

Abstract
Palladium‐catalyzed aminoalkynylation of electronically unbiased olefins with iodoalkynes is reported. The picolinamide auxiliary enables for the first time the syn‐selective aminoalkynylation of mono‐, di‐ and trisubstituted alkenes to afford the corresponding pyrrolidines in up to 97 % yield and as single diastereomers. Furthermore, through a C−H activation approach, the picolinamide allows the rapid synthesis of functionalized olefins, which are suitable cyclization precursors. Facile and orthogonal deprotection of the amides and Si i Pr3‐acetylenes in the products, and a subsequent Pictet–Spengler reaction is demonstrated.
Keywords: Alkenes, Alkynes, C−C Coupling, Nitrogen Heterocycles, Pd Catalysis
A palladium‐catalyzed, syn‐selective aminoalkynylation of mono‐, di‐ and trisubstituted olefins to afford pyrrolidines is developed. The combination of iodoalkynes and a picolinamide as nucleophile is shown to be essential for the observed reactivity. Furthermore, the picolinamide enables the rapid synthesis of cyclization substrates through a C−H activation approach.

Oxidative difunctionalization of alkenes is useful for the rapid synthesis of complex molecular scaffolds from simple and often readily available starting materials. [1] Combining intramolecular alkene heterofunctionalization with subsequent intermolecular carbon–carbon bond‐formation affords valuable functionalized heterocycles in a modular and efficient fashion. [2] For instance, the intramolecular aminoalkynylation of bis‐homoallylic amines gives access to propargyl pyrrolidines suitable for further downstream transformations. Current methods, however, are largely limited to the aminoalkynylation of terminal olefins.[ 3 , 4 ] Consequently, they do not allow access to pyrrolidines with branched substituents at the 2‐position, a motif commonly found in natural products and bioactive compounds. [5] Thus, novel methods for the stereospecific aminoalkynylation of substituted olefins are highly desirable but have been lacking. Herein, we report diastereospecific, Pd‐catalyzed cyclization to access propargylated pyrrolidines from highly substituted olefins bearing a picolinamide group as intramolecular nucleophile (Scheme 1A). The use of disubstituted olefins reveals that the reactions involve stereospecific syn‐aminoalkynylation. Key to the success of the reaction is the combination of picolinamide with iodoalkynes.
Scheme 1.
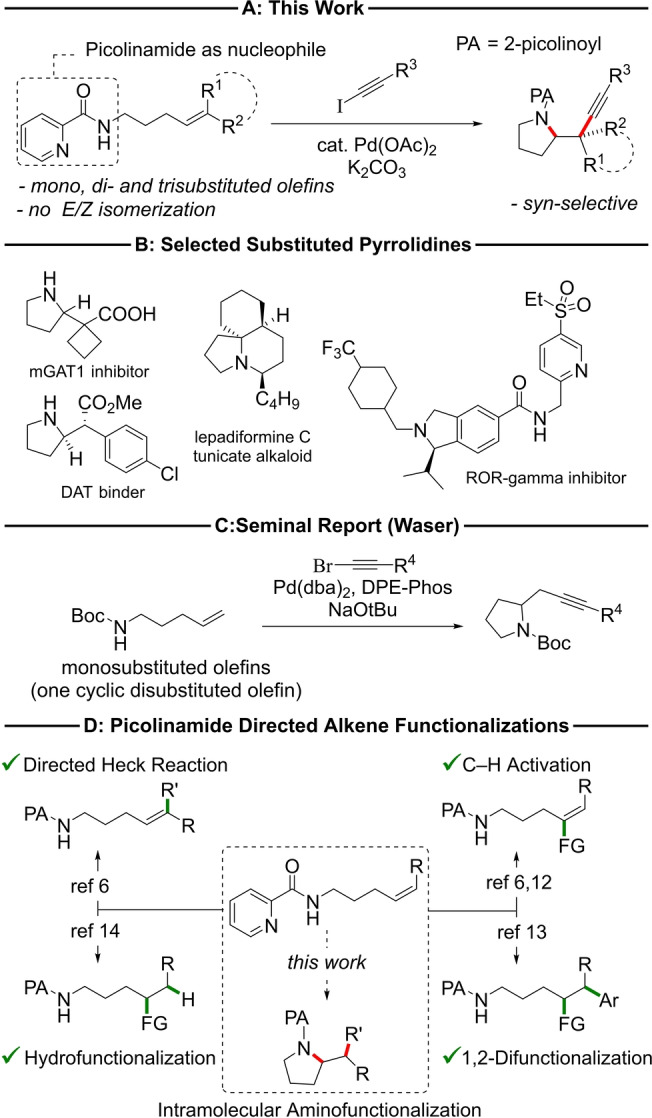
A) Summary of aminoakynylation reported herein. B) Selected bioactive pyrrolidines. C) Waser's aminoalkynylation reaction. D) Transition metal mediated reactivity of bishomoallylic picolinamides.[ 6 , 12 , 13 , 14 ]
During our previous studies on Pd‐catalyzed picolinamide‐directed C−H alkynylation of olefins [6] we observed that the use of bromoalkynes as coupling partners was essential for formation of products resulting from C−H activation (see Supporting Information). Over the course of our investigations, we observed that the use of the corresponding iodoalkyne provided a different set of products, namely the pyrrolidines resulting from an aminoalkynylation reaction. This unexpected halide dependent result led us to explore the reaction further and the development of the aminoalkynylative cyclization described herein.
In a seminal report, Waser and co‐workers described the Pd‐catalyzed reaction of N‐tosyl amides derived from 4‐pentenoic acids with the eponymous reagent, triisopropylsilyl ethynylbenziodoxolone (TIPS‐EBX), to afford propargylated N‐tosyl gamma‐lactams. [3] Waser and co‐workers later extended the method to allow the synthesis of pyrrolidines from N‐Boc‐pent‐4‐en amines and alkynyl bromides (Scheme 1C). [4] While these reports feature a wide range of substituents on the alkyl linker the scope is limited with respect to the olefin used. With one single exception, only terminal olefins are reported to take part in the cyclization reaction, limiting its application in the synthesis of complex targets, especially ones displaying substitution in the propargylic position. The sole internal disubstituted alkene example is a cyclopentene derivative, leaving questions about potential Pd‐catalyzed olefin E/Z‐isomerization and substrate generality unanswered. No examples utilizing trisubstituted alkenes have been reported. This is in line with the accepted notion that nucleopalladation is generally restricted to mono‐ or disubstituted olefins. [7]
There has been intense interest in intramolecular carboamination of olefins involving alkynes as coupling partners, with pioneering contributions from Bower, Waser, Wang, Leonori, Han, and Zhang. [8] These reports can be categorized by the reactive intermediates proposed therein. Cyclization‐reactions involving putative radical intermediates allow for the aminoalkynylation of highly substituted alkenes. However, these transformations are not stereospecific, and cyclization products are obtained as diastereomeric mixtures. On the other hand, reactions not proceeding through putative radical intermediates are currently limited to terminal alkenes. Thus, the stereospecific aminoalkynylation of internal olefins remains an unsolved challenge.
Daugulis and co‐workers introduced and elegantly demonstrated the use of picolinamides to enable the functionalization of unactivated aliphatic C−H bonds. [9] Following the pioneering research of Engle's group on the use of bidentate directing groups in the hydroamination of unactivated alkenes, [10] a multitude of related olefin functionalizations have been reported. [11] Bishomoallylic picolinamides were demonstrated to be suitable substrates for alkene C−H activation,[ 6 , 12 ] directed Heck reactions, [6] 1,2‐difunctionalization, [13] and hydrofunctionalization reactions (Scheme 1D). [14] However, to the best of our knowledge no systematic study utilizing the picolinamide as both an auxiliary and a coupling partner for alkene 1,2‐difunctionalization has been reported to date (Wu's group reports a single example using the picolinamide group during reaction‐optimization of an aminocarbonylation reaction, affording the target compound in 26 % yield [11h] ).
Following up on our initial observations described above, we set out to optimize the reaction, using the picolinamide derivative of 4‐(4‐pyranylidene)butylamine 1 a as a model substrate: Subjecting 1 a and Si i Pr3 protected iodoalkyne 3 to either Pd(PPh3)Cl2 or Pd(OAc)2 and K2CO3 in 1,2‐dichloroethane (DCE) as solvent afforded pyrrolidine 2 a in 85 and 70 % yield, respectively (Table 1, entries 1 and 2). The use of the corresponding bromoalkyne did not lead to product formation (entry 3), highlighting the importance of the halide on the alkyne.
Table 1.
Exploration of reaction conditions.
|
| ||
|---|---|---|
|
|
|
|
|
Entry |
Variation from “standard condtions”[a] |
Yield [%][b] |
|
1 |
None |
85 |
|
2 |
Pd(OAc)2 |
70 |
|
3 |
Si i Pr3‐protected bromoalkyne (3′) |
<5 |
|
4 |
t BuO2C‐ instead of PA (1 a′) |
<5 |
|
5 |
1 a′, 3′ |
<5 |
|
6 |
1 a′, Pd(dba)2, DPE‐Phos, NaOtBu, PhMe |
<5 |
|
7 |
1 a′, 3′, Pd(dba)2, DPE‐Phos, NaOtBu, PhMe |
<5 |
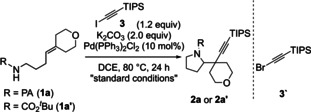
[a] Reactions conducted using 0.1 mmol 1 a at 0.1 M. [b] Yield determined by 1H NMR analysis of unpurified reaction mixture. DPE‐Phos=Bis[(2‐diphenylphosphino)phenyl] ether.
Based on the pioneering studies by Waser and co‐workers, [4a] we examined the reactivity of Boc‐protected 4‐(4‐pyranylidene)butylamine 1 a′. No desired product was observed under our standard reaction conditions using either iodo‐ or bromosubstituted Si i Pr3‐acetylene as coupling partners (entries 4 and 5). Our attempts to employ Waser's reaction conditions using Pd(dba)2, DPE‐Phos, NaO t Bu, and toluene did not afford any desired product with either iodo‐ or bromosubstituted Si i Pr3‐acetylene as coupling partner (entries 6 and 7). These results indicate that the picolinamide directing group is crucial for product formation for highly substituted olefins.
With suitable reaction conditions in hand, we explored the scope of the aminoalkynylation reaction (Scheme 2). Simple pent‐4‐en‐1‐amine derived alkene 1 b furnished the desired pyrrolidine 2 b in good yield (82 %). Alkene 1 c, bearing an ester moiety alpha to the picolinamide group was able to undergo the cyclization, affording 5‐substituted proline derivative 2 c (72 %, 1.4 : 1 dr). Moving on to structurally more complicated substrates, we investigated the scope for disubstituted olefins. Disubstituted alkenes bearing an isopropyl moiety, a cyclohexane, or sterically demanding silyl protected alcohol (1 d–1 f) were well tolerated and furnished the desired pyrrolidines 2 d–2 f (89–96 %). cis‐Substituted styrene derived substrates performed well under the reaction conditions: Alkene 1 g (R1=p‐ClC6H4, R2=H) furnished pyrrolidine 2 g in 97 % yield. Importantly, all products were obtained as single diastereoisomers indicating that the transformation proceeds through a stereospecific mechanism. To exclude the possibility of rapid E/Z‐isomerization of the starting material followed by a slow cyclization step we synthesized trans‐styrene 1 h (R1=H, R2=p‐ClC6H4). Pd‐catalyzed aminoalkynylation afforded pyrrolidine 2 h in 57 % yield. NMR analysis clearly shows 2 h to be a different diastereomer than cis‐styrene derived 2 g, thus implying that E/Z isomerization of the starting material does not occur at a competing rate under the reaction conditions. Bromo styrenes furnished products 2 i (72 %) and 2 j (85 %). Single‐crystal X‐ray analysis of 2 j allowed us to unambiguously determine the relative configuration of the newly formed stereocenters, revealing that the reaction is a formal syn‐carboamination of alkenes. [15] Arenes bearing methoxy groups (1 k, 1 l) underwent the aminoalkynylation in 77–85 % yield. The method also gives access to fused bicyclic systems: Cyclopentene 1 m and cyclohexene 1 n undergo cyclization to pyrrolizidine 2 m (44 %) and octahydroindole 2 n (81 %), respectively.
Scheme 2.

Reaction scope. Yields from 1 (0.3 mmol) given of isolated product after purification, as single diastereomers unless indicated otherwise. [a] Pd(PPh3)2Cl2 (10 mol %). [b] Pd(OAc)2 (10 mol %). [c] Conducted on 1.0 gram scale. [d] Thermal ellipsoids (100 K) at the 70 % probability level; disorder of i Pr removed for clarity.
Next, we investigated the generality of the method towards challenging trisubstituted alkene substrates. Di‐methyl substituted pyrrolidine 2 o was successfully obtained in 82 % yield. Substituted methylene cyclohexane (1 p) or cyclobutane (1 q) cyclized in 74 and 59 % yield, respectively. Pyrrolidines substituted with tetrahydropyran (2 a, 93 %) and oxetane (2 r, 65 %) were furnished under standard reaction conditions. Spiroisoindoline 2 s was accessed in 73 % yield. Collectively these results showcase trisubstituted olefins as suitable substrates.
Following up on these results, we investigated the scope of alkyne coupling partners (Scheme 3). The reaction of 1‐chloro‐4‐(iodoethynyl)benzene with 1 e afforded pyrrolidine 4 in 52 % yield. Sterically encumbered 1,3‐dichloro‐2‐(iodoethynyl)benzene was well tolerated in reactions with 1 a and 1 e, affording pyrrolidines 5 and 6 in 61 % and 92 % yield, respectively. tert‐Butyliodoacetylene gave access to 7 in 70 % yield and (4‐iodobut‐3‐yn‐1‐yl)benzene afforded 8 in 38 % yield. Protected propargylic alcohols were tolerated as demonstrated by formation of 9 and 10 (92 % and 80 %, respectively). N‐Boc‐piperidine derived alkyne 11 was obtained in 61 % yield.
Scheme 3.
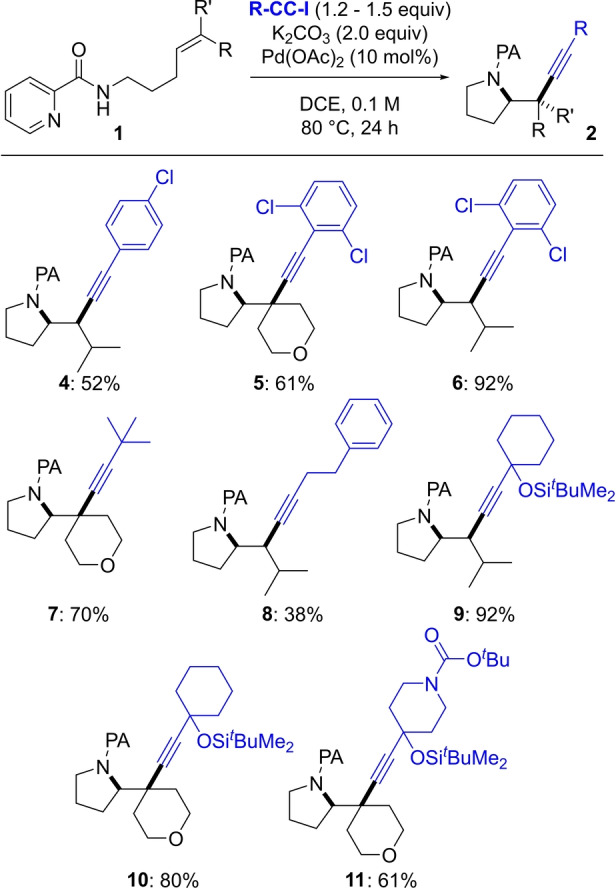
Alkyne scope. Reactions conducted on 0.3 mmol scale. Yields of isolated product after purification, obtained as single diastereomers.
A major advantage of the picolinamide directing group is its utility in the efficient synthesis of diverse and highly functionalized olefins as starting materials for the aminoalkynylation reaction. To demonstrate this, picolinamide‐directed C−H functionalization was used for the preparation of di‐ and trisubstituted olefins (Scheme 4). Through the use of our method developed for C−H alkynylation of olefins, [6] alkene 12 was accessed from 1 g in 76 % yield. Enyne 12 then undergoes aminoalkynylation to yield highly functionalized pyrrolidine 13 as a single diastereomer (72 %).
Scheme 4.
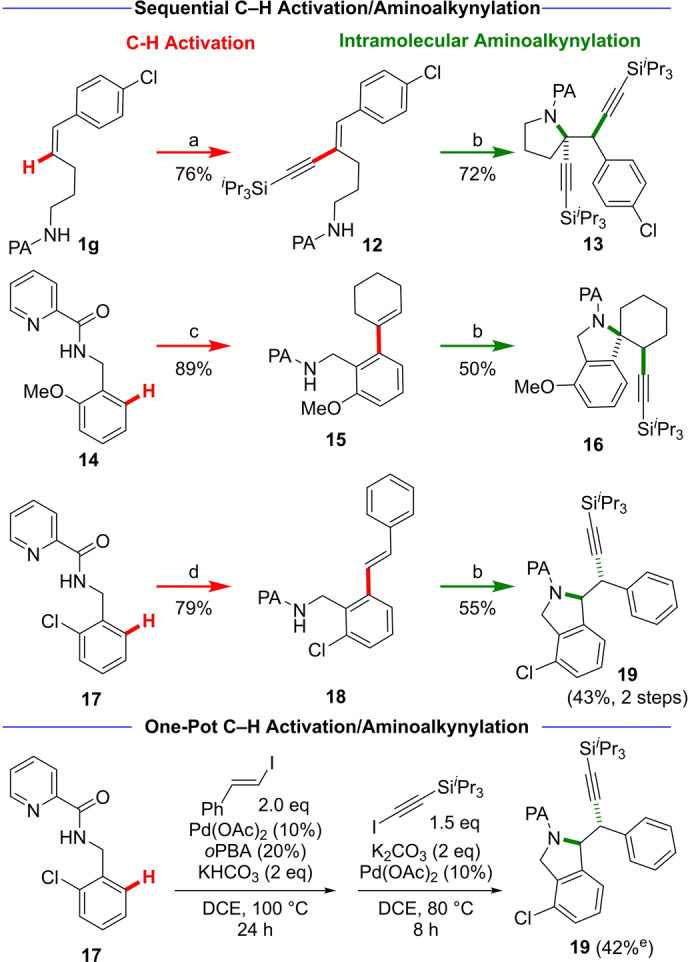
a) 3′ (1.2 equiv), Pd(OAc)2 (5 mol %), K2CO3 (2 equiv), AdCOOH (20 mol %), MeCN, 100 °C, 6 h. b) 3 (1.2 equiv), Pd(OAc)2 (10 mol %), K2CO3 (2 equiv), DCE, 80 °C, 24 h. c) Iodocyclohexene (2 equiv), Pd(OAc)2 (5 mol %), KHCO3 (2 equiv), 2‐Ph‐C6H4CO2H (oPBA, 20 mol %), DCE, 100 °C, 24 h. d) (E)‐(2‐iodovinyl)benzene (2 equiv), Pd(OAc)2 (5 mol %), KHCO3 (2 equiv), oPBA (20 mol %), DCE, 100 °C, 20 h. [e] Yield determined by 1H NMR analysis of unpurified reaction mixture.
Using Chen's method, [16] picolinamide 14 was subjected to ortho‐C−H alkenylation to give 15 as a substrate for subsequent cyclization to afford spirocyclic isoindoline 16 as a single diastereomer (50 % yield). (E)‐Stilbene 18, which was prepared via coupling of picolinamide 17 and (E)‐(2‐iodovinyl)benzene, can successfully undergo cyclization to give substituted isoindoline 19 in 55 % yield. A one‐pot C−H alkenylation/cyclization sequence is feasible, affording 19 in 42 % yield. These examples highlight the merging of C−H functionalization methods employing a picolinamide directing group with aminoalkynylation for the synthesis of highly functionalized building blocks.
The picolinamide auxiliary was removed under reductive conditions (Zn/HCl) [17] to afford amines 20 and 21 in 71 and 76 % yield, respectively (Scheme 5). Treatment of Si i Pr3‐protected alkyne 2 a with Bu4NF effects deprotection of the acetylene in 80 % yield. To showcase that the pyrrolidines obtained can be elaborated to a complex polycyclic scaffold we subjected 2 k to a two‐step sequence involving picolinamide cleavage followed by Pictet–Spengler reaction to afford tetrahydroisoquinoline 23 in 70 % yield.
Scheme 5.
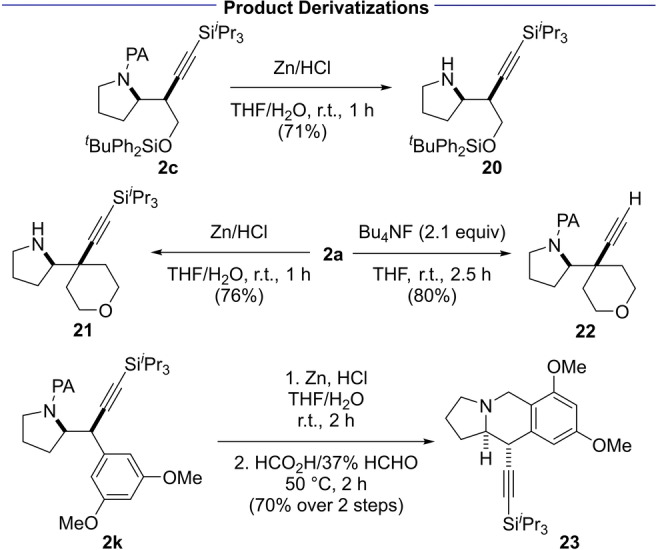
Product derivatizations.
In summary, we disclose a Pd‐catalyzed picolinamide enabled syn‐aminoalkynylation reaction of highly substituted alkenes to afford complex and versatile pyrrolidines bearing a wide range of aliphatic and aromatic substituents. Facile and orthogonal deprotection of the picolinamide auxiliary and the Si i Pr3‐acetylene is possible, and the products were demonstrated to be amenable to further complexity generating reactions. A salient feature of this protocol is the versatility of the picolinamide directing group, which serves an essential function in the C−H activation based synthesis of complex alkene substrates, as well as in the subsequent aminoalkynylation reaction. Thus, we believe this study will serve as a starting point for further research on the complex transition metal mediated reactivity landscape accessible to alkenes bearing bidentate directing groups.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Acknowledgements
ETH Zürich is gratefully acknowledged for financial support. We are grateful to Dr. N. Trapp and M. Solar for X‐ray crystallographic analysis (all ETH Zürich). Open access funding provided by Eidgenössische Technische Hochschule Zürich.
N. Müller, B. S. Schreib, S. U. Leutenegger, E. M. Carreira, Angew. Chem. Int. Ed. 2022, 61, e202204535; Angew. Chem. 2022, 134, e202204535.
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.
References
- 1. Yin G., Mu X., Liu G., Acc. Chem. Res. 2016, 49, 2413. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Wolfe J. P., Topics in Heterocyclic Chemistry, Vol. 32, Springer, Berlin, 2013, p. 1, [Google Scholar]; 10.1007/7081_2012_98; [DOI]
- 2b. Garlets Z. J., White D. R., Wolfe J. P., Asian J. Org. Chem. 2017, 6, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nicolai S., Piemontesi C., Waser J., Angew. Chem. Int. Ed. 2011, 50, 4680; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4776. [Google Scholar]
- 4.
- 4a. Nicolai S., Waser J., Org. Lett. 2011, 13, 6324; [DOI] [PubMed] [Google Scholar]
- 4b. Nicolai S., Sedigh-Zadeh R., Waser J., J. Org. Chem. 2013, 78, 3783. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Sauviat M.-P., Vercauteren J., Grimaud N., Jugé M., Nabil M., Petit J.-Y., Biard J.-F., J. Nat. Prod. 2006, 69, 558; [DOI] [PubMed] [Google Scholar]
- 5b. Davies H. M. L., Hopper D. W., Hansen T., Liu Q., Childers S. R., Bioorg. Med. Chem. Lett. 2004, 14, 1799; [DOI] [PubMed] [Google Scholar]
- 5c. Steffan T., Renukappa-Gutke T., Höfner G., Wanner K. T., Bioorg. Med. Chem. 2015, 23, 1284; [DOI] [PubMed] [Google Scholar]
- 5d.“ISOINDOLINE INHIBITORS OF ROR-GAMMA”: Claremon D. A., Dillard L. W., Dong C., Fan Y., Jia L., Lotesta S. D., Marcus A., Singh S. B., Tice C. M., Yuan J., et al., United States Patent Application: 0160122318, A1, 2016.
- 6. Schreib B. S., Fadel M., Carreira E. M., Angew. Chem. Int. Ed. 2020, 59, 7818; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7892. [Google Scholar]
- 7. Hartwig J. F. Organotransition Metal Chemistry: From Bonding to Catalysis, University Science books, Mill Valley, 2010, p. 429. [Google Scholar]
- 8.
- 8a. Faulkner A., Scott J. S., Bower J. F., J. Am. Chem. Soc. 2015, 137, 7224; [DOI] [PubMed] [Google Scholar]
- 8b. Orcel U., Waser J., Angew. Chem. Int. Ed. 2015, 54, 5250; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5339; [Google Scholar]
- 8c. Orcel U., Waser J., Angew. Chem. Int. Ed. 2016, 55, 12881; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13073; [Google Scholar]
- 8d. Shen K., Wang Q., Chem. Sci. 2017, 8, 8265; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Davies J., Sheikh N. S., Leonori D., Angew. Chem. Int. Ed. 2017, 56, 13361; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13546; [Google Scholar]
- 8f. Han W.-J., Wang Y.-R., Zhang J.-W., Chen F., Zhou B., Han B., Org. Lett. 2018, 20, 2960; [DOI] [PubMed] [Google Scholar]
- 8g. Nicolai S., Orcel U., Muriel B., Greenwood P. D. G., Buzzetti L., Puriņš M., Waser J., Synlett 2021, 32, 472; [Google Scholar]
- 8h. Zhang X., Qi D., Jiao C., Zhang Z., Liu X., Zhang G., Org. Chem. Front. 2021, 8, 6522. [Google Scholar]
- 9.
- 9a. Zaitsev V. G., Shabashov D., Daugulis O., J. Am. Chem. Soc. 2005, 127, 13154; [DOI] [PubMed] [Google Scholar]
- 9b. Daugulis O., Roane J., Tran L. D., Acc. Chem. Res. 2015, 48, 1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gurak J. A., Yang K. S., Liu Z., Engle K. M., J. Am. Chem. Soc. 2016, 138, 5805. [DOI] [PubMed] [Google Scholar]
- 11.Selected examples:
- 11a. Liu Z., Zeng T., Yang K. S., Engle K. M., J. Am. Chem. Soc. 2016, 138, 15122; [DOI] [PubMed] [Google Scholar]
- 11b. Liu Z., Wang Y., Wang Z., Zeng T., Liu P., Engle K. M., J. Am. Chem. Soc. 2017, 139, 11261; [DOI] [PubMed] [Google Scholar]
- 11c. O'Duill M. L., Matsuura R., Wang Y., Turnbull J. L., Gurak J. A., Gao D.-W., Lu G., Liu P., Engle K. M., J. Am. Chem. Soc. 2017, 139, 15576; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11d. Wang H., Bai Z., Jiao T., Deng Z., Tong H., He G., Peng Q., Chen G., J. Am. Chem. Soc. 2018, 140, 3542; [DOI] [PubMed] [Google Scholar]
- 11e. Nimmagadda S. K., Liu M., Karunananda M. K., Gao D.-W., Apolinar O., Chen J. S., Liu P., Engle K. M., Angew. Chem. Int. Ed. 2019, 58, 3923; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3963; [Google Scholar]
- 11f. van der Puyl V. A., Derosa J., Engle K. M., ACS Catal. 2019, 9, 224; [Google Scholar]
- 11g. Shen H.-C., Zhang L., Chen S.-S., Feng J., Zhang B.-W., Zhang Y., Zhang X., Wu Y.-D., Gong L.-Z., ACS Catal. 2019, 9, 791; [Google Scholar]
- 11h. Peng J.-B., Wu F.-P., Li D., Geng H.-Q., Qi X., Ying J., Wu X.-F., ACS Catal. 2019, 9, 2977; [Google Scholar]
- 11i. Jeon J., Ryu H., Lee C., Cho D., Baik M.-H., Hong S., J. Am. Chem. Soc. 2019, 141, 10048; [DOI] [PubMed] [Google Scholar]
- 11j. Pan R., Shi C., Zhang D., Tian Y., Guo S., Yao H., Lin A., Org. Lett. 2019, 21, 8915; [DOI] [PubMed] [Google Scholar]
- 11k. Li Y., Liang Y., Dong J., Deng Y., Zhao C., Su Z., Guan W., Bi X., Liu Q., Fu J., J. Am. Chem. Soc. 2019, 141, 18475; [DOI] [PubMed] [Google Scholar]
- 11l. Deng Y., Zhao C., Zhou Y., Wang H., Li X., Cheng G.-J., Fu J., Org. Lett. 2020, 22, 3524; [DOI] [PubMed] [Google Scholar]
- 11m. Ni H.-Q., Kevlishvili I., Bedekar P. G., Barber J. S., Yang S., Tran-Dubé M., Romine A. M., Lu H.-X., McAlpine I. J., Liu P., Engle K. M., Nat. Commun. 2020, 11, 6432; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11n. Li Y., Ali A., Dong J., Zhang Y., Shi L., Liu Q., Fu J., Org. Lett. 2021, 23, 4072; [DOI] [PubMed] [Google Scholar]
- 11o. Shukla R. K., Chaturvedi A. K., Volla C. M. R., ACS Catal. 2021, 11, 7750; Recent review: [Google Scholar]
- 11p. Jeon J., Lee C., Park I., Hong S., Chem. Rec. 2021, 21, 3613. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Liu M., Yang P., Karunananda M. K., Wang Y., Liu P., Engle K. M., J. Am. Chem. Soc. 2018, 140, 5805; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Schreib B. S., Carreira E. M., J. Am. Chem. Soc. 2019, 141, 8758; [DOI] [PubMed] [Google Scholar]
- 12c. Shen C., Lu X., Zhang J., Ding L., Sun Y., Zhong G., Chem. Commun. 2019, 55, 13582; [DOI] [PubMed] [Google Scholar]
- 12d. Mao C.-L., Zhao S., Zang Z.-L., Xiao L., Zhou C.-H., He Y., Cai G.-X., J. Org. Chem. 2020, 85, 774; [DOI] [PubMed] [Google Scholar]
- 12e. Schreib B. S., Son M., Aouane F. A., Baik M.-H., Carreira E. M., J. Am. Chem. Soc. 2021, 143, 21705. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Liu Z., Chen J., Lu H.-X., Li X., Gao Y., Coombs J. R., Goldfogel M. J., Engle K. M., Angew. Chem. Int. Ed. 2019, 58, 17068; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17224; [Google Scholar]
- 13b. Xie L., Wang S., Zhang L., Zhao L., Luo C., Mu L., Wang X., Wang C., Nat. Commun. 2021, 12, 6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Jeon J., Lee C., Seo H., Hong S., J. Am. Chem. Soc. 2020, 142, 20470; [DOI] [PubMed] [Google Scholar]
- 14b. Lee C., Seo H., Jeon J., Hong S., Nat. Commun. 2021, 12, 5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deposition Number 2108775 (for 2 j) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 16. Zhao Y., He G., Nack W. A., Chen G., Org. Lett. 2012, 14, 2948. [DOI] [PubMed] [Google Scholar]
- 17. O'Donovan H., De Fusco C., Spring D. R., Tetrahedron Lett. 2016, 57, 2962. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the Supporting Information of this article.