| Patients of the literature (n = 15) | Patients with the p.Arg32Leu variant in RPL10 (n = 4) | |
|---|---|---|
| Variant inherited by the mother | 93% (14/15) | 100% (4/4) |
| Microcephaly | ||
| Prenatal | 0% (0/4) | 0% (0/2) |
| Postnatal | 75% (6/8) | 100% (4/4) |
| Short stature | 63% (5/8) | 0% (0/4) |
| Neurodevelopmental features | ||
| DD/ID | 83% (10/12) | 100% (4/4) |
| Hypotonia | 100% (6/6) | 75% (1/4) |
| Seizures | 89% (8/9) | 0% (0/4) |
| ASD | 78% (7/9) | 75% (3/4) |
| Cardiac anomalies | 75% (3/4) | 50% (2/4) |
| Eye defects | 50% (5/10) | 100% (4/4) |
| Hearing loss | 67% (4/6) | 0% (0/4) |
| Genitourinary anomalies | 78% (7/9) | 75% (3/4) |
| Gastrointestinal problems | 100% (5/5) | 25% (1/4) |
Abbreviations: ASD, autism spectrum disorder; DD, developmental delay; ID, intellectual disability.
6. DISCUSSION
Defects of ribosomal proteins have been associated to several neurodevelopmental disorders (Hetman & Slomnicki, 2019). Recently, pathogenic variants in RPL10 gene have been identified in patients with multisystem neurodevelopmental disorder. The core clinical manifestations of RPL10‐related condition include ASD, DD/ID, moderate/severe microcephaly, and seizures (Bourque et al., 2018; Brooks et al., 2014; Chiocchetti et al., 2011; Klauck et al., 2006; Thevenon et al., 2015; Zanni et al., 2015). In this report, we describe four further patients all sharing the same novel missense variant in RPL10 (p.Arg32Leu), that presented with a similar pattern of anomalies, including DD/ID, retinal anomalies and postnatal microcephaly, only partially overlapping to the patients already described in literature bearing different variant in RPL10 gene (n = 15).
In addition to another patient bearing p.Ala64Val variant in RPL10 (Zanni et al., 2015), all the four patients herein presented showed some degree of retinal damage, ranging from mild cones dysfunction to bilateral retinitis pigmentosa, that emerged at early age as difficulties in object fixation, squinting, exotropia, and nystagmus. Retinal anomalies have been described in other ribosomopathies such as SD‐S due to biallelic DNAJC21 mutations (Warren, 2018), and TCS (Goverdhan et al., 2005; Holla et al., 2013). Like other ribosomal genes such as RPS9, RPL7A, RPL21, RPS19, and DBA are expressed in the retina and involved in retinal degeneration (Uechi et al., 2001; Xie et al., 2009), RPL10 is also expressed in the human retina (Pinelli et al., 2016) at 553rd position out of 12,000 expressed genes. The functional consequences of RPL10 dysfunction in retina is unknown to date, but interestingly, a zebrafish model induced by morpholino antisense oligonucleotides to knock down ribosomal protein L10a (rpl10a), showed smaller eyes with reduced pigmentation and edema (Palasin et al., 2019). Translation perturbations as a consequence of the ribosomal dysfunction due to RPL10 mutations, might affect protein abundance and/or accumulation of abnormal proteins in the retina causing retinal damage (Dierschke et al., 2019; O'Connell et al., 2019).
Our case series expand the spectrum of clinical abnormalities of the RPL10‐related disorders to include retinal abnormalities. Whether the retinal dystrophy is an overlooked feature of RPL10 ribosomopathy or is due to specific RPL10 variants, such as the p.Arg32Val or the p.Ala64Val, is currently unknown. Nevertheless, retinal anomalies may be more frequent in patients with RPL10 pathogenic variants and a precocious ophthalmological referral after the molecular diagnosis is warranted. Moreover, the inclusion of RPL10 within NGS panel for syndromic retinopathies might be recommended.
Postnatal microcephaly was found as a common feature in our case series (100%) and was highly prevalent in the other patients reported in the literature (75%) with average SDS of OFC of −3.2. Evolution of disease phenotype has been already reported in other ribosomopathies (De Keersmaecker et al., 2015). Mutations in different ribosomal components and transacting ribosomal biogenesis factors might result into progressive neurodegeneration including progressive microcephaly. Neurogenesis failure due to ribosomal stress and apoptosis, and/or dysregulated protein translation might be the implicated neuropathogenic mechanisms (Brooks et al., 2014; Hetman & Slomnicki, 2019). RPL10‐associated microcephaly was indeed associated to insufficient proliferation and/or loss of neuroprogenitors cells, along with secondary translational deficits, in RPL10‐depleted zebrafish, (Brooks et al., 2014; Hetman & Slomnicki, 2019). Interestingly, Zika virus is a neuroteratogenic factor that target ribosomal biogenesis and causes severe microcephaly and other congenital brain abnormalities in the fetus (Chimelli & Avvad‐Portari, 2018). Recently, KIF11 pathogenic variants have been identified in individuals with syndromic autosomal‐dominant mild‐to‐severe microcephaly associated with lymphedema and/or chorioretinopathy (Ostergaard et al., 2012). The retinal anomalies and the microcephaly might be suggestive of KIF11‐related disorder. However, the prenatal onset of microcephaly, the lymphedema, and the different dysmorphic features differentiate KIF11‐related disorder from RPL10‐related disorder (Jones et al., 2014).
The frequency of DD/ID along with ASD was similar in p.Arg32Leu patients compared to the patients of the literature, while higher prevalence of seizures, hearing loss, hypotonia, and gastrointestinal anomalies was observed in the patients of the literature compared to the p.Arg32Leu group.
Growth retardation was initially reported as a feature of RPL10‐related disorder (Brooks et al., 2014). However, our survey showed that the overall prevalence of short stature was about 42%.
In conclusion, we report four probands from three unrelated families all carrying the same p.Arg32Leu variant in the RPL10 gene that expand the clinical spectrum of RPL10‐related disorder to include retinal anomalies and progressive microcephaly.
AUTHOR CONTRIBUTIONS
Gerarda Cappuccio conceived this research. Nicola Brunetti‐Pierri supervised the writing. Margherita De Bernardi, Alessia Morlando, Cristina Peduto, Iris Scala, Michele Pinelli, Flavio Gioele Gallo, Adriano Magli, Carmen Plaitano, Mercedes Serrano, Leticia Pías, Jaume Català, Mercè Bolasell, and Ginevra Zanni performed clinical assessments. Emanuele Bellacchio, Annalaura Torella, and Vincenzo Nigro performed genetic testing and bioinformatic prediction testing.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Supporting information
Table S1 Clinical and molecular findings of all (n = 19) reported patients with pathogenic variant in RPL10.
ACKNOWLEDGMENTS
The authors are grateful to patients and their parents for their participation to the study. This study was supported by Telethon Foundation, Telethon Undiagnosed Diseases Program (TUDP, GSP15001). This study was in part generated within the European Reference Network ITHACA.
Cappuccio, G. , De Bernardi, M. L. , Morlando, A. , Peduto, C. , Scala, I. , Pinelli, M. , Bellacchio, E. , Gallo, F. G. , Magli, A. , Plaitano, C. , Serrano, M. , Pías, L. , Català, J. , Bolasell, M. , Torella, A. , Nigro, V. , Zanni, G. , & Brunetti‐Pierri, N. (2022). Postnatal microcephaly and retinal involvement expand the phenotype of RPL10 ‐related disorder. American Journal of Medical Genetics Part A, 188A:3032–3040. 10.1002/ajmg.a.62911
Funding information TUDP; Telethon Foundation, Telethon Undiagnosed Diseases Program, Grant/Award Number: GSP15001
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- Bourque, D. K. , Hartley, T. , Nikkel, S. M. , Pohl, D. , Tétreault, M. , Kernohan, K. D. , Care4Rare Canada Consortium , & Dyment, D. A. (2018). A de novo mutation in RPL10 causes a rare X‐linked ribosomopathy characterized by syndromic intellectual disability and epilepsy: A new case and review of the literature. European Journal of Medical Genetics, 61, 89–93. [DOI] [PubMed] [Google Scholar]
- Brooks, S. S. , Wall, A. L. , Golzio, C. , Reid, D. W. , Kondyles, A. , Willer, J. R. , Botti, C. , Nicchitta, C. V. , Katsanis, N. , & Davis, E. E. (2014). A novel ribosomopathy caused by dysfunction of RPL10 disrupts neurodevelopment and causes X‐linked microcephaly in humans. Genetics, 198, 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimelli, L. , & Avvad‐Portari, E. (2018). Congenital Zika virus infection: A neuropathological review. Child's Nervous System, 34, 95–99. [DOI] [PubMed] [Google Scholar]
- Chiocchetti, A. , Pakalapati, G. , Duketis, E. , Wiemann, S. , Poustka, A. , Poustka, F. , & Klauck, S. M. (2011). Mutation and expression analyses of the ribosomal protein gene RPL10 in an extended German sample of patients with autism spectrum disorder. American Journal of Medical Genetics. Part A, 155, 1472–1475. [DOI] [PubMed] [Google Scholar]
- Dierschke, S. K. , Miller, W. P. , Favate, J. S. , Shah, P. , Imamura Kawasawa, Y. , Salzberg, A. C. , Kimball, S. R. , Jefferson, L. S. , & Dennis, M. D. (2019). O‐GlcNAcylation alters the selection of mRNAs for translation and promotes 4E‐BP1‐dependent mitochondrial dysfunction in the retina. The Journal of Biological Chemistry, 294, 5508–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley‐Barnes, K. I. , Ogawa, L. M. , & Baserga, S. J. (2019). Ribosomopathies: Old concepts, New Controversies. Trends in Genetics, 35, 754–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goverdhan, S. V. , Temple, I. K. , Self, J. , Lotery, A. J. , Dixon, M. J. , & Evans, A. R. (2005). Macular degeneration associated with a novel Treacher Collins tcof1 mutation and evaluation of this mutation in age related macular degeneration. The British Journal of Ophthalmology, 89, 1063–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman, M. , & Slomnicki, L. P. (2019). Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. Journal of Neurochemistry, 148, 325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holla, A. , Gonsalves, S. R. J. , & Lobo, G. J. (2013). Treacher Collins syndrome with microcornea and retinal detachment. BMJ Case Reports, 2013, 202425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, G. E. , Ostergaard, P. , Moore, A. T. , Connell, F. C. , Williams, D. , Quarrell, O. , Brady, A. F. , Spier, I. , Hazan, F. , Moldovan, O. , Wieczorek, D. , Mikat, B. , Petit, F. , Coubes, C. , Saul, R. A. , Brice, G. , Gordon, K. , Jeffery, S. , Mortimer, P. S. , … Mansour, S. (2014). Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): Review of phenotype associated with KIF11 mutations. European Journal of Human Genetics, 22, 881–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampen, K. R. , Sulima, S. O. , Vereecke, S. , & De Keersmaecker, K. (2020). Hallmarks of ribosomopathies. Nucleic Acids Research, 48, 1013–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Keersmaecker, K. , Sulima, S. O. , & Dinman, J. D. (2015). Ribosomopathies and the paradox of cellular hypo‐ to hyperproliferation. Blood, 125, 1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauck, S. M. , Felder, B. , Kolb‐Kokocinski, A. , Schuster, C. , Chiocchetti, A. , Schupp, I. , Wellenreuther, R. , Schmötzer, G. , Poustka, F. , Breitenbach‐Koller, L. , & Poustka, A. (2006). Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Molecular Psychiatry, 11, 1073–1084. [DOI] [PubMed] [Google Scholar]
- Konikkat, S. , & Woolford, J. L. (2017). Principles of 60S ribosomal subunit assembly emerging from recent studies in yeast. The Biochemical Journal, 474, 195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys, B. , Nuytinck, L. , Delvaux, I. , De Bie, S. , & De Paepe, A. (2001). Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin‐1 gene FBN1 because of suspected Marfan syndrome. Archives of Internal Medicine, 161, 2447–2454. [DOI] [PubMed] [Google Scholar]
- Lubs, H. A. , Stevenson, R. E. , & Schwartz, C. E. (2012). Fragile X and X‐linked intellectual disability: Four decades of discovery. American Journal of Human Genetics, 90, 579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchia, F. , Ciolfi, A. , Mutarelli, M. , Bruselles, A. , Castello, R. , Pinelli, M. , Basu, S. , Banfi, S. , Casari, G. , Tartaglia, M. , Nigro, V. , & TUDP . (2018). VarGenius executes cohort‐level DNA‐seq variant calling and annotation and allows to manage the resulting data through a PostgreSQL database. BMC Bioinformatics, 19, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman, I. J. , van Montfrans, J. M. , Hoogstraat, M. , Boes, M. L. , van de Corput, L. , Renner, E. D. , van Zon, P. , van Lieshout, S. , Elferink, M. G. , van der Burg, M. , Vermont, C. L. , van der Zwaag, B. , Janson, E. , Cuppen, E. , Ploos van Amstel, J. K. , & van Gijn, M. E. (2014). Targeted next‐generation sequencing: A novel diagnostic tool for primary immunodeficiencies. The Journal of Allergy and Clinical Immunology, 133, 529–534. [DOI] [PubMed] [Google Scholar]
- O'Connell, A. E. , Gerashchenko, M. V. , O'Donohue, M.‐F. , Rosen, S. M. , Huntzinger, E. , Gleeson, D. , Galli, A. , Ryder, E. , Cao, S. , Murphy, Q. , Kazerounian, S. , Morton, S. U. , Schmitz‐Abe, K. , Gladyshev, V. N. , Gleizes, P.‐E. , Séraphin, B. , & Agrawal, P. B. (2019). Mammalian Hbs1L deficiency causes congenital anomalies and developmental delay associated with Pelota depletion and 80S monosome accumulation. PLoS Genetics, 15, e1007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard, P. , Simpson, M. A. , Mendola, A. , Vasudevan, P. , Connell, F. C. , van Impel, A. , Moore, A. T. , Loeys, B. L. , Ghalamkarpour, A. , Onoufriadis, A. , Martinez‐Corral, I. , Devery, S. , Leroy, J. G. , van Laer, L. , Singer, A. , Bialer, M. G. , McEntagart, M. , Quarrell, O. , Brice, G. , … Jeffery, S. (2012). Mutations in KIF11 cause autosomal‐dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. American Journal of Human Genetics, 90, 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palasin, K. , Uechi, T. , Yoshihama, M. , Srisowanna, N. , Choijookhuu, N. , Hishikawa, Y. , Kenmochi, N. , & Chotigeat, W. (2019). Abnormal development of zebrafish after knockout and knockdown of ribosomal protein L10a. Scientific Reports, 9, 18130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinelli, M. , Carissimo, A. , Cutillo, L. , Lai, C.‐H. , Mutarelli, M. , Moretti, M. N. , Singh, M. V. , Karali, M. , Carrella, D. , Pizzo, M. , Russo, F. , Ferrari, S. , Ponzin, D. , Angelini, C. , Banfi, S. , & di Bernardo, D. (2016). An atlas of gene expression and gene co‐regulation in the human retina. Nucleic Acids Research, 44, 5773–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striano, P. , & Zara, F. (2017). ARHGEF9 mutations cause a specific recognizable X‐linked intellectual disability syndrome. Neurology. Genetics, 3, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenon, J. , Michot, C. , Bole, C. , Nitschke, P. , Nizon, M. , Faivre, L. , Munnich, A. , Lyonnet, S. , Bonnefont, J.‐P. , Portes, D. V. , & Amiel, J. (2015). RPL10 mutation segregating in a family with X‐linked syndromic intellectual disability. American Journal of Medical Genetics. Part A, 167, 1908–1912. [DOI] [PubMed] [Google Scholar]
- Uechi, T. , Tanaka, T. , & Kenmochi, N. (2001). A complete map of the human ribosomal protein genes: Assignment of 80 genes to the cytogenetic map and implications for human disorders. Genomics, 72, 223–230. [DOI] [PubMed] [Google Scholar]
- Vissers, L. E. , Gilissen, C. , & Veltman, J. A. (2016). Genetic studies in intellectual disability and related disorders. Nature Reviews. Genetics, 17, 9–18. [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, A. J. (2018). Molecular basis of the human ribosomopathy Shwachman‐diamond syndrome. Advances in Biological Regulation, 67, 109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, M. , Kobayashi, I. , Kiyoshima, T. , Nagata, K. , Ookuma, Y. , Fujiwara, H. , & Sakai, H. (2009). In situ expression of ribosomal protein L21 in developing tooth germ of the mouse lower first molar. Journal of Molecular Histology, 40, 361–367. [DOI] [PubMed] [Google Scholar]
- Zanni, G. , Kalscheuer, V. M. , Friedrich, A. , Barresi, S. , Alfieri, P. , Di Capua, M. , Haas, S. A. , Piccini, G. , Karl, T. , Klauck, S. M. , Bellacchio, E. , Emma, F. , Cappa, M. , Bertini, E. , & Breitenbach‐Koller, L. (2015). A novel mutation in RPL10 (ribosomal protein L10) causes X‐linked intellectual disability, cerebellar hypoplasia, and Spondylo‐epiphyseal dysplasia. Human Mutation, 36, 1155–1158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Clinical and molecular findings of all (n = 19) reported patients with pathogenic variant in RPL10.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.