

Abstract
Aims
Neuromyelitis optica spectrum disorders (NMOSD) is an autoantibody‐mediated, B cell‐driven disease. Inebilizumab is a humanized, affinity‐optimized, afucosylated IgG1 κ monoclonal antibody that binds to the B‐cell specific surface antigen CD19, resulting in rapid, profound and sustained depletion of circulating peripheral B cells in NMOSD subjects (pivotal study). The objective of this study was to conduct population modelling of B‐cell response following inebilizumab treatment in adult subjects with NMOSD, and to assess the impact of drug exposure to outcome.
Methods
A haematopoietic transit model was developed to describe the joint effects of reducing influx from pro‐B cells and accelerating CD20+ B‐cell depletion in the blood by inebilizumab. Furthermore, the relationships between inebilizumab pharmacokinetic (PK) exposure and the primary efficacy endpoint and key secondary efficacy endpoints were evaluated.
Results
At the 300‐mg dose, there was no apparent relationship between efficacy (reduction in disease attack risk, risk of worsening from baseline in Expanded Disability Status Scale, cumulative total active MRI lesions, and the number of NMOSD‐related in‐patient hospitalizations) and PK exposure. Subjects with low, medium and high PK exposure had a similar hazard ratio of NMOSD attack vs. placebo group.
Conclusion
The pharmacodynamic modelling confirmed effective depletion of B cells is achieved with a 300 mg intravenous dose of inebilizumab administered on Day 1 and Day 15 and every 6 months thereafter. The PK variability between patients had no apparent effect on clinical efficacy.
Keywords: B cell, CD19, exposure–response, inebilizumab, NMOSD, pharmacodynamics, population modelling
Abbreviations
- AC
Adjudication Committee
- ADA
anti‐drug antibodies
- AUC
area under the concentration–time curve
- CL
clearance
- NMOSD
neuromyelitis optica spectrum disorder
- OLP
open‐label period
- PD
pharmacodynamic(s)
- PK
pharmacokinetic(s)
- PRED
population predictions
- RCP
randomized controlled period
What is already known about this subject
Inebilizumab depletes peripheral B cells and significantly reduced the onset of neuromyelitis optica spectrum disorder attack.
The pharmacokinetics of inebilizumab were adequately described by a 2‐compartment model with parallel first‐order and time‐dependent nonlinear elimination pathways.
What this study adds
A 300‐mg efficacy plateau dose minimizes the impact of pharmacokinetic variability on inebilizumab efficacy and was determined to be the optimal dosage for the treatment of neuromyelitis optica spectrum disorders.
1. INTRODUCTION
Neuromyelitis optica spectrum disorders (NMOSD) is an autoantibody‐mediated, B cell‐driven disorder of the central nervous system characterized by attacks of predominantly optic neuritis and longitudinally extensive transverse myelitis. An important feature of NMOSD is the presence of serum autoantibodies against aquaporin‐4, such as aquaporin‐4‐IgG, which is detected in about 80–90% of NMOSD patients. 1 Pathogenic aquaporin‐4‐IgG can be produced by a subpopulation of CD19‐positive (CD19+) CD20‐negative (CD20−) B cells showing morphological and phenotypical properties of plasmablasts, which are selectively increased in peripheral blood in NMOSD patients. 2 Results of small, uncontrolled studies of the anti‐CD20 monoclonal antibody rituximab have provided some evidence for the therapeutic effect of B‐cell depletion in NMOSD. 3 , 4 , 5 , 6 , 7 Compared to CD20, CD19 is expressed on a wider lineage of B cells, from pro‐B to plasmablasts and some plasma cells. Direct depletion of CD19+ B cells could be more effective in reducing the risk of NMOSD attack by more effectively depleting plasmablasts producing aquaporin‐4‐IgG.
Inebilizumab is a humanized, affinity‐optimized, afucosylated IgG1 κ monoclonal antibody that binds to the B cell‐specific surface antigen CD19, resulting in the depletion of pro‐B cells 8 and B cells, as well as plasmablasts and some plasma cells. Unlike the anti‐CD20 monoclonal antibody rituximab, inebilizumab does not mediate complement dependent cytotoxicity. 9 The removal of fucose from the monoclonal antibody Fc results in approximately 10‐fold increased affinity for the activating Fc γ receptor IIIA and significantly enhances natural killer cell‐mediated depletion of B cells via antibody‐dependent cellular cytotoxicity and antibody‐dependent cellular phagocytosis mechanisms.
In a multicentre, double‐blind, randomized, placebo‐controlled Phase 2/3 study in adults with NMOSD (NCT02200770, N‐MOmentum), inebilizumab treatment significantly depleted circulating B cells compared with placebo, and reduced the onset of NMOSD attack (hazard ratio 0.272, P < .0001). 10 The objectives of this analysis were: (i) to develop a population pharmacodynamic (PD) model describing the depletion of the peripheral B‐cell count by inebilizumab; and (ii) to evaluate the relationship between inebilizumab pharmacokinetic (PK) exposure and efficacy endpoints in subjects with NMOSD.
2. METHODS
2.1. Patients and trial designs
Peripheral B‐cell count data were obtained from 2 randomized, double‐blind, placebo‐controlled Phase 1 studies (MI‐CP200 [Study CP200] and CD‐IA‐MEDI‐551‐1102 [Study 1102]) and 1 multicentre, double‐blind, randomized, placebo‐controlled Phase 2/3 study (N‐MOmentum). Summary of all clinical studies and the descriptive statistics of baseline categorical and continuous covariates are listed in Tables 2 and 3, respectively.
TABLE 2.
Descriptive statistics of baseline categorical and continuous covariates for subjects who received inebilizumab treatment
| Study CP200 | Study 1102 | Study 1155 | Total | |
|---|---|---|---|---|
| n = 24 | n = 15 | n = 174 | n = 213 | |
| Sex, n (%) | ||||
| Male | 7 (29.2) | 6 (40) | 15 (8.6) | 28 (13.1) |
| Female | 17 (70.8) | 9 (60) | 159 (91.4) | 185 (86.9) |
| Race, n (%) | ||||
| White | 20 (83.3) | 13 (86.7) | 92 (52.9) | 125 (58.7) |
| Black | 3 (12.5) | 1 (6.7) | 15 (8.6) | 19 (8.9) |
| Asian | 0 | 0 | 39 (22.4) | 39 (18.3) |
| American Indian or Alaskan | 0 | 0 | 14 (8) | 14 (6.6) |
| Other | 1 (4.2) | 1 (6.7) | 14 (8) | 16 (7.5) |
| Anti‐drug antibody, n (%) | ||||
| Positive | 4 (16.7) | 0 | 17 (9.8) | 21 (9.9) |
| Age (y) | ||||
| Median | 48.5 | 44.0 | 43.0 | 44.0 |
| Range | 31.0–64.0 | 28.0–60.0 | 18.0–73.0 | 18.0–73.0 |
| Weight (kg) | ||||
| Mean (SD) | 73.6 (17.7) | 78.3 (21.9) | 68.3 (17.4) | 69.6 (17.9) |
| Median | 73.2 | 72.0 | 65.0 | 66.2 |
| Range | 41.1–114 | 54.0–122 | 38.0–148 | 38.0–148 |
| BMI (kg m −2 ) | ||||
| Mean (SD) | 26.3 (5.91) | 26.7 (5.90) | 25.2 (5.50) | 25.4 (5.57) |
| Median | 26.8 | 26.0 | 24.5 | 24.7 |
| Range | 15.7–37.9 | 19.8–38.6 | 15.6–52.8 | 15.6–52.8 |
| Total bilirubin (μmol L −1 ) | ||||
| Mean (SD) | 4.56 (2.35) | 9.13 (3.78) | 8.20 (5.23) | 7.86 (5.03) |
| Median | 4.28 | 8.00 | 7.00 | 7.00 |
| Range | 1.71–12.0 | 4.00–17.0 | 3.00–40.0 | 1.71–40.0 |
| Alkaline phosphatase (U L −1 ) | ||||
| Mean (SD) | 74.1 (20.6) | 79.7 (28.8) | 67.0 (25.5) | 68.7 (25.4) |
| Median | 73.5 | 90.0 | 63.0 | 66.0 |
| Range | 36.0–118 | 33.0–129 | 26.0–188 | 26.0–188 |
| Aspartate transaminase (U L −1 ) | ||||
| Mean (SD) | 22.1 (8.37) | 20.5 (6.74) | 22.4 (18.9) | 22.3 (17.4) |
| Median | 21.0 | 21.0 | 19.0 | 19.0 |
| Range | 9.00–53.0 | 12.0–33.0 | 7.00–164 | 7.00–164 |
| Creatinine clearance (mL min −1 ) | ||||
| Mean (SD) | 129 (55.1) | 126 (47.4) | 119 (39.7) | 121 (42.2) |
| Median | 122 | 108 | 110 | 110 |
| Range | 51.5–282 | 84.1–245 | 50.9–247 | 50.9–282 |
| Estimated glomerular filtration rate (mL min −1 1.73 m 2 ) | ||||
| Mean (SD) | 113 (50.7) | 96.1 (19.6) | 103 (26.5) | 103 (29.9) |
| Median | 107 | 93.1 | 97.0 | 96.6 |
| Range | 42.8–292 | 67.6–128 | 56.9–226 | 42.8–292 |
| CD20 (cells μL −1 ) | ||||
| Mean (SD) | 161 (143) | 187 (66.6) | 205 (129) | 198 (128) |
| Median | 108 | 182 | 183 | 174 |
| Range | 22.0–624 | 93.4–319 | 6.28–676 | 6.28–676 |
SD, standard deviation.
TABLE 3.
Summary of pharmacodynamic parameter estimates
| Parameter | Population estimate | Standard error |
|---|---|---|
| Slope | 0.0140 | 0.0006 |
| Effect of baseline CD20 on slope a | 0.272 | 0.051 |
| Lifespan (d) | 391 | 50.2 |
| Baseline CD20 (cells μL−1) | 135 | 6.08 |
| kCD19 (d−1) | 0.0751 | 0.0103 |
| Interindividual variability b | ||
| ηSlope | 51.8 | 3.3 |
| ηLifespan | 126 | 10 |
| ηBaseline CD20 | 59.2 | 3.9 |
| ηkCD19 | 110 | 17 |
| Residual variability | ||
| Proportional error b | 60.8 | 1.6 |
kCD19, rate constant representing the maturation of pro‐B to CD20+ B cells in the circulation; η, interindividual variability.
Natural exponents.
Displayed as %CV.
Each Phase 1 study involved with 28 adult subjects with either systemic sclerosis (SSc; Study CP200) who had at least moderate skin thickening in an area suitable for repeat biopsy or with relapsing forms of multiple sclerosis (MS; Study 1102). Subjects with SSc were administered with single intravenous dose (0.1, 0.3, 1.0, 3.0 and 10.0 mg kg−1) of inebilizumab or placebo; subjects with relapsing MS were either administered with 2 doses of inebilizumab (30, 100 or 600 mg) or placebo on Day 1 and 15 intravenously or administered a single dose of inebilizumab (60 or 300 mg) or placebo subcutaneously.
In a Phase 2/3 study (N‐MOmentum), comprised of a randomized controlled period (RCP) with an open label period (OLP), a total of 230 adult subjects with NMOSD were intravenously administered inebilizumab (300 mg) or placebo on study Day 1 and 15 in the RCP. Subjects who experienced an Adjudication Committee (AC)‐determined attack in the RCP, or who completed the Day 197 visit without an attack, exited the RCP and had the option to enroll into the OLP, and initiate or continue treatment with inebilizumab. The trial design schematic is shown in Figure 1.
FIGURE 1.
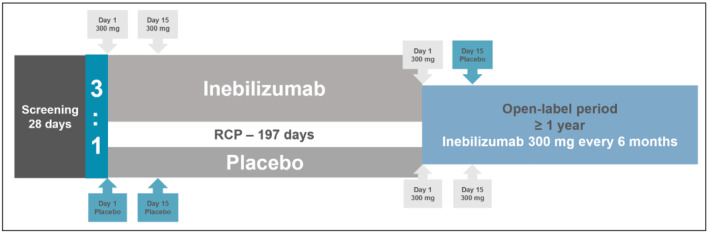
N‐MOmentum study design. RCP, randomized controlled period
2.2. PD modelling
The PD effect of inebilizumab on B cells is assayed by measuring CD20+ B‐cell counts, as inebilizumab interferes with CD19‐based detection methods. A haematopoietic transit model was developed to describe the depletion of circulating B‐cell count data by inebilizumab in adults. The PD effect of inebilizumab was exerted by joint effects of reducing influx from pro‐B cells and accelerating CD20+ B‐cell depletion in the blood, as the following differential equations demonstrate:
| (1) |
| (2) |
| (3) |
In the above equations, n is the number of transit compartments and Λ represents the longevity (lifespan) of blood CD20+ B cells. S0 is the influx rate of pro‐B cells (B0), and the total CD20+ B‐cell count in the blood (BCD20) is calculated as the sum of CD20+ B cells in all ageing compartments (Bi, i = 1 to n):
| (4) |
The initial conditions are shown in Equations 5 and 6:
| (5) |
| (6) |
The first‐order elimination rate constant kCD19 represents the maturation of pro‐B to CD20+ B cells in the circulation. Initially, kinebi, the accelerated removal of CD19+ pro‐B and mature B cells by inebilizumab, was described using an asymptotic Emax function. However, the estimated EC50 (inebilizumab concentration corresponding to half‐maximal B‐cell depletion) was less than the assay lower limit of quantitation due to the high potency of inebilizumab. As such, a log‐linear relationship was used to describe the effect of inebilizumab on depletion of pro‐B and mature B cells in blood:
| (7) |
Where C represents the serum concentration of inebilizumab. From population modelling, the PD model with dual activity of inebilizumab to deplete pro‐B and mature B cells was superior to the transit model where inebilizumab only depleted the circulating CD20+ mature B cells. The final PD model structure is shown in Figure 2.
FIGURE 2.
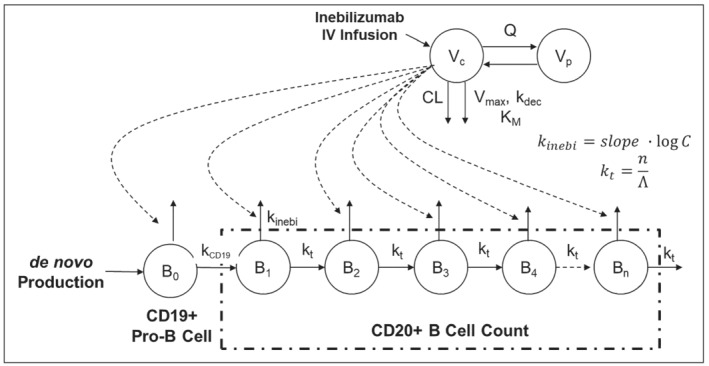
A diagram of the pharmacodynamic model of inebilizumab in adults. B0, pro‐B cell; Bi, CD20+ B cells in each aging compartment; C, serum concentration of inebilizumab; CL, clearance; IV, intravenous; kCD19, rate constant representing the maturation of pro‐B to CD20+ B cells in the circulation; kdec, first‐order rate constant describing the decrease of Vmax over time; kinebi, accelerated removal of CD19+ pro‐B and mature B cells by inebilizumab; KM, concentration to achieve the half of Vmax; Vc, volume of distribution in the central compartment; n, number of transit compartments; Q, intercompartmental clearance; Vp, volume of distribution in the peripheral compartment; Vmax, maximum velocity of Michaelis–Menten equation; Λ, longevity (lifespan) of blood CD20+ B cells; kt, transit rate constant defined as the ratio of n and Λ
Pooled CD20+ B‐cell data from adult subjects previously diagnosed with SSc (Study CP200), MS (Study 1102) and NMOSD (RCP of N‐MOmentum) were simultaneously modelled using first‐order conditional estimation with interaction (FOCEI) method in NONMEM (version 7.3, ICON, Hanover, MD, USA). Goodness‐of‐fit plots were generated using an R package XPOSE 4. 11
2.3. Exposure–response relationship
As the subjects were randomized in a 3:1 ratio to receive inebilizumab or placebo treatment in the RCP of N‐MOmentum study, inebilizumab‐treated subjects were grouped by tertiles based on the PK exposure (low, medium and high) to achieve a comparable sample size to the placebo group for time‐to‐event analysis. Three PK metrics, namely area under the concentration–time curve following the first dose (AUC0−14d), the cumulative AUC to the last observation in the RCP (AUCcumulative), and the systemic clearance (CL) of the first‐order elimination pathway, were obtained from the population PK modelling. The relationship between PK exposure and the primary efficacy endpoint (AC‐determined NMOSD attack) was evaluated by comparing the efficacy outcome of inebilizumab‐treated subjects of low, medium and high PK exposure with placebo in RCP. In addition, a statistically significant improvement with inebilizumab compared to placebo was demonstrated for 3 key secondary endpoints: worsening from baseline to the last visit of the RCP in Expanded Disability Severity Scale; cumulative total active MRI lesions during the RCP; and number of NMOSD‐related in‐patient hospitalizations during the RCP.
Potential impacts of body weight, a known PK covariate for therapeutic monoclonal antibodies, and the presence of anti‐drug antibodies (ADA) were also assessed on the efficacy outcomes of the pivotal study.
The exposure–response analysis was performed using SAS (V9.4, SAS Institute, Cary, NC, USA).
3. RESULTS
3.1. Subjects
In a randomized, double‐blind, placebo‐controlled Phase 2/3 trial (N‐MOmentum), 56 subjects with NMOSD received intravenous infusions of placebo and 174 subjects received inebilizumab (300 mg) on study days 1 and 15 in the randomized controlled period (RCP, Figure 1). In the N‐MOmentum study, at 300‐mg dose level, circulating B cells were significantly depleted compared with the placebo group at all time points from day 8 onwards. 10 Therefore, to aid the PD evaluation, peripheral CD20+ B‐cell count data from 2 dose‐ranging Phase 1 studies with lower dose cohorts in inebilizumab‐treated subjects with SSc (n = 24) or MS (n = 15) was pooled with that in subjects with NMOSD for population PD modelling. Overview of studies and descriptive statistics of baseline demographics of these subjects are summarized in Tables 1 and 2, respectively.
TABLE 1.
Overview of inebilizumab clinical studies
| Study no. | Study design | Dose(s) | Number of subjects | Serum sampling schedule |
|---|---|---|---|---|
| MI‐CP200: Study CP200 | A phase 1, randomized, double‐blind, placebo‐controlled study of the safety and tolerability of inebilizumab in SSc | Single IV dose of inebilizumab (0.1, 0.3, 1.0, 3.0 or 10.0 mg kg−1) or placebo. | 28 subjects were enrolled into the study, with 5 cohorts of subjects receiving 1 of 5 single IV doses of inebilizumab (0.1, 0.3, 1.0, 3.0 or 10.0 mg kg−1) or placebo |
PK at predose, 30 min after the end of the infusion on d 1, and on d 3, 8, 15, 29, 57 and 85 ADA on d 1, 29, 57, 85 |
|
CD‐IA‐MEDI‐551‐1102: Study 1102 |
A phase 1, multicentre, multinational, randomized, blinded, placebo‐controlled, dose‐escalation study to evaluate the safety and tolerability of inebilizumab in adult subjects with relapsing forms of MS |
IV infusion of inebilizumab (30, 100 or 600 mg) or placebo on d 1 and 15, or Single s.c. administration of inebilizumab (60 or 300 mg) or placebo on d 1 |
28 subjects were enrolled in the study. Among them 21 received inebilizumab (15 IV and 6 s.c.) and 7 received placebo. |
PK at predose and 15 min after the end of infusion (IV Cohorts only) on d 1 and d 15, and on d 4 (SC Cohorts only), 8, 29, 57, 85, 113, 141 and 169 ADA on d 1 (predose), 29, 85 and 169, and at 3, 6, 9, 12, 15 and 18 mo during the LTFU period |
| CD‐IA‐MEDI‐551‐1155: Study 1155 (N‐MOmentum study) | A phase 2/3, randomized, double‐masked, placebo‐controlled study followed by open label period to evaluate the efficacy, safety and tolerability of inebilizumab in adult subjects with NMOSD | IV infusion of inebilizumab (300 mg) or placebo on d 1 and 15 of RCP | 230 subjects with NMOSD were enrolled, randomized, and dosed in a 3:1 ratio to receive inebilizumab or placebo for 197 d (RCP). |
PK at predose and 15 min after the end of infusion on d 1 and d 15, and on d 8, 29, 57, 85, 113, 155 and 197 of RCP ADA on d 1 (predose), 29, 85 and 197 of RCP, and at wk 0, 13, 26, 39 and 52 during the OLP then every 3 mo for 1 y post‐last dose in SFP |
ADA, anti‐drug antibody; IV, intravenous; LTFU, long‐term follow‐up; MS, multiple sclerosis; No., number; MEDI‐551, inebilizumab; NMO, neuromyelitis optica; NMOSD, neuromyelitis optica spectrum disorders; OLP, open label period; PK, pharmacokinetic; RCP, randomized controlled period; s.c., subcutaneous; SFP, safety follow‐up period; SSc, systemic sclerosis.
3.2. PD modelling
The PD of inebilizumab were assessed with an assay for peripheral CD20+ B cells, since inebilizumab interferes with the CD19+ B‐cell assay. A haematopoietic transit model in which inebilizumab reducing influx from pro‐B cells and accelerating CD20+ B‐cell depletion in the blood was developed to describe the PD effect (Figure 2). The parameter estimates from the final model are summarized in Table 3. The estimated typical baseline CD20+ B‐cell count and lifespan were 135 cells per μL and 391 days, respectively. Only baseline B‐cell count was identified as a relevant PD covariate: higher baseline count was associated with more prominent B‐cell depletion by inebilizumab. Other covariates including age had no impact on the PD of inebilizumab.
The basic goodness‐of‐fit plots of the final PD model are shown in Figure S1. The conditional weighted residuals over the population predictions (PRED) are shown in Figure S1A to evaluate the model fit. The observations (dependent variables) over the PRED and individual predictions are shown in Figure S1B, C, respectively, to diagnose any misspecification of the structural model. The spread of the conditional weighted residuals is relatively consistent across the PRED range, while the regression lines in Figure S1B, C are close to the line of identity, indicating appropriate specification of the model structure and good model fit of the data.
The visual predictive check plots by study are presented in Figure 3. The symbol represents the observed CD20+ B‐cell count; the black solid and dotted lines represent the observed median at the 5th and 95th percentiles, the shaded area represents the 90% confidence interval of predicted median, 5th and 95th percentiles from 200 clinical simulations.
FIGURE 3.
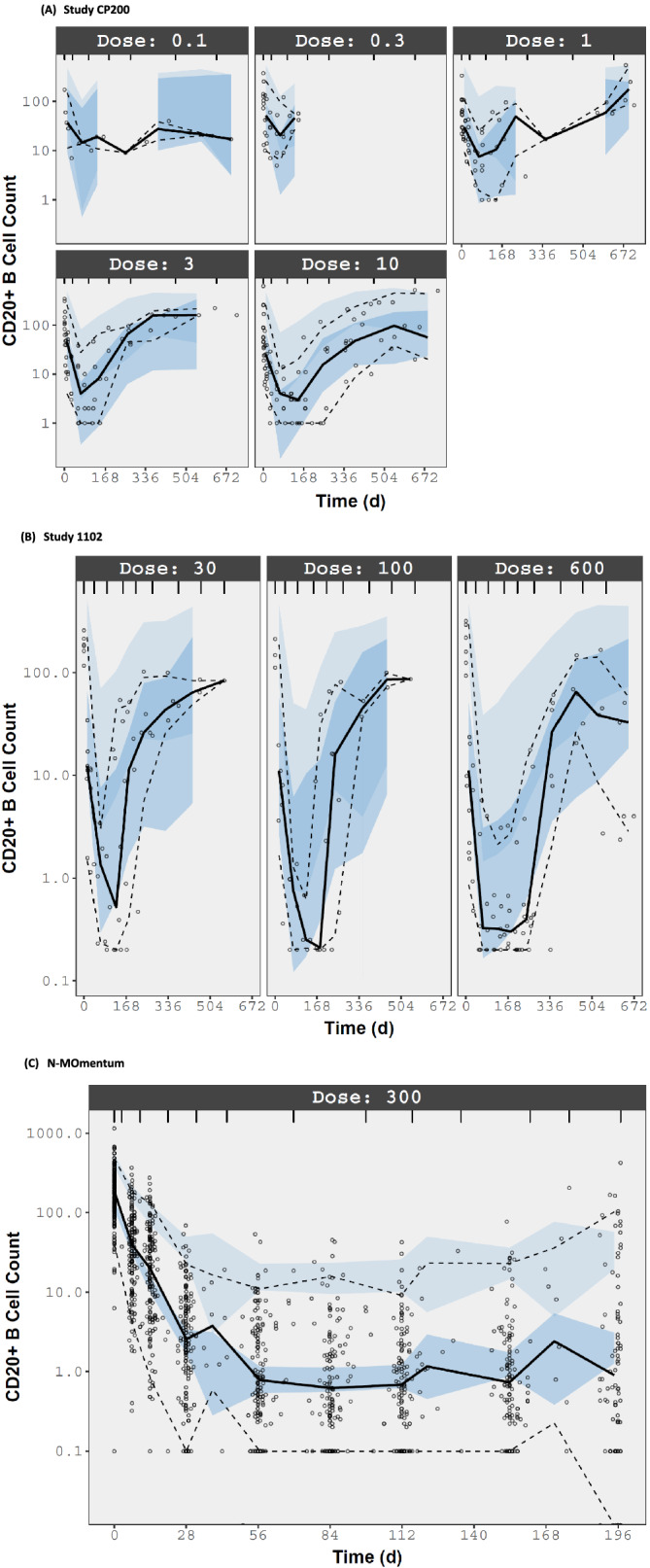
Visual predictive check of inebilizumab PD model. All dose units in study CP200 are mg kg−1 and in studies 1102 and N‐MOmentum are mg. Symbol: observed CD20+ B‐cell count; Black solid and dotted lines: observed median, 5th and 95th percentiles. Shaded area: 90% confidence interval of predicted median, 5th and 95th percentiles from 200 clinical simulations
3.3. Exposure–response assessment for NMOSD attack
The Kaplan–Meier plot of AC‐determined NMOSD attacks during the RCP in placebo and inebilizumab‐treated subjects with low (first‐tertile), medium (second tertile) and high (third tertile) AUC0−14d is shown in Figure 4. Subjects with low and medium AUC0−14d shown higher probability on attack‐free period compared to subjects with high AUC0−14d and placebo. The AUC following the first dose was selected for the exposure–response assessment since the steady‐state PK data were unavailable. Per protocol, subjects experiencing NMOSD attack exited the RCP and had the option to enroll into the OLP to initiate or continue to receive inebilizumab treatment, resulting in incomplete PK profiles following the second dose in those who discontinued treatment in the RCP. There was no apparent relationship between the hazard ratio of NMOSD attack for inebilizumab relative to placebo during the RCP with the first dose AUC of inebilizumab.
FIGURE 4.
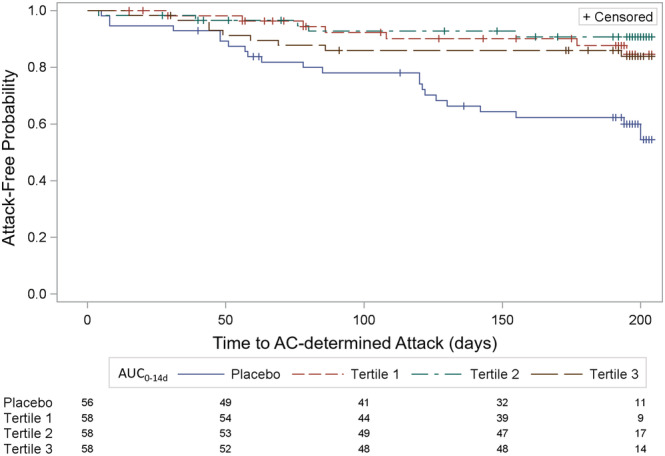
Kaplan–Meier (survival) plot of Adjudication Committee (AC)‐determined neuromyelitis optica spectrum disorder attack during the randomized controlled period in subjects with low, medium and high AUC0−14d. AUC0−14d, area under the concentration–time curve from Time 0 to 14 days postdose; Tertile 1, 300 mg inebilizumab‐treated subjects with low AUC0−14d; Tertile 2, 300 mg inebilizumab‐treated subjects with medium AUC0−14d; Tertile 3, 300 mg inebilizumab‐treated subjects with high AUC0−14d. The numbers under the legend represent the corresponding number of subjects for placebo, Tertile 1, Tertile 2 and Tertile 3 at the X axis time to AC‐determined attack (d). The hazard ratio and 95% confidence interval were 0.289 (0.123–0.679) for AUC0−14d Tertile 1, 0.189 (0.072–0.501) for AUC0−14d Tertile 2 and 0.341 (0.157–0.741) for AUC0−14d Tertile 3
Although body weight was identified as a PK covariate from the population modelling, 12 with the 300‐mg dose residing on the efficacy plateau, there were no clear trends in efficacy across the different quartiles of body weight (Figure 5). Based on the analysis of the primary efficacy endpoint, the adjustment of inebilizumab fixed dose (300 mg) by body weight is not warranted. The presence of ADA was detected in 25 subjects (8 of 56 in the placebo group, and 17 of 174 in the inebilizumab group) at baseline or during the RCP. From population modelling, the development of ADA had no significant effect on the PK of inebilizumab. No apparent effect of the presence of ADA on B‐cell depletion by inebilizumab treatment in the RCP or the primary efficacy endpoint was observed.
FIGURE 5.
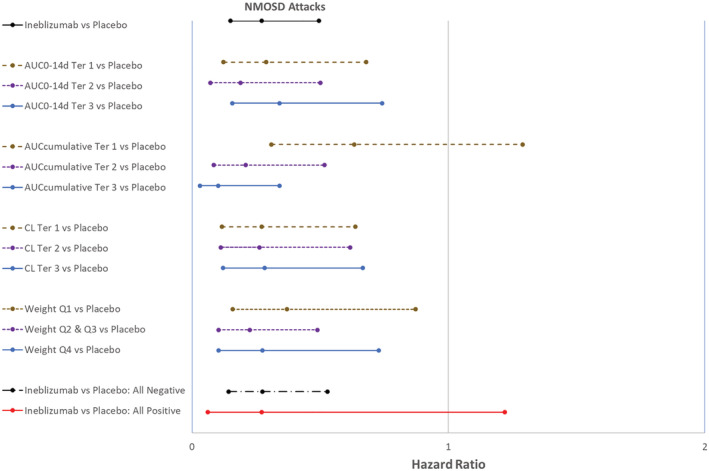
Forest plot of reduction of Adjudication Committee‐determined neuromyelitis optica spectrum disorder attack by exposure, clearance, weight and ADA subgroups during the RCP. ADA, anti‐drug antibody; AUC0−14d, area under the concentration–time curve from Time 0 to 14 days postdose; AUC0−14d Ter 1, 300 mg inebilizumab‐treated subjects with low AUC0−14d; AUC0−14d Ter 2, 300 mg inebilizumab‐treated subjects with medium AUC0−14d; AUC0−14d Ter3, 300 mg inebilizumab‐treated subjects with high AUC0−14d; AUCcumulative, cumulative area under the concentration–time curve from time 0 of Dose 1 to the last measurable concentration in RCP; AUCcumulative Ter 1, 300 mg inebilizumab‐treated subjects with low AUCcumulative; AUCcumulative Ter 2, 300 mg inebilizumab‐treated subjects with medium AUCcumulative; AUCcumulative Ter 3, 300 mg inebilizumab‐treated subjects with high AUCcumulative; CL, systemic clearance; CL Ter 1, inebilizumab‐treated subjects with low CL; CL Ter 2, inebilizumab‐treated subjects with medium CL; CL Ter 3, inebilizumab‐treated subjects with high CL; RCP, randomized, controlled period; Weight Q1, inebilizumab‐treated subjects with lowest quartile body weight; Weight Q2 & Q3, inebilizumab‐treated subjects with interquartile range (2nd and 3rd quartile) of body weight; Weight Q4, inebilizumab‐treated subjects with highest quartile of body weight. All Negative, only subjects who were determined ADA negative; All Positive, only subjects who were determined ADA positive. Hazard ratio and 95% confidence interval <1 supports treatment with inebilizumab was better than placebo
3.4. Exposure–response assessment for key secondary endpoints
Results of the subgroup analysis in each of the 3 key secondary endpoints, worsening from baseline to the last visit of the RCP in Expanded Disability Severity Scale (Figure S2), cumulative total active MRI lesions during the RCP (Figure S3) and number of NMOSD‐related in‐patient hospitalizations during the RCP (Figure S4), by PK exposure (AUC0−14d or CL) were generally consistent with that of the primary endpoint: no apparent relationship between the PK exposure and efficacy outcome in the RCP was observed. In addition, body weight had no apparent effect on key secondary endpoints. The presence of ADA had no observed impact on PK of inebilizumab 11 or the B‐cell depletion in subjects with NMOSD. Despite the small number of subjects who tested positive for ADA during the RCP, there was no apparent effect of ADA on the key secondary endpoints.
4. DISCUSSION
Inebilizumab is an affinity‐optimized, afucosylated, CD19‐directed cytolytic antibody for the treatment of NMOSD, a rare autoimmune disease. Following cell surface binding to B lymphocytes, inebilizumab results in antibody‐dependent cellular cytolysis. In the Phase 2/3 N‐MOmentum study, 300 mg inebilizumab intravenously administered 2 weeks apart resulted in profound and persistent peripheral CD20+ B‐cell depletion and significant reduction in NMOSD attack during the 28‐week RCP.
The PK of intravenously administered inebilizumab were adequately described by a 2‐compartment model with parallel first‐order and time‐dependent nonlinear elimination pathways. 11 As for other therapeutic monoclonal antibodies, the CL of inebilizumab increased with body weight: subjects with high body weight tended to have lower PK exposure following fixed‐dose administration. This analysis focused on the evaluation of PD of inebilizumab using a population modelling approach, and exposure–response assessments to evaluate the impact of PK exposure, body weight and the presence of ADA on efficacy endpoints for the treatment of NMOSD.
The pooled CD20+ B‐cell data from 2 dose‐ranging Phase 1 studies of inebilizumab in subjects with SSc and MS were initially utilized for exploration of various PD models. Then the B‐cell data from the pivotal NMOSD study were included for the development of the base model. Further assessments were performed to identify and evaluate demographic covariate effects on B‐cell response to inebilizumab treatment.
Compared to CD20, CD19 is expressed on a wider lineage of B cells, from pro‐B to plasmablasts and some plasma cells. A mechanistic haematopoietic transit model was developed to describe the depletion of peripheral B‐cell count in adults following inebilizumab treatment. In this model, inebilizumab depletes CD20+ B cells in each aging compartment as well as pro‐B cells in blood. Given the high potency of inebilizumab, EC50 (inebilizumab concentration corresponding to half‐maximal B‐cell depletion) could not be reliably estimated. Instead, the PD effect of inebilizumab was described using a log‐linear model (Equation 7), which approximates an asymptotic exposure–response relationship when the PK exposure exceeds EC50.
The estimated kCD19 was 0.0751 day−1, corresponding to a 13‐day mean residence time of CD19+ pro‐B cells before maturation to CD20+ B cells in the circulation. By contrast, mature CD20+ B cells have a rather long lifespan, with an estimated typical value of 391 days in humans. However, there was a large interindividual variability of CD20+ B‐cell lifespan across subjects.
Although SSc subjects in a Phase 1 study (Study CP200) tended to have lower estimate of B‐cell depletion slope than MS and NMOSD subjects, from population analysis the difference was not statistically significant. By contrast, the effect of inebilizumab on slope increased with baseline CD20+ B‐cell count: the effect of inebilizumab was greater in subjects with higher CD20+ B‐cell count at baseline. ADA, age and other demographic covariates had no impact on PD of inebilizumab.
The goodness‐of‐fit and visual predictive check plots were used to evaluate the appropriateness of the model structure in predicting the clinical data. Due to the small sample size of each dose group of Phase 1 studies (Studies CP200 and 1102), the prediction bands although captured the depletion and recovering trends of CD20+ B‐cell response, in some regions deviated from the observations (Figure 3). Nevertheless, when the sample size is adequate (N‐MOmentum), the B‐cell response in adult subjects with NMOSD was well described by the PD model.
There was no apparent relationship of PK exposure (AUC0−14d) to inebilizumab and the outcome of the primary or key secondary efficacy endpoints (Figures 4, 5, S2–S4). Results of the exposure–response analyses confirmed that the fixed dose of 300 mg inebilizumab resides at the efficacy plateau for the treatment of NMOSD. In fact, the efficacy was slightly lower in subjects with high AUC0−14d, probably due to random variability across subgroups (Figures 4, 5). The relationship of AUCcumulative and time to onset of NMOSD attack during the RCP was skewed since subjects experiencing NMOSD attack exited the RCP and had the option to enter the OLP to receive active inebilizumab treatment per protocol (Figure 5). The early withdrawal from the RCP led to a lower AUCcumulative in subjects who experienced NMOSD attack. In addition, subjects with slow, medium and fast CL of inebilizumab had nearly identical outcomes, confirming that the 300‐mg fixed dose of inebilizumab resides at the efficacy plateau (Figure 5).
Although body weight has an impact on PK based on the population PK analysis, it did not affect the inebilizumab efficacy. The 300‐mg dose, residing at the efficacy plateau, minimized the impact of PK variability on efficacy outcome. As such, dose‐adjustment by body weight is not warranted.
Consistent with the absence of significant impact of ADA on inebilizumab PK, from population modelling and exposure–response assessment, the presence of ADA had no impact on PD (B‐cell depletion) or efficacy endpoints in subject with NMOSD. However, this conclusion should be taken with caution due to the small number of subjects who tested positive for ADA during the RCP.
The population PD modelling and results of the exposure–response analysis of the primary and key secondary endpoints demonstrate that the 300‐mg fixed dose of inebilizumab is adequate to achieve an effective B‐cell depletion and reduced risk for AC‐determined NMOSD attack.
CONTRIBUTORS
L.Y., B.W. and W.R. wrote the manuscript. E.K., D.S., W.R., D.C. and L.Y. designed the research. L.Y., B.W., D.S., W.R., E.K. and D.C. interpreted the data. L.Y., B.W., B.M., R.C. and D.S. analysed the data.
COMEPTING INTERESTS
L.Y., D.S., D.C., E.K. and W.A.R. are employees of Horizon Therapeutics and hold Horizon stock. B.W., B.M. and R.C. are employed by Amador Bioscience. Amador Bioscience reports payment for consultation from Viela Bio (now Horizon Therapeutics).
CLINICAL TRIAL REGISTRATION
The clinical trials mentioned in the manuscript are registered with www.clintrials.gov.
Supporting information
FIGURE S1 Basic goodness‐of‐fit plots of inebilizumab PD model. CWRES, conditional weighted residual; DV, dependent variable (observation); IPRED, individual prediction; PRED, population prediction. The red solid lines are loess smoothing lines
FIGURE S2 Forest plot of worsening from baseline in EDSS during the RCP. ADA, anti‐drug antibody; AUC0−14d, area under the concentration–time curve from Time 0 to 14 days postdose; AUC0−14d Ter 1, 300 mg inebilizumab‐treated subjects with low AUC0−14d; AUC0−14d Ter 2, 300 mg inebilizumab‐treated subjects with medium AUC0−14d; AUC0−14d Ter 3, 300 mg inebilizumab‐treated subjects with high AUC0−14d; AUCcumulative, cumulative area under the concentration–time curve from time 0 of Dose 1 to the last measurable concentration in RCP; AUCcumulative Ter 1, 300 mg inebilizumab‐treated subjects with low AUCcumulative; AUCcumulative Ter 2, 300 mg inebilizumab‐treated subjects with medium AUCcumulative; AUCcumulative Ter 3, 300 mg inebilizumab‐treated subjects with high AUCcumulative; CL, systemic clearance; CL Ter 1, inebilizumab‐treated subjects with low CL; CL Ter 2, inebilizumab‐treated subjects with medium CL; CL Ter 3, inebilizumab‐treated subjects with high CL; EDSS, Expanded Disability Status Scale; RCP, randomized controlled period; Weight Q1, inebilizumab‐treated subjects with lowest quartile body weight; Weight Q2 & Q3, inebilizumab‐treated subjects with interquartile range (2nd and 3rd quartile) of body weight; Weight Q4, inebilizumab‐treated subjects with highest quartile of body weight. All Negative, only subjects who were determined ADA negative; All Positive, only subjects who were determined ADA positive. Odds ratio and 95% confidence interval < 1 supports treatment with inebilizumab was better than placebo.
FIGURE S3 Forest plot of the cumulative number of active MRI lesions during the RCP. ADA, anti‐drug antibody; AUC0−14d, area under the concentration–time curve from time 0 to 14 days post dose; AUC0−14d Ter 1, 300 mg inebilizumab‐treated subjects with low AUC0−14d; AUC0−14d Ter 2, 300 mg inebilizumab‐treated subjects with medium AUC0−14d; AUC0−14d Ter3, 300 mg inebilizumab‐treated subjects with high AUC0−14d; AUCcumulative, cumulative area under the concentration–time curve from time 0 of Dose 1 to the last measurable concentration in RCP; AUCcumulative Ter 1, 300 mg inebilizumab‐treated subjects with low AUCcumulative; AUCcumulative Ter 2, 300 mg inebilizumab‐treated subjects with medium AUCcumulative; AUCcumulative Ter 3, 300 mg inebilizumab‐treated subjects with high AUCcumulative; CL, systemic clearance; CL Ter 1, inebilizumab‐treated subjects with low CL; CL Ter 2, inebilizumab‐treated subjects with medium CL; CL Ter 3, inebilizumab‐treated subjects with high CL; MRI, magnetic resonance imaging; RCP, randomized controlled period; Weight Q1, inebilizumab‐treated subjects with lowest quartile body weight; Weight Q2 & Q3, inebilizumab‐treated subjects with interquartile range (2nd and 3rd quartile) of body weight; Weight Q4, inebilizumab‐treated subjects with highest quartile of body weight. All Negative, only subjects who were determined ADA negative; All Positive, only subjects who were determined ADA positive. Rate ratio and 95% confidence interval < 1 supports treatment with inebilizumab was better than placebo.
FIGURE S4 Forest plot of number of NMOSD‐related in‐patient hospitalizations during the RCP. ADA, anti‐drug antibody; AUC0−14d, area under the concentration–time curve from time 0 to 14 days post dose; AUC0−14d Ter 1, 300 mg inebilizumab‐treated subjects with low AUC0−14d; AUC0−14d Ter 2, 300 mg inebilizumab‐treated subjects with medium AUC0−14d; AUC0−14d Ter 3, 300 mg inebilizumab‐treated subjects with high AUC0−14d; AUCcumulative, cumulative area under the concentration–time curve from time 0 of Dose 1 to the last measurable concentration in RCP; AUCcumulative Ter 1, 300 mg inebilizumab‐treated subjects with low AUCcumulative; AUCcumulative Ter 2, 300 mg inebilizumab‐treated subjects with medium AUCcumulative; AUCcumulative Ter 3, 300 mg inebilizumab‐treated subjects with high AUCcumulative; CL, systemic clearance; CL Ter 1, inebilizumab‐treated subjects with low CL; CL Ter 2, inebilizumab‐treated subjects with medium CL; CL Ter 3, inebilizumab‐treated subjects with high CL; NMOSD, neuromyelitis optica spectrum disorder; RCP, randomized, controlled period; Weight Q1, inebilizumab‐treated subjects with lowest quartile body weight; Weight Q2 & Q3, inebilizumab‐treated subjects with interquartile range (2nd and 3rd quartile) of body weight; Weight Q4, inebilizumab‐treated subjects with highest quartile of body weight. All Negative, only subjects who were determined ADA negative; All Positive, only subjects who were determined ADA positive. Rate ratio and 95% confidence interval < 1 supports treatment with inebilizumab was better than placebo.
ACKNOWLEDGEMENTS
The authors would like to thank all the patients and investigators that participated in the studies. The author would like to thank John N. Ratchford for his contributions to the inebilizumab programme. The authors would like to acknowledge Kamille O'Connor, Xiaoying Yu and Rouletta Blowers of Amador Bioscience (Pleasanton, CA, USA) for assistance with preparation of this manuscript. The clinical studies of inebilizumab were funded by Medimmune and Viela Bio; the PD modelling and exposure–response assessment included in this manuscript was funded by Viela Bio.
Yan L, Wang B, She D, et al. Pharmacodynamic modelling and exposure–response assessment of inebilizumab in subjects with neuromyelitis optica spectrum disorders. Br J Clin Pharmacol. 2022;88(8):3803‐3812. doi: 10.1111/bcp.15332
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
REFERENCES
- 1. Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6(7):383‐392. doi: 10.1038/nrneurol.2010.72 [DOI] [PubMed] [Google Scholar]
- 2. Chihara N, Aranami T, Sato W, et al. Interleukin 6 signaling promotes anti‐aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci. 2011;108(9):3701‐3706. doi: 10.1073/pnas.1017385108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA. Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler. 2011;17(10):1225‐1230. doi: 10.1016/j.msard.2020.102683 [DOI] [PubMed] [Google Scholar]
- 4. Cree BAC, Wingerchuk DM. Acute transverse myelitis: Is the “idiopathic” form vanishing? Neurology. 2005;65(12):1857‐1858. doi: 10.1212/01.wnl.0000194615.51750.f8 [DOI] [PubMed] [Google Scholar]
- 5. Ip VH, Lau AY, Au LW, et al. Rituximab reduces attacks in Chinese patients with neuromyelitis optica spectrum disorders. J Neurol Sci. 2013;324(1‐2):38‐39. doi: 10.1016/j.jns.2012.09.024 [DOI] [PubMed] [Google Scholar]
- 6. Jacob CMA, Pastorino AC, Fahl K, Carneiro‐Sampaio M, Monteiro RC. Autoimmunity in IgA Deficiency: Revisiting the Role of IgA as a Silent Housekeeper. J Clin Immunol. 2008;28(S1):56‐61. doi: 10.1007/s10875-007-9163-2 [DOI] [PubMed] [Google Scholar]
- 7. Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol. 2011;68(11):1412‐1420. doi: 10.1001/archneurol.2011.154 [DOI] [PubMed] [Google Scholar]
- 8. Gallagher S, Yusuf I, McCaughtry TM, et al. MEDI‐551 Treatment Effectively Depletes B Cells and Reduces Serum Titers of Autoantibodies in Mice Transgenic for Sle1 and Human CD19. Arthritis Rheumatol. 2016;68(4):965‐976. doi: 10.1002/art.39503 [DOI] [PubMed] [Google Scholar]
- 9. Herbst R, Wang Y, Gallagher S, et al. J Pharmacol Exp Ther. 2010;335(1):213‐222. doi: 10.1124/jpet.110.168062 [DOI] [PubMed] [Google Scholar]
- 10. Cree BAC, Bennett JL, Kim HJ, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N‐MOmentum): a double‐blind, randomised placebo‐controlled phase 2/3 trial. Lancet. 2019;394(10206):1352‐1363. doi: 10.1016/s0140-6736(19)31817-3 [DOI] [PubMed] [Google Scholar]
- 11. Jonsson EN, Karlsson MO. Xpose‐‐an S‐PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed. 1999;58(1):51‐64. doi: 10.1016/s0169-2607(98)00067-4 [DOI] [PubMed] [Google Scholar]
- 12. Yan L, Kimko H, Wang B, Cimbora D, Katz E, Rees WA. Population pharmacokinetic modeling of inebilizumab in subjects with Neuromyelitis Optica Spectrum Disorders, Systemic Sclerosis or relapsing Multiple Sclerosis. Clin Pharmacokinet. 2021;61(3):387‐400. doi: 10.1007/s40262-021-01071-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Basic goodness‐of‐fit plots of inebilizumab PD model. CWRES, conditional weighted residual; DV, dependent variable (observation); IPRED, individual prediction; PRED, population prediction. The red solid lines are loess smoothing lines
FIGURE S2 Forest plot of worsening from baseline in EDSS during the RCP. ADA, anti‐drug antibody; AUC0−14d, area under the concentration–time curve from Time 0 to 14 days postdose; AUC0−14d Ter 1, 300 mg inebilizumab‐treated subjects with low AUC0−14d; AUC0−14d Ter 2, 300 mg inebilizumab‐treated subjects with medium AUC0−14d; AUC0−14d Ter 3, 300 mg inebilizumab‐treated subjects with high AUC0−14d; AUCcumulative, cumulative area under the concentration–time curve from time 0 of Dose 1 to the last measurable concentration in RCP; AUCcumulative Ter 1, 300 mg inebilizumab‐treated subjects with low AUCcumulative; AUCcumulative Ter 2, 300 mg inebilizumab‐treated subjects with medium AUCcumulative; AUCcumulative Ter 3, 300 mg inebilizumab‐treated subjects with high AUCcumulative; CL, systemic clearance; CL Ter 1, inebilizumab‐treated subjects with low CL; CL Ter 2, inebilizumab‐treated subjects with medium CL; CL Ter 3, inebilizumab‐treated subjects with high CL; EDSS, Expanded Disability Status Scale; RCP, randomized controlled period; Weight Q1, inebilizumab‐treated subjects with lowest quartile body weight; Weight Q2 & Q3, inebilizumab‐treated subjects with interquartile range (2nd and 3rd quartile) of body weight; Weight Q4, inebilizumab‐treated subjects with highest quartile of body weight. All Negative, only subjects who were determined ADA negative; All Positive, only subjects who were determined ADA positive. Odds ratio and 95% confidence interval < 1 supports treatment with inebilizumab was better than placebo.
FIGURE S3 Forest plot of the cumulative number of active MRI lesions during the RCP. ADA, anti‐drug antibody; AUC0−14d, area under the concentration–time curve from time 0 to 14 days post dose; AUC0−14d Ter 1, 300 mg inebilizumab‐treated subjects with low AUC0−14d; AUC0−14d Ter 2, 300 mg inebilizumab‐treated subjects with medium AUC0−14d; AUC0−14d Ter3, 300 mg inebilizumab‐treated subjects with high AUC0−14d; AUCcumulative, cumulative area under the concentration–time curve from time 0 of Dose 1 to the last measurable concentration in RCP; AUCcumulative Ter 1, 300 mg inebilizumab‐treated subjects with low AUCcumulative; AUCcumulative Ter 2, 300 mg inebilizumab‐treated subjects with medium AUCcumulative; AUCcumulative Ter 3, 300 mg inebilizumab‐treated subjects with high AUCcumulative; CL, systemic clearance; CL Ter 1, inebilizumab‐treated subjects with low CL; CL Ter 2, inebilizumab‐treated subjects with medium CL; CL Ter 3, inebilizumab‐treated subjects with high CL; MRI, magnetic resonance imaging; RCP, randomized controlled period; Weight Q1, inebilizumab‐treated subjects with lowest quartile body weight; Weight Q2 & Q3, inebilizumab‐treated subjects with interquartile range (2nd and 3rd quartile) of body weight; Weight Q4, inebilizumab‐treated subjects with highest quartile of body weight. All Negative, only subjects who were determined ADA negative; All Positive, only subjects who were determined ADA positive. Rate ratio and 95% confidence interval < 1 supports treatment with inebilizumab was better than placebo.
FIGURE S4 Forest plot of number of NMOSD‐related in‐patient hospitalizations during the RCP. ADA, anti‐drug antibody; AUC0−14d, area under the concentration–time curve from time 0 to 14 days post dose; AUC0−14d Ter 1, 300 mg inebilizumab‐treated subjects with low AUC0−14d; AUC0−14d Ter 2, 300 mg inebilizumab‐treated subjects with medium AUC0−14d; AUC0−14d Ter 3, 300 mg inebilizumab‐treated subjects with high AUC0−14d; AUCcumulative, cumulative area under the concentration–time curve from time 0 of Dose 1 to the last measurable concentration in RCP; AUCcumulative Ter 1, 300 mg inebilizumab‐treated subjects with low AUCcumulative; AUCcumulative Ter 2, 300 mg inebilizumab‐treated subjects with medium AUCcumulative; AUCcumulative Ter 3, 300 mg inebilizumab‐treated subjects with high AUCcumulative; CL, systemic clearance; CL Ter 1, inebilizumab‐treated subjects with low CL; CL Ter 2, inebilizumab‐treated subjects with medium CL; CL Ter 3, inebilizumab‐treated subjects with high CL; NMOSD, neuromyelitis optica spectrum disorder; RCP, randomized, controlled period; Weight Q1, inebilizumab‐treated subjects with lowest quartile body weight; Weight Q2 & Q3, inebilizumab‐treated subjects with interquartile range (2nd and 3rd quartile) of body weight; Weight Q4, inebilizumab‐treated subjects with highest quartile of body weight. All Negative, only subjects who were determined ADA negative; All Positive, only subjects who were determined ADA positive. Rate ratio and 95% confidence interval < 1 supports treatment with inebilizumab was better than placebo.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.