

Abstract
Accurate and consistent interpretation of sequence variants is integral to the delivery of safe and reliable diagnostic genetic services. To standardize the interpretation process, in 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published a joint guideline based on a set of shared standards for the classification of variants in Mendelian diseases. The generality of these standards and their subjective interpretation between laboratories has prompted efforts to reduce discordance of variant classifications, with a focus on the expert specification of the ACMG/AMP guidelines for individual genes or diseases. Herein, we describe our experience as a ClinGen Variant Curation Expert Panel to adapt the ACMG/AMP criteria for the classification of variants in three globin genes (HBB, HBA2, and HBA1) related to recessively inherited hemoglobinopathies, including five evidence categories, as use cases demonstrating the process of specification and the underlying rationale.
Keywords: ACMG/AMP criteria, ClinGen VCEP, globin gene variants, hemoglobinopathy, variant classification
1. INTRODUCTION
Burgeoning demand for genetic screening and correspondingly expanded diagnostic sequencing efforts have dramatically increased the number of sequence variants, many of unknown significance, which require clinical annotation. The collection, assessment, and evaluation of variant evidence required to determine clinical actionability is a resource‐intensive process, influenced by expert opinion and differences in methodologies and thresholds across clinical laboratories (Harrison et al., 2017). Toward an effort to establish a common framework for variant classification based on a standardized and transparent assessment of different lines of evidence, in 2015, the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published joint recommendations for the interpretation of variants in genes associated with Mendelian disorders (Richards et al., 2015). The ACMG/AMP framework defined 28 evidence criteria, organized by type and strength, and developed a five‐tier scheme to classify variants as pathogenic, likely pathogenic, of uncertain significance, likely benign, or benign. This framework was designed for general use across different genes, diseases, and inheritance patterns, thus necessitating the application of expert judgment when evaluating and weighing evidence for the interpretation of variants. In response to the need for standardized evidence‐based methods to characterize the clinical relevance of gene‐ and disease‐specific sequence variants, the Clinical Genome Resource (ClinGen) assembles Variant Curation Expert Panels (VCEPs) to develop specifications for the ACMG/AMP framework (Rehm et al., 2015). In addition, ClinGen established the Sequence Variant Interpretation Working Group (SVI WG) to provide general refinement of the ACMG/AMP guidelines for criteria that are applicable across diverse domains and to harmonize guideline specifications made by the individual VCEP (Harrison et al., 2019). The SVI WG systematically reviews the ACMG/AMG guidelines and has already published recommendations for the specification of multiple evidence types (https://www.clinicalgenome.org/svi/).
The ClinGen Hemoglobinopathy VCEP (www.clinicalgenome.org/affiliation/50052/) was created collaboratively between the ITHANET Portal (https://www.ithanet.eu/) and the Global Globin Network of the Human Variome Project (http://www.humanvariomeproject.org/gg2020/) to perform gene‐ and disease‐specific modifications to the ACMG/AMP framework for variants related to hemoglobinopathies. Hemoglobinopathies are the commonest monogenic disorders worldwide, with an extremely diverse clinical spectrum of conditions of varying severity and can be broadly classified into the thalassemia syndromes, characterized by a reduction in protein synthesis, and the structural hemoglobin variants, characterized by changes in protein stability and structure. Hemoglobinopathies are caused by both short nucleotide variants (SNVs) and copy number variants in the two globin gene clusters, namely the α‐globin locus (NG_000006), including genes HBA1, HBA2, and HBZ, and the β‐globin locus (NG_000007), including genes HBB, HBD, HBG1, HBG2, and HBE1, and locus‐specific regulatory elements (Higgs et al., 2012). They predominantly have a recessive mode of inheritance, although dominantly inherited phenotypes have also been described and a large number of genetic modifiers are known to affect disease expressivity and penetrance (Stephanou et al., 2019). An epidemiological complication of hemoglobinopathies in regions historically endemic for malaria is the resistance of heterozygotes for established pathogenic variants to malaria (heterozygote advantage), which over time has led to an atypical enrichment of disease‐causing alleles in corresponding populations (Kountouris et al., 2014; Roberts & Williams, 2003).
Normal adults have a complement of four alpha (αα/αα) and two beta (β/β) globin genes, which encode the globin chains constituting the main adult tetrameric α2β2 hemoglobin molecule. Correspondingly, the underlying HBA1, HBA2, and HBB genes are primary pathology determinants in adults and are, therefore, the focus of initial guideline specification efforts for the hemoglobinopathies. The spectrum of phenotypes and disease severity depend on the properties of the protein variant in the case of structural defects (Thom et al., 2013), and on the number of genes that are lost, abnormally expressed, or, in some cases, duplicated in the case of the thalassemias (Farashi & Harteveld, 2018; Thein, 2013). For the thalassemias, the severity of the co‐inherited mutant alleles, from mild (++) to moderate (+) to absolute (0) deficiency of globin expression, affects survival (Kountouris et al., 2020) and determines disease severity through anemia, that is, an overall reduced level of hemoglobin, and through the toxicity of homotetramers formed by unaffected, excess globin chains. The degree of globin chain imbalance is thus central to thalassemia pathophysiology, with most thalassemia alleles causing observable changes in the hematological indices of heterozygotes (Kohne, 2011). These gene‐disease characteristics pose challenges for current ACMG/AMP variant interpretation guidelines and require customized criteria for accurate variant interpretation.
The Hemoglobinopathy VCEP is tasked with providing expert review of all globin gene variants and resolution of conflicting interpretations in the ClinVar variant database using the specified ACMG/AMP guidelines. Table 1 shows current summary data for hemoglobinopathy variants available on ClinVar (accessed on May 14, 2021). Accordingly, a total of 794 sequence variants affecting HBB are annotated in ClinVar, and a smaller number of 199 and 242 sequence variants affecting HBA1 and HBA2, respectively. Most importantly, only a fraction of these variants has a review status of two stars in ClinVar, denoting two or more submissions with assertion criteria and evidence (or a public contact) providing the same interpretation. Specifically, the percentage of these variants with a two‐star review status in ClinVar is 26.4%, 8%, and 10.7%, for HBB, HBA1, and HBA2, respectively, highlighting the need for expert review of variants in these genes.
Table 1.
Number of annotated globin gene variants in ClinVar (accessed on May 14, 2021)
| HBB (Two‐star status) | HBA1 (Two‐star status) | HBA2 (Two‐star status) | |
|---|---|---|---|
| Pathogenic | 407 (101) | 103 (3) | 120 (12) |
| Likely pathogenic | 70 (42) | 4 (1) | 12 (3) |
| Variant of uncertain significance | 163 (28) | 42 (4) | 50 (4) |
| Likely benign | 114 (23) | 21 (4) | 17 (3) |
| Benign | 40 (16) | 29 (4) | 43 (4) |
| Conflicting interpretations | 52 | 10 | 16 |
The Hemoglobinopathy VCEP specifications were approved by ClinGen in April 2021 (Step 2 approval), which initiated the process of further validation and adaptation with known globin gene variants in a pilot study (toward Step 3 approval). Correspondingly, this report avoids detailed presentation of the current specifications and instead uses the perspective of the Hemoglobinopathy VCEP to describe the process of ACMG/AMP guideline adaptation for SNVs with recessive inheritance in HBB, HBA2, and HBA1. Owing to the involvement of two loci, the unusual epidemiology, and the complexity of allele interaction and phenotypes for hemoglobinopathies, our observations help amplify the challenges generally encountered during variant curation and interpretation, and during the specification of ACMG/AMP guidelines for future VCEPs.
The ACMG/AMP framework constitutes a classification system for Mendelian variants based on evidence criteria that assess variant frequency in the general population, variant types with disease causality, protein domains and mutational hotspots implicated in disease, disease/trait phenotypes in probands and families with observed segregation, in silico predictions and functional evidence. Evidence criteria are divided into those that support a benign and a pathogenic classification, with intermediate categories being likely pathogenic, of uncertain significance and likely benign, each with a suggested measure of strength, namely supporting, moderate, strong, or very strong (Richards et al., 2015). The Hemoglobinopathy VCEP has a total of 31 unique evidence codes, shown in Table 2, some of which are assigned at different levels of strength depending on the amount of evidence that is available. These criteria can be broadly grouped into five categories that are discussed in the subsequent sections of this article.
Table 2.
An overview of the ClinGen Hemoglobinopathy VCEP‐specified ACMG/AMP criteria
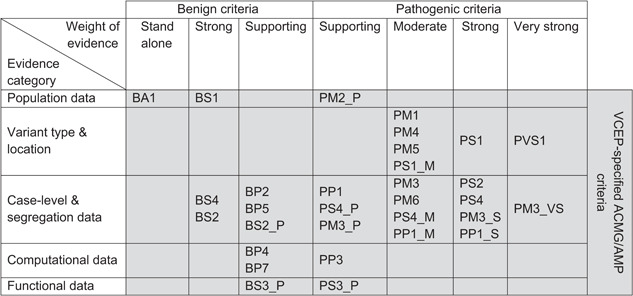
|
Abbreviations: ACMG, American College of Medical Genetics and Genomics; AMP, Association for Molecular Pathology; VCEP, Variant Curation Expert Panel.
2. POPULATION DATA (PM2, BA1, AND BS1)
The frequency of a variant in the general population can be informative for its pathogenicity, as variants of high frequency in any large general population or control cohort are unlikely to be disease‐causing. For this reason, a stand‐alone benign criterion (BA1) was introduced in the original ACMG/AMP guidelines for all variants with a frequency of at least 5% in a general or control population. This threshold is very conservative, is selected for use across different genes and diseases, and is adjusted by VCEPs to reflect known allele frequencies in the genes of interest. In addition, BS1 is used for variants with a frequency higher than expected for the disorder and it is also VCEP‐specific (Ghosh et al., 2018). A statistical framework has been developed to facilitate the estimation of these thresholds and is available at the Allele Frequency App (Whiffin et al., 2017). The framework accounts for disease prevalence, genetic and allelic heterogeneity, inheritance mode, penetrance, and sampling variance in reference data sets. Currently, gnomAD is the largest available population database and is widely used as a reference data set for the calculation of these thresholds (Karczewski et al., 2020).
In the case of hemoglobinopathies, the high frequencies of some established pathogenic variants in historical malaria regions directly affect the application of criteria BA1 and BS1. Hence, to ensure the effective use of minor allele frequency evidence, the VCEP compiled a list of established variants that are excluded from criteria requiring population data (i.e., BA1, BS1, and PM2_Supporting) based on their frequency in different populations globally. The variant frequencies were derived from the IthaMaps database (Kountouris et al., 2014, 2017), which manually curates relative allele frequencies of specific globin gene variants at the country and regional level.
Extremely low frequency of genetic variants in the general population is considered moderate evidence for pathogenicity, based on the original ACMG/AMP guidelines (PM2). After further analysis and modeling, the ClinGen SVI subsequently recommended downgrading the strength of evidence to supporting. The threshold for PM2_Supporting is often defined to an order of magnitude lower than the BS1 threshold, while it can be alternatively defined by analyzing the frequency of established pathogenic or benign variants for the gene of interest and calculation of likelihood ratios for different thresholds. Using variants with established pathogenicity, the Hemoglobinopathy VCEP has selected the threshold that maximizes the likelihood ratio for globin gene variants and this approach will be validated and adjusted as required during the ongoing pilot study.
3. VARIANT TYPE AND LOCATION (PVS1, PS1, PM1, PM4, AND PM5)
The interpretation of a sequence variant requires an understanding of its effect on the structure and function of the gene product and prior knowledge of the molecular mechanism of disease. While some variants have a deleterious impact on protein production and/or function, others may cause partial or no discernible changes in phenotype. In contrast to variants that can lead to loss of function owing to premature termination of translation and protein synthesis, missense variants are difficult to assess for their pathogenicity, which largely depends on the variant position in the protein sequence and the biochemical consequence of the amino acid change. In addition, variants may act as benign bystanders to disease, or they may contribute to disease in the presence of another variant in the same gene. The molecular consequences of sequence variants depending on the variant type and the genomic location are evaluated by several rules in the ACMG/AMP framework (i.e., PVS1, PS1, PM4, and PM5).
Loss of function is an established primary disease mechanism for hemoglobinopathies, hence the PVS1 criterion for null variants (e.g., nonsense, frameshift, canonical ±1,2 splice sites, initiation codon, single‐exon, or multi‐exon deletion) would apply, particularly in the case of the thalassemia syndromes. In fact, there is currently no null globin gene variant with a benign or likely benign effect reported in ClinVar, further highlighting the role of loss of function as a primary disease mechanism. Nevertheless, in line with ClinGen SVI recommendations (Abou Tayoun et al., 2018), the Hemoglobinopathy VCEP is currently working on PVS1 modification for different null variant types. Specifically, the VCEP will evaluate existing evidence on variant pathogenicity for each null variant type as well as alternative splicing and alternate routes of nonsense‐mediated decay for globin genes (Peixeiro et al., 2011).
In addition, PM4 (protein length changing variant) will be applied for in‐frame deletions/insertions and losses of stop codons that disrupt protein function, such as the widespread stop‐loss variant NM_000517.4:c.427T>C (Hb Constant Spring). Furthermore, as the globin genes are affected by both pathogenic and benign missense variants, standard ACMG/AMP criteria PP2 (missense variants are a common cause of disease with little benign variation) and BP1 (truncating variants are the only known mechanism of variant pathogenicity) do not apply to hemoglobinopathies. Likewise, globin genes do not contain a repetitive region without known function, as would be a prerequisite for applying BP3 (in‐frame indels in a repetitive region without known function).
The location of a variant within a protein can impart changes to the protein structure, function, and other properties. Expert opinion is necessary for specifying the important regions of a protein in the context of the molecular mechanisms of disease, also acknowledging that these regions must have a low rate of benign variants. The hemoglobin molecule is among the best‐characterized proteins, for which several structure‐ and function‐critical domains, such as α1β1 and α1β2 interfaces, the heme‐binding pocket, and other biologically relevant sites, have already been associated with molecular mechanisms, including the Bohr effect, 2,3‐DPG binding or AHSP binding (Thom et al., 2013). The ACMG/AMP criteria PM1 (variant in a critical domain/mutational hotspot), PS1 (variant creates same amino acid change as a known pathogenic variant), and PM5 (novel missense variant at the same position as known pathogenic variant) evaluate the variant location and similarly argue that variants affecting critical domains are more likely to cause functional disruption and, thus, a pathogenic effect. In hemoglobinopathies, the HBA2 and HBA1 genes are paralogous with identical structure and function, and can thus be incorporated in these criteria to provide additional evidence for variant hotspots in critical functional domains (Moradkhani et al., 2009). The ACMG/AMP criteria can be further adapted to accommodate variants found in regions that affect the expression or splicing of globin genes.
4. COMPUTATIONAL DATA (PP3, BP4, AND BP7)
Computational (in silico) tools predicting the effect of sequence variants can also facilitate variant interpretation. A plethora of tools is available with important differences in their algorithmic approach and the type of sequence variants they can predict. Some tools, such as SIFT (Kumar et al., 2009) and PolyPhen‐2 (Adzhubei et al., 2010), predict the impact of missense variants, and others, such as MaxEntScan (Yeo & Burge, 2004) and SpliceAI (Jaganathan et al., 2019), are focused on predicting the variant effect on splicing, while more recent tools can predict the effect of both coding and noncoding variants (Kircher et al., 2014). With the accuracy of in silico tools in the range of 65%–80% (Thusberg et al., 2011), the ACMG/AMP framework recommends the use of these predictions as supporting evidence for variant interpretation (PP3, BP4, and BP7). Nevertheless, the original framework does not recommend the use of specific in silico tools and, therefore, ClinGen VCEPs use different approaches to provide gene‐specific recommendations based on the predictive performance of multiple tools. Some VCEPs require an agreement in variant effect prediction among multiple tools. Other VCEPs opt for simplicity by using a meta‐predictor, such as REVEL (Ioannidis et al., 2016), which combines the results of multiple in silico tools, thus, providing a single prediction for each sequence variant. Rather than using the default threshold recommended by the in silico tool, several VCEPs have used data from established pathogenic and benign variants to identify the threshold that maximizes the predictive performance in the genes of interest. More recently, a quantitative approach (Johnston et al., 2021) has been proposed to calibrate different thresholds for benign and pathogenic computational evidence by using a Bayesian Classification Framework (Tavtigian et al., 2018).
The Hemoglobinopathy VCEP recognizes the varying strengths and weaknesses of computational tools and recommends the use of REVEL for evaluating the effect of missense variants in the globin genes. For assessing the impact of variants in splicing, the specified criteria require concordant predictions across at least 50% of the tested tools. The Hemoglobinopathy VCEP is currently conducting a large‐scale study with over 1000 annotated globin gene variants to compare the performance of computational tools, adjust the prediction thresholds and, thus, further specify the criteria that use computational data (manuscript under preparation). Notably, the use of computational evidence is not allowed for loss‐of‐function variants that meet the PVS1 rule, to avoid accounting for the same evidence in different criteria.
5. CASE LEVEL/SEGREGATION DATA (PS2, PS4, PM3, PM6, PP1, BP2, BP5, BS2, AND BS4)
Case‐level data capture information about individuals who carry the variant of interest and can satisfy several components of the ACMG/AMP framework, such as PP4 (phenotype specific for disease), PS2/PM6 (de novo with/without parental testing), PM3/BP2 (in‐trans or in‐cis with a pathogenic variant), PP1/BS4 (cosegregation in affected family members, or lack thereof), PS4/BS2 (variant observation in cases or controls), and BP5 (alternate locus observations).
Disease‐specific phenotype information is necessary to ensure that all affected individuals meet uniform diagnostic criteria. The inactivation of two β‐globin genes (β‐thalassemia) or three α‐globin genes (Hb H disease) results in disease with both hematological and clinical phenotypes. The absence of all four α‐globin genes causes the hydrops fetalis syndrome, which results in death in utero or shortly after birth. Furthermore, the inactivation of one β‐globin gene or two α‐globin genes characterizes the trait state of β‐ and α‐thalassemia, respectively, and produces a hematological phenotype that includes the change of red blood cell indices, hemoglobin pattern, and globin chain synthesis ratio. Such readily detectable clinical signs are usually uncovered by routine laboratory diagnostic screening since trait individuals are clinically asymptomatic and, thus, often unaware of their carrier status. Accordingly, prevention strategies for thalassemias and other hemoglobinopathies depend in large part on population and newborn screening, which has allowed diagnostic laboratories across different countries to amass evidence about common and rare variants in the heterozygous state. To utilize this large volume of data on heterozygous trait individuals in hemoglobinopathies as a resource for variant annotation, the Hemoglobinopathy VCEP has adapted the ACMG/AMP framework to capture the phenotype of heterozygous trait individuals as additional evidence for variant pathogenicity. In light of many rare variants only ever being detected in the compound heterozygous or carrier state, this step made many more globin variants accessible to formal annotation. Due to the lack of case‐control studies with hemoglobinopathy variants, PS4 has been adapted to count individuals with the trait phenotype. In contrast to β‐thalassemia, α‐thalassemia cannot be discerned from iron deficiency based on hematological parameters, which will prompt differential strength‐level adjustments for corresponding data in both major thalassemias. In addition, the ACMG/AMP criterion that pertains to phenotypic correlation (PP4) will not be applied as it would double‐count evidence collected in PS4 (observation in heterozygotes) and PM3 (in‐trans occurrence in an individual with disease). The Hemoglobinopathy VCEP follows ClinGen SVI recommendations for elevating the weight of in‐trans occurrence (PM3) and implements quantitative thresholds to modulate the strength of segregation evidence based on the number of individuals examined (PP1). Furthermore, specifications are provided to guide expert curation of variants with alternate locus observations, such as a β‐thalassemia phenotype caused by heterozygous β‐thalassemia in combination with duplication of the α‐globin locus and a correspondingly aggravated imbalance of the α‐globin/β‐globin ratio (Clark et al., 2018).
6. FUNCTIONAL DATA (PS3 AND BS3)
Functional assays are powerful tools to provide variant‐level evidence of the effect on protein function and splicing to meet PS3 (damaging effect) or BS3 (no effect). The Hemoglobinopathy VCEP reviewed functional assays used by multiple investigators and selected those that reflect the pathophysiological mechanism of disease for the assessment of thalassemia and structural hemoglobin variants. Well‐recognized assays include those that evaluate globin chain biosynthesis, red cell inclusions (e.g., denatured β4 tetramers), and the stability, solubility, and oxygen affinity of the hemoglobin molecule. Functional criteria are also applied for evidence of abnormal RNA or protein expression of the variant allele as a consequence of a null or splicing effect. Other assays involve in vitro transcription assays, which are mainly used in research and, thus, do not conform to diagnostic laboratory standards. Functional evidence has a strong level of strength in the ACMG/AMP framework, yet not all assays are consistent predictors of a certain variant effect or uniformly evaluated across clinical laboratories. The SVI WG provides recommendations based on the validation, reproducibility, and robustness of data for individual assays, so as to advise on the appropriate level of strength of evidence to apply (Brnich et al., 2019). However, as validation controls and replicates are rarely documented for functional assays in hemoglobinopathies, the functional data will initially be considered as supporting level evidence in favor of pathogenicity or of benign interpretation in the ongoing pilot study, pending evaluation by the Hemoglobinopathy VCEP of applying increased weight during annotation of variants with established pathogenicity.
7. A PILOT FOR SPECIFIED CRITERIA AND THEIR EVALUATION
Table 2 lists the draft VCEP‐specified criteria organized by evidence type and strength, which are currently being tested in a pilot variant curation study comprising an informative mixture of established structural and thalassemia mutations in the HBA1, HBA2, and HBB genes. In the process, the ACMG/AMP framework provides rules for combining criteria to arrive at a classification; however, it does not guide the interpretation of variants with conflicting evidence. By contrast, interpretation within the Bayesian Classification Framework (Tavtigian et al., 2018) provides a quantitative approach to the combination of rules and, thus, allows refining the strength of evidence and combining rules for the classification of variants that have contradictory benign and pathogenic evidence. In the evaluation and refinement of its draft criteria, the Hemoglobinopathy VCEP will use the standard ACMG/AMP framework in parallel to the application of the Bayesian Classification Framework, thus additionally testing the impact of a quantitative approach in sequence variant interpretation.
8. CONCLUSION
The ClinGen Hemoglobinopathy VCEP is a group of experts and biocurators with diverse specialties tasked with the adaptation of the 2015 ACMG/AMP guidelines for the classification of genetic variants in the HBA1, HBA2, and HBB genes for hemoglobinopathies. This report provides insights into the challenges and considerations of specifying the ACMG/AMP criteria to evaluate all available evidence relevant to hemoglobinopathies and the globin genes, with the aim to standardize the curation and interpretation of variants in different conditions. The current test by the ClinGen Hemoglobinopathy VCEP of its specifications in a small set of globin gene variants with known pathogenicity will lead to further specifications and minor adjustments of the rules described in this report. Once approved by ClinGen, the resulting final set of classification rules will be the first standardized framework for the interpretation of sequence variants in the globin genes. Most importantly, following the 2018 recognition of ClinGen by the Food and Drug Administration, assertions in the framework of ClinGen VCEPs are considered to be valid scientific evidence and can be used for test development and validation processes. An ever‐accelerating accumulation of diagnostic sequencing data for the globin loci, with global relevance of reliable variant interpretation for genetic counseling, diagnosis, and prognosis for hemoglobinopathies, means that both diligence and speed are of the essence in the current refinement and application of specified Hemoglobinopathy VCEP criteria.
WEB RESOURCES
ITHANET Portal: https://www.ithanet.eu/
Genome Aggregation Database (gnomAD): https://gnomad.broadinstitute.org/
ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
Allele Frequency App: https://cardiodb.org/allelefrequencyapp/
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
We are grateful to Helen Merrideth Robinson for her pioneering liaison efforts critical to establishing the Hemoglobinopathy VCEP, and to Sir John Burn for ongoing advice and support as Executive Chairman of the Human Variome Project. We also thank Steven M. Harrison, Leslie G. Biesecker, and the ClinGen SVI WG for reviewing the specified VCEP rules and for providing valuable feedback. This study is co‐funded by the European Regional Development Fund and the Republic of Cyprus through the Research and Innovation Foundation (Project: EXCELLENCE/1216/256).
Kountouris, P. , Stephanou, C. , Lederer, C. W. , Traeger‐Synodinos, J. , Bento, C. , Harteveld, C. L. , Fylaktou, E. , Koopmann, T. T. , Halim‐Fikri, H. , Michailidou, K. , Nfonsam, L. E. , Waye, J. S. , Zilfalil, B. A. , Kleanthous, M. , & On behalf of ClinGen Hemoglobinopathy Variant Curation Expert Panel. (2022). Adapting the ACMG/AMP variant classification framework: A perspective from the ClinGen Hemoglobinopathy Variant Curation Expert Panel. Human Mutation, 43, 1089–1096. 10.1002/humu.24280
Petros Kountouris and Coralea Stephanou are equal contributors and share joint first authorship.
The full membership of author group ClinGen Hemoglobinopathy Variant Curation Expert Panel is provided in Table S1.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Abou Tayoun, A. N. , Pesaran, T. , DiStefano, M. T. , Oza, A. , Rehm, H. L. , Biesecker, L. G. , & Harrison, S. M. (2018). Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Human Mutation, 39(11), 1517–1524. 10.1002/humu.23626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , Kondrashov, A. S. , & Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brnich, S. E. , Abou Tayoun, A. N. , Couch, F. J. , Cutting, G. R. , Greenblatt, M. S. , Heinen, C. D. , Luo, X. , McNulty, S. M. , Starita, L. M. , Tavtigian, S. V. , Wright, M. W. , Harrison, S. M. , Biesecker, L. G. , Berg, J. S. & the Clinical Genome Resource Sequence Variant Interpretation Working Group . (2019). Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Medicine, 12(1), 3. 10.1186/s13073-019-0690-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, B. , Shooter, C. , Smith, F. , Brawand, D. , Steedman, L. , Oakley, M. , Rushton, P. , Rooks, H. , Wang, X. , Drousiotou, A. , Kyrri, A. , Hadjigavriel, M. , Will, A. , Fisher, C. , Higgs, D. R. , Phylipsen, M. , Harteveld, C. , Kleanthous, M. , & Thein, S. L. (2018). Beta thalassaemia intermedia due to co‐inheritance of three unique alpha globin cluster duplications characterised by next generation sequencing analysis. British Journal of Haematology, 180(1), 160–164. 10.1111/bjh.14294 [DOI] [PubMed] [Google Scholar]
- Farashi, S. , & Harteveld, C. L. (2018). Molecular basis of α‐thalassemia. Blood Cells, Molecules, and Diseases, 70, 43–53. 10.1016/j.bcmd.2017.09.004 [DOI] [PubMed] [Google Scholar]
- Ghosh, R. , Harrison, S. M. , Rehm, H. L. , Plon, S. E. , & Biesecker, L. G. (2018). Updated recommendation for the benign stand alone ACMG/AMP criterion. Human Mutation, 39(11), 1525–1530. 10.1002/humu.23642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, S. M. , Biesecker, L. G. , & Rehm, H. L. (2019). Overview of specifications to the ACMG/AMP variant interpretation guidelines. Current Protocols in Human Genetics, 103(1), e93. 10.1002/cphg.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, S. M. , Dolinsky, J. S. , Knight Johnson, A. E. , Pesaran, T. , Azzariti, D. R. , Bale, S. , Chao, E. C. , Das, S. , Vincent, L. , & Rehm, H. L. (2017). Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 19(10), 1096–1104. 10.1038/gim.2017.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs, D. R. , Engel, J. D. , & Stamatoyannopoulos, G. (2012). Thalassaemia. The Lancet, 379(9813), 373–383. 10.1016/S0140-6736(11)60283-3 [DOI] [PubMed] [Google Scholar]
- Ioannidis, N. M. , Rothstein, J. H. , Pejaver, V. , Middha, S. , McDonnell, S. K. , Baheti, S. , Musolf, A. , Li, Q. , Holzinger, E. , Karyadi, D. , Cannon‐Albright, L. A. , Teerlink, C. C. , Stanford, J. L. , Isaacs, W. B. , Xu, J. , Cooney, K. A. , Lange, E. M. , Schleutker, J. , Carpten, J. D. , … Sieh, W. (2016). REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. The American Journal of Human Genetics, 99(4), 877–885. 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan, K. , Kyriazopoulou Panagiotopoulou, S. , McRae, J. F. , Darbandi, S. F. , Knowles, D. , Li, Y. I. , Kosmicki, J. A. , Arbelaez, J. , Cui, W. , Schwartz, G. B. , Chow, E. D. , Kanterakis, E. , Gao, H. , Kia, A. , Batzoglou, S. , Sanders, S. J. , & Farh, K. K. (2019). Predicting splicing from primary sequence with deep learning. Cell, 176(3), 535–548. 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Johnston, J. J. , Dirksen, R. T. , Girard, T. , Gonsalves, S. G. , Hopkins, P. M. , Riazi, S. , Saddic, L. A. , Sambuughin, N. , Saxena, R. , Stowell, K. , Weber, J. , Rosenberg, H. , & Biesecker, L. G. (2021). Variant curation expert panel recommendations for RYR1 pathogenicity classifications in malignant hyperthermia susceptibility. Genetics in Medicine, 23, 1–8. 10.1038/s41436-021-01125-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski, K. J. , Francioli, L. C. , Tiao, G. , Cummings, B. B. , Alföldi, J. , Wang, Q. , Collins, R. L. , Laricchia, K. M. , Ganna, A. , Birnbaum, D. P. , Gauthier, L. D. , Brand, H. , Solomonson, M. , Watts, N. A. , Rhodes, D. , Singer‐Berk, M. , England, E. M. , Seaby, E. G. , Kosmicki, J. A. , … MacArthur, D. G. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher, M. , Witten, D. M. , Jain, P. , O'Roak, B. J. , Cooper, G. M. , & Shendure, J. (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohne, E. (2011). Hemoglobinopathies. Deutsches Ärzteblatt international, 108(31–32), 532–540. 10.3238/arztebl.2011.0532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kountouris, P. , Lederer, C. W. , Fanis, P. , Feleki, X. , Old, J. , & Kleanthous, M. (2014). IthaGenes: An interactive database for haemoglobin variations and epidemiology. PLOS One, 9(7), 103020. 10.1371/journal.pone.0103020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kountouris, P. , Michailidou, K. , Christou, S. , Hadjigavriel, M. , Sitarou, M. , Kolnagou, A. , Kleanthous, M. , & Telfer, P. (2020). Effect of HBB genotype on survival in a cohort of transfusion‐dependent thalassemia patients in Cyprus. Haematologica, 106, 2458–2468. 10.3324/haematol.2020.260224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kountouris, P. , Stephanou, C. , Bento, C. , Fanis, P. , Elion, J. , Ramesar, R. S. , Zilfalil, B. A. , Robinson, H. M. , Traeger‐Synodinos, J. , Human Variome Project Global Globin 2020 Challenge , Lederer, C. W. , & Kleanthous, M. (2017). ITHANET: Information and database community portal for haemoglobinopathies [Preprint]. Bioinformatics, 12, 189–191. 10.1101/209361 [DOI] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Moradkhani, K. , Préhu, C. , Old, J. , Henderson, S. , Balamitsa, V. , Luo, H.‐Y. , Poon, M. C. , Chui, D. H. , Wajcman, H. , & Patrinos, G. P. (2009). Mutations in the paralogous human α‐globin genes yielding identical hemoglobin variants. Annals of Hematology, 88(6), 535–543. 10.1007/s00277-008-0624-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixeiro, I. , Silva, A. L. , & Romão, L. (2011). Control of human β‐globin mRNA stability and its impact on beta‐thalassemia phenotype. Haematologica, 96(6), 905–913. 10.3324/haematol.2010.039206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm, H. L. , Berg, J. S. , Brooks, L. D. , Bustamante, C. D. , Evans, J. P. , Landrum, M. J. , Ledbetter, D. H. , Maglott, D. R. , Martin, C. L. , Nussbaum, R. L. , Plon, S. E. , Ramos, E. M. , Sherry, S. T. , & Watson, M. S. (2015). ClinGen—The Clinical Genome Resource. The New England Journal of Medicine, 372(23), 2235–2242. 10.1056/NEJMsr1406261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance, C. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, D. J. , & Williams, T. N. (2003). Haemoglobinopathies and resistance to malaria. Redox Report, 8(5), 304–310. 10.1179/135100003225002998 [DOI] [PubMed] [Google Scholar]
- Stephanou, C. , Tamana, S. , Minaidou, A. , Papasavva, P. , Kleanthous, M. , & Kountouris, P. (2019). Genetic modifiers at the crossroads of personalised medicine for haemoglobinopathies. Journal of Clinical Medicine , 8(11). 10.3390/jcm8111927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian, S. V. , Greenblatt, M. S. , Harrison, S. M. , Nussbaum, R. L. , Prabhu, S. A. , Boucher, K. M. , & Biesecker, L. G. (2018). Modeling the ACMG/AMP Variant Classification guidelines as a Bayesian classification framework. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 20(9), 1054–1060. 10.1038/gim.2017.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thein, S. L. (2013). The molecular basis of β‐thalassemia. Cold Spring Harbor Perspectives in Medicine, 3(5), 011700. 10.1101/cshperspect.a011700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom, C. S. , Dickson, C. F. , Gell, D. A. , & Weiss, M. J. (2013). Hemoglobin variants: Biochemical properties and clinical correlates. Cold Spring Harbor Perspectives in Medicine, 3(3), 011858. 10.1101/cshperspect.a011858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg, J. , Olatubosun, A. , & Vihinen, M. (2011). Performance of mutation pathogenicity prediction methods on missense variants. Human Mutation, 32(4), 358–368. 10.1002/humu.21445 [DOI] [PubMed] [Google Scholar]
- Whiffin, N. , Minikel, E. , Walsh, R. , O'donnell‐Luria, A. H. , Karczewski, K. , Ing, A. Y. , Barton, P. , Funke, B. , Cook, S. A. , MacArthur, D. , & Ware, J. S. (2017). Using high‐resolution variant frequencies to empower clinical genome interpretation. Genetics in Medicine, 19(10), 1151–1158. 10.1038/gim.2017.26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo, G. , & Burge, C. B. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Journal of Computational Biology, 11(2–3), 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.