

Abstract
Background
Most reported patients carrying GNAO1 mutations showed a severe phenotype characterized by early‐onset epileptic encephalopathy and/or chorea.
Objective
The aim was to characterize the clinical and genetic features of patients with mild GNAO1‐related phenotype with prominent movement disorders.
Methods
We included patients diagnosed with GNAO1‐related movement disorders of delayed onset (>2 years). Patients experiencing either severe or profound intellectual disability or early‐onset epileptic encephalopathy were excluded.
Results
Twenty‐four patients and 1 asymptomatic subject were included. All patients showed dystonia as prominent movement disorder. Dystonia was focal in 1, segmental in 6, multifocal in 4, and generalized in 13. Six patients showed adolescence or adulthood‐onset dystonia. Seven patients presented with parkinsonism and 3 with myoclonus. Dysarthria was observed in 19 patients. Mild and moderate ID were present in 10 and 2 patients, respectively.
Conclusion
We highlighted a mild GNAO1‐related phenotype, including adolescent‐onset dystonia, broadening the clinical spectrum of this condition. © 2022 The Authors. Movement Disorders published by Wiley Periodicals LLC on behalf of International Parkinson and Movement Disorder Society
Keywords: dystonia, GNAO1, phenotypes, mutation
GNAO1 mutations have been associated with two phenotypes: a severe, early‐infantile epileptic encephalopathy with burst‐suppression (EIEE17, OMIM 615473 1 ) and a neurodevelopmental disorder with involuntary movements (NEDIM, OMIM 6174932, 3, 4), with or without seizures. GNAO1 encodes the α‐subunit of a heterotrimeric guanine nucleotide‐binding protein (Gαo), which is widely expressed in the central nervous system, playing an important role in signal transduction through AMPc metabolism in the striatum.2, 5, 6 As the number of reports increased, it became evident that GNAO1‐related encephalopathies encompass a continuous spectrum of neurological syndromes featuring variable association of movement disorders, psychomotor delay, intellectual disability (ID), and different types of epilepsy.2, 7 GNAO1‐related movement disorder usually starts in infancy. Choreoathetosis is usually described with spontaneous or trigger‐induced exacerbations, potentially leading to status dystonicus, as a hallmark of the disease. 2 Most patients reported so far showed a severe phenotype, with recurrent exacerbations and significant disability. However, in a few atypical, milder cases, with movement disorder onset in late childhood or adolescence, no acute exacerbation and less‐severe disability have been identified using next‐generation sequencing techniques.8, 9, 10 In this study, we characterized the clinical and genetic features of a cohort of patients with mild GNAO1‐related phenotype characterized by prominent movement disorders, further expanding the spectrum of this condition.
Patients and Methods
Patients
Patients carrying causative heterozygous variants in GNAO1 and exhibiting mild phenotypes were included from 18 neurology and neuropediatric movement disorders reference centers from the United States, France, Israel, Switzerland, the United Kingdom, and Italy. Mild phenotype was defined by (1) lack of severe or profound ID, (2) lack of early‐onset epileptic encephalopathy, (3) late‐onset (ie, after age 2 years) appearance of movement disorders, and (4) acquisition of walk. Patients were recruited through an international collaboration mediated by the online platform Genematcher. 11 All patients were assessed by neurologists or neuro‐pediatricians with an expertise in movement disorders. Patients' phenotypes from family 6 and family 4, which were previously reported elsewhere, were added in the cohort as further clinical data were obtained.
Genetic Analysis
CGH‐array, gene panel, exome, and genome sequencing were performed as previously reported.9, 10, 12, 13, 14, 15 Detailed procedures of the sequencing, including library preparation and bioinformatic analysis, are available in Supplementary Data. Variants were considered as causative if they fulfilled the following criteria: (1) known disease mutation reported in ClinVar; (2) loss‐of‐function variant, including protein truncating variants, frameshift indel, large deletion, and splice site changes predicted to cause aberrant splicing; or (3) missense variant with a CADD score >20, absent in GnomAD and predicted to be deleterious by at least two additional algorithms (Polyphen‐2, SIFT and Mutation taster). In addition, variant class of pathogenicity was reported according to the American College of Medical Genetics and Genomics (ACMG) guidelines. 16
Ethics
All patients and relatives provided written informed consent before genetic analysis. Strasbourg University Hospital review board gave approval for the exome sequencing of families 4 and 6 that was performed in a research framework. Genetic analysis for other families was performed for diagnostic purposes.
Results
We included 24 patients (15 women) and 1 asymptomatic carrier from 20 different families. Patients' clinical characteristics are provided in Table 1. Mean age at inclusion was 23.8 years (range: 5–66), mean age at disease onset was 6.6 years (range: 0.25–47), and mean age of dystonia onset was 10.1 years (range: 2–47). Initial manifestations included dystonia in 10 (41%), myoclonus or seizure in 1, developmental delay in 13, language delay in 4, motor delay in 9, and hypotonia in 4 patients. Seven patients were from three unrelated families showing autosomal dominant inheritance, while all others were sporadic cases due to de novo mutations. Pedigrees of these three families and videos of patients are available in Supplementary Data.
TABLE 1.
Clinical and genetic features of GNAO1 mutation carriers
| Patient ID | Ancestry | Gender | Age at last assessment | Age at first symptoms | First symptom | Dystonia | Parkinsonism | Myoclonus | Chorea | Hypotonia | Intellectual disability | Seizures | Speech | Other | Treatment response | GNAO1 variant | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dystonia age of onset | Topography | Progression | Acute exacerbations | ||||||||||||||||
|
Family 1 |
North African | Female | 28 y | 4 y | Seizure | 12 y | Segmental: face, neck, upper limbs | No | No | Akinetic‐rigid syndrome | No | No | No | Mild | Yes | Normal | None | Mild response to levodopa, moderate improvement with trihexyphenidyl | [NM_020988.3]:c.68 T > C; p.L23P, htz |
| Case A | |||||||||||||||||||
|
Family 2 |
North African | Female | 5 y | By 1 y | Developmental delay (motor delay) |
2 y |
Generalized: oromandibular, trunk. Dystonic gait | Yes | No | No | No | No | Yes | No | No | Dysarthria | None | NA | [NM_020988.3]:c.137A > G; p.K46R, htz |
| Case A | 4 mo | ||||||||||||||||||
|
Family 3 |
European | Male | 19 y | 3 mo | Developmental delay (motor delay, hypotonia) | 12 y | Generalized: left upper limb, cervical and axial dystonia. BFMDRS: 16 | Yes | No | No | No | No | Yes | Mild | Yes | Dysarthria | None | No response to levodopa and tetrabenazine, minimal improvement with gabapentin and trihexyphenidyl | [NM_020988.3]:c.535A > G; p.R179G, htz |
| Case A | |||||||||||||||||||
|
Family 4 |
European | Male | 24 y | 15 y | Dystonia | 15 y | Segmental: oromandibular and cervical dystonia | No | No | No | No | No | No | No | No | Dysarthria | None | No response to levodopa and tetrabenazine, mild worsening by Gpi‐DBS | [NM_020988.3]:c.617G > A; p.R206Q, htz |
| Case A | |||||||||||||||||||
|
Family 4 |
European | Female | 53 y | 47 y | Dystonia | 47 y | Focal: cervical | No | No | No | No | No | No | No | No | Normal | None | NA | |
| Case B | |||||||||||||||||||
|
Family 4 |
European | Female | 57 y | 30 y | Dystonia | 30 y | Multifocal: upper and lower limbs, laryngeal dystonia with dysarthria | No | No | No | No | No | No | No | No | Dysarthria | None | NA | |
| Case C | |||||||||||||||||||
|
Family 5 |
European | Female | 5 y 11 mo | 3 mo | Developmental delay (motor delay, hypotonia) | 5 y | Multifocal: four limbs | No | No | No | No | No | Yes | No | No | Dysarthria language delay | None | NA | [NM_020988.3]:c.622G > C; p.E208N, htz |
| Case A | |||||||||||||||||||
|
Family 6 |
European | Male | 31 y | 3 y | Dystonia | 3 y | Generalized: left lower limb dystonia, bilateral upper limbs dystonia, laryngeal dystonia, abnormal axial posture. BFMDRS: 24.5 | Yes | No | No | No | No | Yes | Mild | No | Dysarthria | None | Mild response to levodopa, mild response to trihexyphenidyl | [NM_020988.3]:c.644G > A; p.C215Y, htz |
| Case A | |||||||||||||||||||
|
Family 6 |
European | Female | 66 y | 5 y | Dystonia | 5 y | Generalized: laryngeal dystonia, facial dystonia, axial dystonia, abnormal posture of the left hand and bilateral pes valgus. BFMDRS: 23.5 | Yes | No | No | Yes | No | No | No | No | Dysarthria | Pyramidal syndrome | Subjective response to levodopa | |
| Case B | |||||||||||||||||||
|
Family 7 |
European | Male | 48 y | 6 y | Dystonia | 6 y | Generalized: oromandibular, cervical, trunk, upper limbs, left foot | Yes | No | No | No | No | No | Mild | No | Dysarthria | None | Subjective response to levodopa; no effect of trihexyphenidyl | |
| Case A | |||||||||||||||||||
|
Family 8 |
European | Female | 16 y | 6 y | Dystonia | 6 y | Segmental: oropharyngeal, neck and trapezius | Yes | Yes | No | No | No | No | Mild | No | Severe dysarthria | MDD | Mild improvement with gabapentin, no benefit with trihexyphenidyl | |
| Case A | |||||||||||||||||||
|
Family 9 |
African American | Male | 13 y | 7 mo | Developmental delay (language delay) | 5 y | Generalized: initially axial (opisthotonic with neck involvement) with secondary limbs and oromandibular involvement | Yes | No | No | No | No | No | Moderate | No | Dysarthria | None | No response to levodopa and trihexyphenidyl; rash with clonazepam; minimal improvement with baclofen | [NM_020988.3]:c.724‐8G > A, htz |
| Case A | |||||||||||||||||||
| Family 10 Case A |
Moroccan |
Female | 15 y | 1 y | Developmental delay (motor delay) | 5 y | Generalized dystonia. BFMDRS: 48.5 | No | No | No | No | No | No | Mild | No | Dysarthria | Exaggerated startle reflex | No response to levodopa | |
| Syrian Jewish | |||||||||||||||||||
| Family 11 Case A | European | Female | 18 y 6 mo | By 1 y | Developmental delay (motor delay) | 7 y | Generalized: cervical extension, dystonic forward trunk lean. BFMDRS: 85 | Yes | No | Severe akinetic rigid syndrome | No | No | Yes | Mild | No | Anarthria | None | No response to levodopa, trihexyphenidyl, and baclofen; sustained response to bilateral Gpi‐DBS | |
| Family 12 Case A | European | Female | 21 y | By 1 y | Developmental delay (motor delay) | 7 y | Generalized dystonia with severe cervical neck extension and oromandibular dystonia. BFMDRS: 64.5 | Yes | No | Akinetic rigidity, facial akinesia | No | No | No | Moderate | No | Normal | None | No response to levodopa, carbamazepine, trihexyphenidyl, and baclofen; good response to Gpi‐DBS | |
| Family 13 Case A | Caucasian | Male | 20 y 2 mo | By 1 y | Developmental delay (motor delay, language delay) | 11 y | Generalized: bilateral upper limb, axial, trunk, cervical, oro‐linguo‐pharygolarynx dystonia with speech and swallowing impairment; dystonic gait; BFMDRS: 45.5 | Yes | Yes | Akinetic‐rigid syndrome | No | No | Yes | No | No | Severe dysarthria | ADHD | No response to amantadine and levodopa; moderate and transient response to methylphenidate and trihexyphenidyl; good response to Gpi‐DBS | |
| Family 14 Case A | Chinese European | Male | 13 y | 3 y | Developmental delay (language delay) myoclonus | 11 y | Segmental: bilateral upper‐limb dystonia | No | No | No | Yes (upper limbs) | No | Yes | Mild | No | Normal | ASD | No response to acetazolamide and amantadine; improvement in dystonia with trihexyphenidyl | |
| Family 15 Case A | Mixed European | Female | 11 y 8 mo | 10 mo | Developmental delay | 2 y | Generalized: upper and lower limbs, axial, dystonia gait | Yes | No | No | Yes (upper limbs) | Yes (left sided) | Yes | No | No | Dysarthria | ADHD | Moderate response to tetrabenazine on chorea, good response to trihexyphenidyl | |
| Family 16 Case A | Northern European | Female | 5 y 6 mo | 3 mo | Developmental delay (motor delay, hypotonia) | 5 y | Segmental: dystonic posturing of fingers and hands | No | No | No | No | No | Yes | Mild | No | Dysarthria | None | Good response to levodopa | |
| Family 17 Case A | Caucasian | Male | 18 y | 2 y | Developmental delay (language delay) | 2 y | Segmental: laryngeal, right upper limb (handwriting) and cervical | No | No | No | No | No | No | No | No | Dysarthria | None | Response to anticholinergic and levodopa | [NM_020988.3]:c.725A > C; p.N242T, htz |
| Family 18 Case A | European | Female | 20 y 4 mo | 6 y | Dystonia | 6 y | Generalized dystonia. BFMDRS: 67.5 | Yes | Yes | Bradyknesia‐akinesia | No | Yes generalized | Yes | Mild | No | Anarthria | None | No response to haloperidol, tetrabenazine, and trihexyphenidyl; moderate response to catapressan; excellent response to GPi‐DBS | [NM_020988.3]:c.737A > T, p.E246V, htz |
| Family 19 Case A | European | Female | 13 y | 6 y | Dystonia | 6 y | Multifocal: bilateral upper limb, cervical, and oromandibular dystonia | No | No | Mild akinetic rigid syndrome | No | No | No | No | Yes | Dysarthria | None | NA | [NM_020988.3]:c.765dupT; p.N256*, htz |
| Family 19 Case B | European | Female | 39 y | 16 y | Dystonia | 16 y | Multifocal: bilateral upper limb, cervical, and oromandibular dystonia | No | No | Mild akinetic rigid syndrome | No | No | No | No | No | Dysarthria | None | No response to clonazepam; sustained response to Gpi‐DBS | |
| Family 20 Case A | Caucasian | Male | 9 y | By 1 y | Developmental delay (motor delay, hypotonia) | 9 y | Generalized: upper‐limb and trunk dystonia, dystonic gait | Yes | No | No | No | No | Yes | No | No | Normal | None | No response to levodopa, clonazepam, or baclofen | Heterozygous deletion in 16q12.2 (273–375 kb) encompassing GNAO1 |
| Summary |
Female 15 Male 9 |
Mean age at last assessment: 23.8 y | Mean age at disease onset: 6.6 y |
Developmental delay: 13 Motor delay: 9 Language delay: 4 Dystonia: 10 Hypotonia: 4 Seizure: 1 Myoclonus: 1 |
Mean age at dystonia onset: 10.1 y |
Focal: 1 Segmental: 6 Multifocal: 4 Generalized: 13 |
Progression 13 |
Exacerbation 3 |
Parkinsonism 7 |
Myoclonus 3 |
Chorea 2 |
Hypotonia 11 |
Intellectual disability 12 |
Seizures 3 |
Dysarthria: 19 |
Pyramidal syndrome: 1 MDD: 1 ADHD: 2 ASD: 1 Exaggerated startle reflex: 1 |
|||
Abbreviations: htz: heterozygous, NA, not available; BFMDRS: Burke Fahn Marsden Dystonia Rating Scale, dystonia score; GPi‐DBS: globus pallidus internal deep brain stimulation; MDD, major depressive disorder; ADHD, attention deficit with hyperactivity disorder; ASD, autism spectrum disorder; ID, intellectual disability.
Dystonia was the main movement disorder in all patients, prominently affecting multiple segments of the upper body part in 21 patients or being limited to the cervical segment in 1 patient. Dystonia was isolated, namely not associated to any other symptom, in 7 patients (29%). Dystonia was the only movement disorder in 14 patients and was combined with other movement disorders in 10, namely myoclonus in 3, chorea in 2, and parkinsonism in 7 (with 2 patients combining three movement disorders). Only 3 patients presented an acute exacerbation of dystonia. Dystonia course was nonprogressive for 11 patients. Dystonia topography was generalized in 13 patients (54%), multifocal in 4 patients, segmental in 6 patients, and focal in 1 patient. Dystonia was associated with dysarthria/anarthria in 19 patients. Early‐onset hypotonia preceded dystonia in 11 patients. Dystonia was associated with ID in 12 patients (mild for 10 and moderate for 2). Seizures occurred in 3 patients between age 4 and 19 years. Magnetic resonance imaging was unremarkable for all patients.
Movement disorders response to medication, including anticholinergic drugs, levodopa, tetrabenazine, amantadine, clonazepam, or methylphenidate, was variable. Six patients received pallidal deep brain stimulation (DBS), with significant improvement for 5 of them. Detailed treatment outcomes are available in Supplementary Data.
Mutations carried by the patients are presented in Table 1. Details regarding pathogenicity assessment are available in Supplementary Data. Apart from the p.R206Q, all variants were classified as pathogenic (class V) according to the ACMG criteria. In family 4, we identified 3 patients with the R206Q variant, which was classified as a variant of uncertain significance due to the presence of an unaffected carrier, 4‐D, son of 4‐C, despite meeting other criteria of pathogenicity (absent from GnomAD, unanimously predicted damaging by in silico tools, affecting a highly conserved residue located in a hot spot without benign variation) (criteria PM1, PM2, PP2, and PP3). A recurring splicing variant (c.724‐8G > A), previously reported in ClinVar, was identified in 8 patients (33%) showing late‐onset and/or segmental dystonia. Previous report showed this variant to damage the natural splice acceptor site and create a stronger cryptic splice acceptor site in intron 6, resulting in the insertion of two amino acids leading to protein mislocation. 17 Two patients were carrying a nonsense variant (p.N256*), and one was carrying a large deletion encompassing GNAO1. Other mutations (p.L23P; p.K46R; p.R179G; p.R206Q; p.E208N; p.C215Y; and p.N242T and p.E246V) were all missense variants absent from GnomAD. The previously reported pathogenic p.C215Y variant 8 was found in three unrelated families. All variants were close to known mutational hot spots (Fig. 1).
FIG. 1.
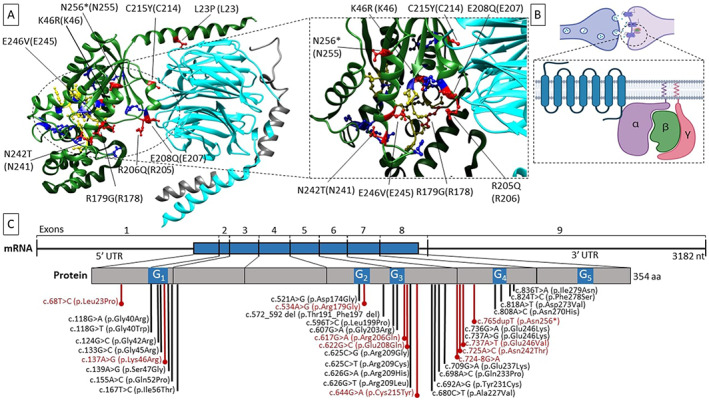
Impact of the mutations on the protein. (A) Position of the variant sites on the heterotrimeric complex containing the Gα subunit. The heterotrimer is depicted in the resting state (GDP‐bound, PDBcode 1GG2). Subunits α, β, and γ are colored in green, cyan, and gray, respectively. Affected residues in this cohort are in red, and their position is indicated both on the human Gαo1 and on rat Gαi1 (UniProtKB ID P10824, between brackets). GDP‐binding residues are colored in yellow. Previously reported GNAO1 variants are in blue. On the right, a focused view of the GDP‐binding site is shown. (B) Cartoon model of the heterotrimeric‐αβγ G‐protein coupled‐receptor on the synaptic cleft. (C) Schematic representation of the disease‐causing variants on GNAO1 transcript (NM_020988.3) and protein (UniprotKB ID P09471‐1). The amino acids impacted by the mutations identified in this work are in red, whereas previously reported variants are in black. The blue bar on the transcript indicates the translated region. The blue segments in the protein sequence indicate the G‐motifs (containing the nucleotide binding residues)—numbered from 1 to 5. Molecular graphics are realized with UCSF Chimera (http://www.rbvi.ucsf.edu/chimera), developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41‐GM103311. The cartoon has been created with BioRender.com. aa, amino acids; nt, nucleotides; UTR, untranslated region. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
Here, we report a large cohort of patients with mild GNAO1‐related phenotypes, experiencing prominent movement disorders without severe chronic encephalopathy. The typical phenotype was a nonprogressive generalized or focal/segmental upper‐body dystonia appearing beyond infancy, associated with dysarthria. Acute exacerbation occurred only in 3 patients, and 29% of patients showed isolated dystonia without additional neurological manifestation. Our inclusion criteria were able to identify these phenotypes that were in contrast with most of the previously reported patients with GNAO1‐related movement disorders, who showed severe hyperkinetic encephalopathy with recurrent dystonic exacerbations,18, 19 and profound developmental delay3, 7 with or without epilepsy in the first year of life.1, 7
Dystonia distribution was segmental or focal in 7 patients, and clinical course was nonprogressive in 11 patients, while most of the previously reported patients with GNAO1‐related movement disorders had generalized and rapidly progressive dystonia. 2 Dystonia topography revealed prominent upper‐body distribution in most of our patients, reminiscent of the clinical pictures associated with other dystonia‐related genes, such as GNAL 20, 21 or ANO3.22, 23 Seven patients also exhibited mild parkinsonism, which is consistent with the role of Gαo in the signal transduction within the striatal projection neurons downstream of the dopamine receptors.6, 24
We identified 3 autosomal dominant families where multiple symptomatic relatives carried heterozygous variants, which was in contrast with all the previously reported patients who showed de novo mutations. 2 One p.R206Q carrier did not present any clinical sign evocative of GNAO1‐related disorders. The similarities between this family's phenotype and the other cases—all showing upper‐body distribution—argue for the implication of the variant, while no other class IV to V variant in a dystonia‐related gene was identified. In addition, a family member carrying this variant had disease onset in his 40s, meaning the 30‐year‐old asymptomatic carrier could be potentially presymptomatic. Future identification of autosomal dominant family with GNAO1‐related dystonia and follow‐up of this patient might confirm whether incomplete penetrance is possible in GNAO1‐related disorders.
Response to medication was variable in our cohort. No significant response to levodopa was identified in our cohort, but 3 patients had partial response to anticholinergic drugs, which was in accordance with previous findings from the literature.2, 4, 25 Conversely, the outcome was good in 5 of 6 patients who received DBS, further confirming its efficacy in GNAO1‐related dystonia. 26
Most of the variants identified in the present work were not reported among previously published cases showing severe phenotype, and two variants recurred in multiple families, suggesting that mild phenotypes could be related to specific mutations. However, the variants we identified were close to previously reported hot spots (Fig. 1), leading to amino‐acid substitution in the same functional domains. Further studies are needed to elucidate if these different variants have a milder impact on protein function. In addition, we identified two putative loss‐of‐function variants (a nonsense variant and a whole‐gene deletion), presumably affecting protein expression and possibly causing GNAO1 haploinsufficiency. All 3 carriers were presenting late‐childhood/adolescence onset dystonia without ID. Thus far, no report described the phenotype of patients harboring GNAO1‐nonsense variants. A few patients with chromosome 16q deletions encompassing GNAO1 have been described, all harboring significantly larger deletions compared to our case and showing variable associations of dysmorphisms, microcephaly, seizures, and developmental delay. 27 Although previous research suggested that loss‐of‐function variants were mainly responsible for epileptic encephalopathy while gain‐of‐function mutations were mostly associated with a movement disorders prominent phenotype, 5 recent evidence suggests that pathogenic variants exert their effect through a combination of dominant‐negative and loss‐of‐function mechanisms, and each mutation likely produces circuit‐selective effects through a peculiar mechanism of signaling disruption. 28 The expanding spectrum of associated phenotypes and disease‐causing variants provides further evidence that genotype–phenotype correlations are nuanced, and GNAO1‐related disorders shape a continuous spectrum of overlapping phenotypes rather than distinct entities. 28 Our study carries some limitations, including the retrospective design and the lack of formal assessment in several cases. Here, we highlighted the milder GNAO1‐related phenotypes, broadening this condition‐clinical spectrum. GNAO1 mutations should be considered as a cause of adolescent or adult‐onset nonprogressive dystonia, particularly in the presence of a speech involvement even in the absence of seizures or ID.
Author Roles
Research project: A. Conception, B. Design, C. Acquisition of data, D. Analysis and interpretation of data; (2) Manuscript: A. Writing of the first draft, B. Review and critique; (3) Other: A. Subject recruitment, B. Clinical assessment of patients, C. Study supervision.
T.W.: 1A, 1B, 1C, 1D, 2A, 3A, 3B, 3C
G.G: 1C, 1D, 2A, 3A, 3B
M.A.K: 1C, 1D, 2A, 3A
A.P.: 1C, 2B, 3A, 3B
F.M.: 1C, 1D, 2B, 3A
A.T.: 1C, 1D, 2B, 3A
N.D.: 1C, 2B, 3A, 3B
G.R.: 1C, 2B, 3A, 3B
J.C.: 1C, 2B, 3A, 3B
W.M.: 1C, 2B, 3A, 3B
L.B.: 1C, 2B, 3A, 3B
D.De: 1C, 2B, 3A, 3B
P.C.: 1C, 2B, 3A, 3B
M.C.‐J.: 1C, 2B, 3A, 3B
S.J.: 1C, 2B, 3A, 3B
J.G.: 1C, 2B, 3A, 3B
J.B.: 1C, 2B, 3A, 3B
J.‐M.F.: 1C, 2B, 3A, 3B
Lu.B.: 1C, 2B, 3A, 3B
M.H.: 1C, 2B, 3A, 3B
M.P.: 1C, 2B, 3A, 3B
C.R.: 1C, 2B, 3A, 3B
N.C.: 1C, 2B, 3A, 3B
G.P.: 1C, 2B.
A.H.N.: 1C, 2B, 3A, 3B
Ma.S.: 1C, 2B, 3A, 3B
A.B.: 1C, 2B, 3A, 3B
C.E.: 1C, 2B, 3A, 3B
A.M.: 1C, 2B, 3A, 3B
E.R.: 1C, 2B, 3A, 3B
C.M.: 1C, 2B, 3A, 3B
C.B.: 1C, 2B, 3A, 3B
F.A.: 1C, 2B, 3A, 3B
C.N.: 1C, 2B, 3A, 3B
W.G.W.: 1C, 2B, 3A, 3B
D.S.: 1C, 2B, 3A, 3B
A.C.: 1C, 2B, 3A, 3B
M.V.: 1C, 2B, 3A, 3B
J.‐P.L.: 1C, 2B, 3A, 3B
C.T.: 1C, 2B, 3A, 3B
L.C.: 1C, 2B, 3A, 3B
D.Dou.: 1A, 1B, 1C, 1D, 2B, 3A, 3B, 3C
M.A.: 1A, 1B, 1C, 1D, 2B, 3A, 3B, 3C
Full financial disclosures for the previous 12 months
T.W. received grants from the Revue Neurologique, the Fondation Planiol, and the APTES organizations and travel funding from LVL medical. F.M. and A.T. are employees of GeneDx, Inc. D.S. received salary from NIHR, M.K. lab received funding from Jules Thorn Trust and Rosetrees Trust. A.M. received speaker honoraria from AbbVie. E.R. received honorarium for speech from Orkyn, Aguettant, and Elivie and for being on the advisory board of allergan; E.R. received research support from Merz‐Pharma, Orkyn, Aguettant, Elivie, Ipsen, Allergan, Everpharma, Fondation Desmarest, AMADYS, ADCY5.org, Agence Nationale de la Recherche, Societé Française de Médecine Esthétique, and Dystonia Medical Research Foundation. The rest of the authors declare no conflicts of interest.
Supporting information
Supplementary Figure S1. Conservation of affected residues across evolution. All coding sequence variants caused substitution or deletion of evolutionarily conserved amino acids. Homologous sequences were aligned using the Clustal Omega program (see Analysis Tool Web Services from the EMBL‐EBI. [2013] McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Nucleic acids research 2013 July; 41[Web Server issue]: W597‐600 doi:10.1093/nar/gkt376) on UniprotKB (https://www.uniprot.org/uniprot/).
Supplementary Table S1. In silico tools prediction and variants class of pathogenicity according to the American College of Medical Genetics and Genomics recommendations.
Video S1. GNAO1‐associated movements disorders in proband A family 3.
Video S2. GNAO1‐associated movements disorders in proband A family 4.
Video S3. GNAO1‐associated movements disorders in proband A family 6.
Video S4. GNAO1‐associated movements disorders in proband B family 6.
Video S5. GNAO1‐associated movements disorders in proband A family 8.
Video S6. GNAO1‐associated movements disorders in proband A family 13, pre‐ and postoperative assessment.
Acknowledgments
We thank the patients and their families for participation in this study and Bernard Jost from the IGBMC Microarray and Sequencing Platform, the France Parkinson organization, and the Revue Neurologique for their support. Open access funding enabled and organized by Projekt DEAL.
Thomas Wirth and Giacomo Garone contributed equally to this work and should be considered as co‐first authors.
Laura Cif, Diane Doummar, and Mathieu Anheim contributed equally to this work and should be considered as co‐last authors.
Relevant conflicts of interest/financial disclosures: None.
Funding agencies: T.W. was funded by a grant from the Revue Neurologique for this work. The study was partly supported by a grant provided by France Parkinson.
Data Availability Statement
Anonymized data pertaining to the research presented will be made available upon reasonable request from external investigators.
References
- 1. Nakamura K, Kodera H, Akita T, Shiina M, Kato M, Hoshino H, et al. De novo mutations in GNAO1, encoding a Gαo subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet 2013;93(3):496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schirinzi T, Garone G, Travaglini L, Vasco G, Galosi S, Rios L, et al. Phenomenology and clinical course of movement disorder in GNAO1 variants: results from an analytical review. Parkinsonism Relat Disord 2019;61:19–25. [DOI] [PubMed] [Google Scholar]
- 3. Kulkarni N, Tang S, Bhardwaj R, Bernes S, Grebe TA. Progressive movement disorder in brothers carrying a GNAO1 mutation responsive to deep brain stimulation. J Child Neurol 2016;31(2):211–214. [DOI] [PubMed] [Google Scholar]
- 4. Danti FR, Galosi S, Romani M, Montomoli M, Carss KJ, Raymond FL, et al. GNAO1 encephalopathy: broadening the phenotype and evaluating treatment and outcome. Neurol Genet 2017;3(2):e143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Feng H, Khalil S, Neubig RR, Sidiropoulos C. A mechanistic review on GNAO1‐associated movement disorder. Neurobiol Dis 2018;116:131–141. [DOI] [PubMed] [Google Scholar]
- 6. Delorme C, Giron C, Bendetowicz D, Méneret A, Mariani L‐L, Roze E. Current challenges in the pathophysiology, diagnosis, and treatment of paroxysmal movement disorders. Expert Rev Neurother 2021;21(1):81–97. [DOI] [PubMed] [Google Scholar]
- 7. Kelly M, Park M, Mihalek I, Rochtus A, Gramm M, Pérez‐Palma E, et al. Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate‐binding region. Epilepsia 2019;60(3):406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carecchio M, Invernizzi F, Gonzàlez‐Latapi P, Panteghini C, Zorzi G, Romito L, et al. Frequency and phenotypic spectrum of KMT2B dystonia in childhood: a single‐center cohort study. Mov Disord 2019;34(10):1516–1527. [DOI] [PubMed] [Google Scholar]
- 9. Graziola F, Garone G, Stregapede F, Bosco L, Vigevano F, Curatolo P, et al. Diagnostic yield of a targeted next‐generation sequencing gene panel for pediatric‐onset movement disorders: a 3‐year cohort study. Front Genet 2019;10:1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wirth T, Tranchant C, Drouot N, Keren B, Mignot C, Cif L, et al. Increased diagnostic yield in complex dystonia through exome sequencing. Parkinsonism Relat Disord 2020;74:50–56. [DOI] [PubMed] [Google Scholar]
- 11. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015;36(10):928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Guillen Sacoto MJ, Tchasovnikarova IA, Torti E, Forster C, Andrew EH, Anselm I, et al. De novo variants in the ATPase module of MORC2 cause a neurodevelopmental disorder with growth retardation and variable craniofacial Dysmorphism. Am J Hum Genet 2020;107(2):352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valence S, Cochet E, Rougeot C, Garel C, Chantot‐Bastaraud S, Lainey E, et al. Exome sequencing in congenital ataxia identifies two new candidate genes and highlights a pathophysiological link between some congenital ataxias and early infantile epileptic encephalopathies. Genet Med Off J Am Coll Med Genet 2019;21(3):553–563. [DOI] [PubMed] [Google Scholar]
- 14. Cif L, Demailly D, Lin J‐P, Barwick KE, Sa M, Abela L, et al. KMT2B‐related disorders: expansion of the phenotypic spectrum and long‐term efficacy of deep brain stimulation. Brain J Neurol 2020;143(11):3242–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smol T, Thuillier C, Boudry‐Labis E, Dieux‐Coeslier A, Duban‐Bedu B, Caumes R, et al. Neurodevelopmental phenotype associated with CHD8‐SUPT16H duplication. Neurogenetics 2020;21(1):67–72. [DOI] [PubMed] [Google Scholar]
- 16. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet 2015;17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miyamoto S, Nakashima M, Fukumura S, Kumada S, Saitsu H. An intronic GNAO1 variant leading to in‐frame insertion cause movement disorder controlled by deep brain stimulation. Neurogenetics 2022;23(2):129‐35. [DOI] [PubMed] [Google Scholar]
- 18. Ananth AL, Robichaux‐Viehoever A, Kim Y‐M, Hanson‐Kahn A, Cox R, Enns GM, et al. Clinical course of six children with GNAO1 mutations causing a severe and distinctive movement disorder. Pediatr Neurol 2016;59:81–84. [DOI] [PubMed] [Google Scholar]
- 19. Honey CM, Malhotra AK, Tarailo‐Graovac M, van Karnebeek CDM, Horvath G, Sulistyanto A. GNAO1 mutation‐induced pediatric dystonic storm rescue with Pallidal deep brain stimulation. J Child Neurol 2018;33(6):413–416. [DOI] [PubMed] [Google Scholar]
- 20. Fuchs T, Saunders‐Pullman R, Masuho I, Luciano MS, Raymond D, Factor S, et al. Mutations in GNAL cause primary torsion dystonia. Nat Genet 2013;45(1):88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Geoghegan AR, Al Hussona M, Beauchamp NJ, Hutchinson M, Sean O'Riordan MB, Lynch T, et al. A novel GNAL mutation in familial dystonia presenting with childhood tremor and myoclonus. Mov Disord Off J Mov Disord Soc 2019;34(6):923–924. [DOI] [PubMed] [Google Scholar]
- 22. Charlesworth G, Plagnol V, Holmström KM, Bras J, Sheerin U‐M, Preza E, et al. Mutations in ANO3 cause dominant Craniocervical dystonia: Ion Channel implicated in pathogenesis. Am J Hum Genet 2012;91(6):1041–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lange LM, Junker J, Loens S, Baumann H, Olschewski L, Schaake S, et al. Genotype‐phenotype relations for isolated dystonia genes: MDSGene systematic review. Mov Disord Off J Mov Disord Soc 2021;36(5):1086–1103. [DOI] [PubMed] [Google Scholar]
- 24. Corvol J‐C, Muriel M‐P, Valjent E, Féger J, Hanoun N, Girault J‐A, et al. Persistent increase in olfactory type G‐protein alpha subunit levels may underlie D1 receptor functional hypersensitivity in Parkinson disease. J Neurosci Off J Soc Neurosci 2004;24(31):7007–7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamashita Y, Ogawa T, Ogaki K, Kamo H, Sukigara T, Kitahara E, et al. Neuroimaging evaluation and successful treatment by using directional deep brain stimulation and levodopa in a patient with GNAO1‐associated movement disorder: a case report. J Neurol Sci 2020;411:116710 [DOI] [PubMed] [Google Scholar]
- 26. Koy A, Cirak S, Gonzalez V, Becker K, Roujeau T, Milesi C, et al. Deep brain stimulation is effective in pediatric patients with GNAO1 associated severe hyperkinesia. J Neurol Sci 2018;15(391):31–39. [DOI] [PubMed] [Google Scholar]
- 27. Apuzzo D, Cappuccio G, Vaisanen T, Alagia M, Pignataro P, Genesio R, et al. Two cases of 16q12.1q21 deletions and refinement of the critical region. Eur J Med Genet 2020;63(6):103878 [DOI] [PubMed] [Google Scholar]
- 28. Muntean BS, Masuho I, Dao M, Sutton LP, Zucca S, Iwamoto H, et al. Gαo is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep 2021;34(5):108718 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. Conservation of affected residues across evolution. All coding sequence variants caused substitution or deletion of evolutionarily conserved amino acids. Homologous sequences were aligned using the Clustal Omega program (see Analysis Tool Web Services from the EMBL‐EBI. [2013] McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, Cowley AP, Lopez R. Nucleic acids research 2013 July; 41[Web Server issue]: W597‐600 doi:10.1093/nar/gkt376) on UniprotKB (https://www.uniprot.org/uniprot/).
Supplementary Table S1. In silico tools prediction and variants class of pathogenicity according to the American College of Medical Genetics and Genomics recommendations.
Video S1. GNAO1‐associated movements disorders in proband A family 3.
Video S2. GNAO1‐associated movements disorders in proband A family 4.
Video S3. GNAO1‐associated movements disorders in proband A family 6.
Video S4. GNAO1‐associated movements disorders in proband B family 6.
Video S5. GNAO1‐associated movements disorders in proband A family 8.
Video S6. GNAO1‐associated movements disorders in proband A family 13, pre‐ and postoperative assessment.
Data Availability Statement
Anonymized data pertaining to the research presented will be made available upon reasonable request from external investigators.