

Abstract
Introduction
Befovacimab (formerly BAY 1093884) is a fully human monoclonal antibody able to bind to tissue factor pathway inhibitor (TFPI) and developed as a non‐replacement therapy for individuals with haemophilia A/B, with or without inhibitors.
Aim
To assess the safety of multiple escalating doses of befovacimab in individuals with severe haemophilia A/B with or without inhibitors.
Methods
In this non‐randomised, open‐label Phase 2 study (NCT03597022), adult males with <1% factor VIII or <2% factor IX and ≥4 bleeds in the previous six months were enrolled in three dose cohorts (100/225/400 mg). Participants received befovacimab subcutaneously once weekly. The primary endpoint was safety; secondary endpoints included annualised bleeding rate (ABR) and pharmacokinetics/pharmacodynamics (PK/PD) of befovacimab.
Results
A total of 24 participants (n = 8 in each dose cohort) were treated for 2–47 weeks. Patients treated with 100 mg and 225 mg doses of befovacimab demonstrated improved bleeding control compared with pre‐study bleeding rates, with a dose‐dependent effect. Dosing was suspended and the study prematurely terminated following three drug‐related thrombotic serious adverse events (SAEs): two at the 225 mg dose and one at the 400 mg dose. These occurred in the absence of bleeding episodes or concomitant use of replacement/bypass therapies. No laboratory abnormalities were observed, and PK/PD data did not show correlation between SAE occurrence and levels of circulating befovacimab or free TFPI.
Conclusion
Despite favourable initial results from preclinical and clinical studies, a positive safety profile of befovacimab was not confirmed. The lack of SAE‐related laboratory abnormalities or differentiating PK/PD characteristics in participants experiencing SAEs raises concerns about the predictability of thrombosis following befovacimab treatment and emphasises the need for further investigation into the therapeutic window of anti‐TFPI treatment.
Keywords: blood coagulation factor inhibitors, haemophilia A, haemophilia B, safety, thrombosis, tissue factor pathway inhibitor
1. INTRODUCTION
Individuals with severe haemophilia A or B experience frequent bleeding episodes, which most commonly affect the joints and muscles; over time, recurrent joint bleeding can cause irreversible damage to cartilage and bone, which translates into the development of chronic arthropathy. 1 , 2 , 3 Prophylaxis with replacement factors is the standard of care for individuals with severe haemophilia; however, the burden associated with frequent infusions can lead to reduced adherence and thus poor clinical outcomes and reduced quality of life (QoL). 1 , 2 In addition, approximately one third of individuals with haemophilia A and 3%–10% of individuals with haemophilia B develop neutralising alloantibodies (inhibitors) against replacement factors, further impacting upon clinical outcomes/QoL. 4 , 5 , 6 , 7 This highlights the need for new approaches to manage and treat haemophilia for individuals both with and without inhibitors, in order to provide reliable protection from bleeds and improve QoL. 1 , 8
Befovacimab (formerly BAY 1093884) is a fully human monoclonal antibody able to bind tissue factor pathway inhibitor (TFPI), developed as a subcutaneous, non‐factor replacement prophylactic treatment for individuals with haemophilia A or B, with or without inhibitors. TFPI is an anticoagulant protein, which inhibits the TF:FVIIa complex, a primary element of the coagulation cascade. 9 TFPI exerts its effect on both the extrinsic and intrinsic coagulation pathways via three Kunitz‐type serine protease inhibitor domains; K1 inhibits activated factor VII (FVIIa), K2 inhibits activated factor X (FXa) and K3 binds Protein S, a cofactor that promotes the interaction between TFPI and FXa. 10 , 11 , 12 , 13 There are three alternatively spliced isoforms of TFPI, each of which has a distinct structure and C‐terminus region: TFPIα, the full‐length form, which contains all three Kunitz‐type domains and a basic C‐terminus; TFPIβ, which is the predominant isoform expressed in humans and comprises K1, K2 and a C‐terminal glycosylphosphatidylinositol anchor and TFPIδ, which consists of K1 and K2 only. 14 , 15
Within the extrinsic coagulation pathway, TFPI binds and inhibits FXa and tissue factor (TF)‐bound FVIIa, resulting in the formation of the FXa‐TFPI‐TF‐FVIIa quaternary complex. 12 , 14 , 15 , 16 The prothrombinase complex, which consists of activated factor V (FVa) and FXa, converts prothrombin to thrombin as a final step in blood clot formation in the intrinsic pathway. TFPI blocks thrombin generation by inhibiting the prothrombinase complex. 17 This interaction only occurs with the TFPIα isoform, as a result of its basic C‐terminus region binding to the acidic B‐domain of FVa. 15 Together, the effect of TFPI on the intrinsic and extrinsic pathways limits the initiation and amplification phases of coagulation, and decreases thrombin generation. 12 , 14 , 15 , 16 , 17 Based on this mechanism of action, therapeutic inhibition of TFPI is predicted to improve the haemostatic response, by restoring coagulation, increasing thrombin generation, and promoting clot formation regardless of the type of haemophilia (A or B) or the inhibitor status, and hence has become an attractive target for therapeutic intervention development (Figure 1). 12 , 15 , 18
FIGURE 1.
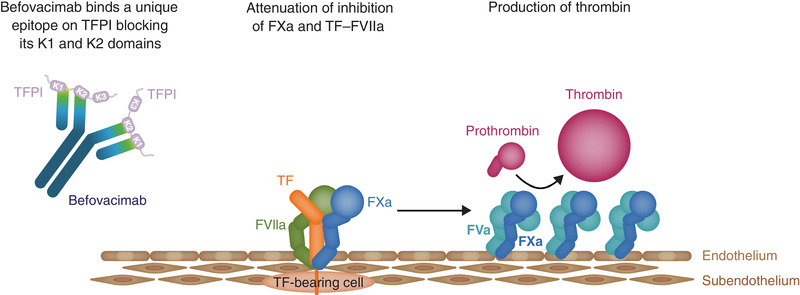
Mechanism by which befovacimab inhibits TFPI and consequently allows thrombin production. FVa, factor Va; FXa, factor Xa; FVIIa, factor VIIa; TF, tissue factor; TFPI, tissue factor pathway inhibitor
Befovacimab has been found to bind human TFPI with high affinity (<10 pM) via the K1 and K2 domains, inhibiting its interactions with both FXa and FVIIa and thus restoring their activity. 19 , 20 , 21 Previous preclinical data have suggested a favourable efficacy and safety profile for befovacimab, with improved thrombin generation and shortened clotting time observed in haemophilia A plasma samples in vitro 19 and dose‐dependent decreases in TFPI concentration and clotting time in female cynomolgus monkeys in vivo. 18 A favourable safety profile was also suggested by the first‐in‐human Phase 1 study of befovacimab, which was undertaken from October 2015 to October 2018 (NCT02571569) in participants with severe haemophilia A or B, with or without inhibitors. 22 The study design included five single‐dose cohorts (n = 28), where participants received one dose of befovacimab either intravenously (0.3 or 1 mg/kg) or subcutaneously (1, 3, or 6 mg/kg), as well as a multiple‐dose cohort (n = 4), where participants received 150 mg of befovacimab subcutaneously once weekly for six weeks. No serious adverse events (SAEs), adverse events of special interest (AESI) or discontinuations due to adverse events (AEs) were reported during the study. In addition, no significant laboratory abnormalities, such as decreased levels of fibrinogen, platelets, antithrombin or FV, were observed in any cohort. Finally, PD analyses showed dose‐dependent suppression of circulating TFPI, as well as greater thrombin generation, sustained over the 6‐week multiple‐dosing period, and improved clot formation following befovacimab treatment. 22
The objective of the Phase 2 study (NCT03597022) was to further evaluate the safety of befovacimab in participants with severe haemophilia A or B, with or without inhibitors, using a multiple‐dose, dose‐escalating study design.
2. MATERIALS AND METHODS
2.1. Participants
An overview of participant eligibility criteria is provided in Table 1. Full inclusion and exclusion criteria are available in the Supplementary Appendix. Eligible participants also had to be willing to interrupt ongoing prophylaxis or immune tolerance induction, if applicable. Following the first reported SAE, the inclusion/exclusion criteria in the protocol were amended to exclude patients with mild haemophilia, as per DMC recommendations.
TABLE 1.
Amended inclusion and exclusion criteria following first reported SAE
| Inclusion criteria |
|
| Exclusion criteria |
|
2.2. Study design
This was a multi‐centre, non‐randomised, open‐label, dose‐escalation Phase 2 study of befovacimab (NCT03597022), undertaken from July 2018 to October 2019. Ethics approval, individual informed consent, and protocol approval by the Independent Ethics Committee or Institutional Review Board at each study site were obtained prior to enrolment.
Eligible participants were enrolled into one of three dose cohorts and received befovacimab once weekly via subcutaneous injection: Cohort 1 (low‐dose), 100 mg; Cohort 2 (intermediate‐dose), 225 mg; and Cohort 3 (high‐dose), 400 mg (Figure 2). The 100 mg dose was selected for Cohort 1 based on safety and PK/PD data from the Phase 1 study, and the 225 and 400 mg doses were defined sequentially based on an adequate demonstration of safety and tolerability for previous doses. 22 In Part A, eight befovacimab‐naïve participants were enrolled in each dose cohort and received 12 weeks of treatment. The escalating dose cohorts only opened following recommendations from a Data Monitoring Committee (DMC), which reviewed safety data from the first six participants in each cohort after six weeks of treatment. For example, to open Cohort 2, adequate safety and tolerability data had to be demonstrated for at least six participants in Cohort 1. Participants were required to remain at the same dose for a period of at least 12 weeks to allow for primary safety evaluations and PK/PD modelling, as determined by laboratory assessments and reporting of bleeding and AEs. Participants in Cohorts 1 and 2 could increase to the next higher dose if they met the dose escalation criteria (>2 bleeds in Part A). In Part B, each participant received treatment for additional 12‐week cycles; at the end of each cycle, participants could continue the same dose for an additional 12 weeks or increase to the next higher dose if they met the dose escalation criteria (>1 bleed in any 12‐week cycle after the initial 12 weeks of treatment at a particular dose). The primary study completion was the date when the last participant in Cohort 3 completed the first 12 weeks of Part B. The extension phase was initiated only after DMC evaluation of safety data from six participants after 6 weeks of treatment in Cohort 3. During the extension, participants were evaluated every 12 weeks and escalated to the next higher dose, as in Part B. Throughout Part B and the extension, safety and efficacy data, including number of bleeds, were assessed every 12 weeks for possible dose escalation. The extension was due to continue until marketing authorisation, pending submission of safety and preliminary efficacy data to the relevant health authorities every 12 months.
FIGURE 2.
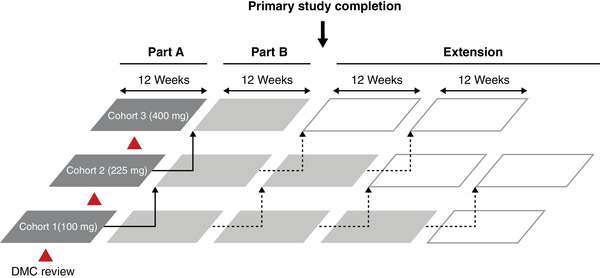
Overview of study design. Dark grey boxes represent Part A (initial 12 weeks of treatment with befovacimab – no escalation allowed). Light grey boxes represent Part B (subsequent 12‐week treatment cycles with befovacimab – first escalation allowed if >2 bleeds in Part A, as indicated by solid line arrows). White boxes represent extension (subsequent 12‐week treatment cycles with befovacimab – escalations allowed if >1 bleed in 12 weeks, as indicated by dotted line arrows). Arrows represent option for dose escalation. DMC, Data Monitoring Committee
FVIII/FIX (inhibitor) antibody testing was performed according to the Nijmegen modified Bethesda assay, with a positive inhibitor test defined as ≥0.6 Bethesda units (BU)/ml; FVIII levels and FVIII inhibitors were measured at screening. All assays were performed at a central laboratory. Befovacimab PK parameters were assessed at baseline, then weeks 1, 3, 6 and 9 and then every 12 weeks. At 12 weeks, to support dose escalation, pre‐dose PK sampling was carried out, then PK parameters were checked 1 and 6 week after dose escalation. Individual befovacimab plasmatic concentrations were not assessed. Befovacimab plasma level was determined by ligand binding assay with electrochemiluminescence readout using biotinylated recombinant human TFPI molecule as capture and a Sulfo‐tag‐labelled recombinant human TFPI molecule as detection reagent. Blood sampling for PK, PD and exploratory biomarkers were also taken at final or early termination visit. Inhibitors refer to anti‐FVIII antibodies and were assessed using the Nijmegen modified Bethesda assay at the same timepoints.
In all cohorts and for each escalated dose, the first injection of every new dose level was administered under medical supervision. Any additional prophylaxis infusions, with either the study drug or any other treatment, even to accommodate physical activities, were not permitted. Any breakthrough bleeding events occurring during the study or extension requiring additional intervention to achieve control were treated using individual patients’ previously assigned treatment (prior to enrolment), with the exception of patients previously treated with emicizumab. Patients who had previously received emicizumab were eligible to be enrolled in the study and, in cases of breakthrough bleeding events, were treated with rFVIIa. Where necessary, it was recommended to use the lowest approved dose of prescribed replacement factor or bypassing agent, subject to the investigators’ judgement, to avoid potential additive effects of the replacement factor and concomitant medication. Immunomodulatory agents as concomitant therapy were prohibited.
2.3. Endpoints
The primary endpoint was to assess the safety and tolerability of multiple escalating doses of befovacimab, as assessed by the frequency of drug‐related AEs, SAEs and AESIs, including hypersensitivity reactions. Participants were also monitored for clinically relevant laboratory abnormalities; variables included complete blood counts, coagulation parameters (D‐dimer, platelets, fibrinogen, TFPI, antithrombin, Protein C, lactate dehydrogenase [LDH]), FVIII/FIX inhibitors and anti‐drug antibodies (aTFPI‐Ab). Secondary endpoints included annualised bleeding rate (ABR) and assessment of PD/PK parameters. The number of spontaneous bleeds requiring additional treatment was also recorded to explore the effect of different doses of befovacimab.
3. RESULTS
A total of 24 male participants from 17 centres in 11 countries received befovacimab (n = 8 in each dose cohort). Participants were treated for 2–47 weeks. Demographic and clinical characteristics are shown in Table 2.
TABLE 2.
Participant demographics and baseline characteristics (safety population)
| Characteristic | Cohort 1 (100 mg) (n = 8) | Cohort 2 (225 mg) (n = 8) | Cohort 3 (400 mg) (n = 8) | Total (N = 24) |
|---|---|---|---|---|
| Age, years | ||||
| Median (range) | 47.0 (21−59) | 41.0 (24−76) | 42.0 (35−55) | 43.5 (21–76) |
| Race, n (%) | ||||
| Asian | 2 (25.0) | 2 (25.0) | 2 (25.0) | 6 (25.0) |
| Black or African American | 0 | 0 | 1 (12.5) | 1 (4.2) |
| White | 6 (75.0) | 6 (75.0) | 5 (62.5) | 17 (70.8) |
| Body Mass Index, kg/ m 2 | ||||
| Median (range) | 23.81 (19.2−33.0) a | 22.51 (16.2−34.8) | 24.20 (19.5−34.3) | 23.46 (16.2–34.8) |
| Haemophilia subtype, n (%) | ||||
| A with inhibitors | 1 (12.5) | 2 (25.0) | 1 (12.5) | |
| A without inhibitors | 6 (75.0) | 4 (50.0) | 5 (62.5) | |
| B with inhibitors | 1 (12.5) | 2 (25.0) | 1 (12.5) | |
| B without inhibitors | 0 | 0 | 1 (12.5) |
Data are shown for each participant based on their initial dose cohort.
BMI recorded for n = 7 participants in Cohort 1.
3.1. Safety
The majority of participants (21/24, 87.5%) reported at least one treatment‐emergent adverse event (TEAE) (Table 3). TEAEs considered to be related to the study drug were reported for 10 participants (42%). The study drug‐related TEAEs of injection site erythema and injection site pruritus were each reported in two (25.0%) participants receiving the 400 mg dose. All other drug‐related TEAEs occurred in six individual participants across all dose cohorts. Four participants experienced TEAEs in the 225 mg cohort and five participants experienced TEAEs in the 400 mg cohort. Three (12.5%) of these events were considered to be both SAEs and adverse events of special interest (AESIs) (Figure 3). These SAEs/AESIs occurred in the absence of bleeding episodes or concomitant use of replacement factors or bypass agents. Two events were reported by participants in the intermediate‐dose cohort (225 mg) and one in the high‐dose cohort (400 mg).
TABLE 3.
Treatment‐emergent adverse events (safety population)
| Participants with TEAEs, n | Cohort 1 (100 mg) (n = 8) | Cohort 2 (225 mg) (n = 8) | Cohort 3 (400 mg) (n = 8) | Total (N = 24) |
|---|---|---|---|---|
| Any TEAE | 7 (87.5) | 8 (100.0) | 6 (75.0) | 21 (87.5) |
| Any study drug‐related TEAE (≥1) | 1 (12.5) | 4 (50.0) | 5 (62.5) | 10 (41.7) |
| Grade 1 | 1 (12.5) | 2 (25.0) | 0 | 3 (12.5) |
| Grade 2 | 0 | 1 (12.5) | 4(50.0) | 5 (20.8) |
| Grade 3 | 0 | 0 | 1 (12.5) | 1 (4.2) |
| Grade 4 | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Study drug‐related TEAE by system organ class a | ||||
| Blood and lymphatic system disorders | 0 | 0 | 1 (12.5) | 1 (4.2) |
| Hypofibrinogenaemia | 0 | 0 | 1 (12.5) | 1 (4.2) |
| Eye disorders | 0 | 0 | 1 (12.5) | 1 (4.2) |
| Retinal artery thrombosis | 0 | 0 | 1 (12.5) | 1 (4.2) |
| Gastrointestinal disorders | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Nausea | 0 | 1 (12.5) | 0 | 1 (4.2) |
| General disorders and administration site conditions | 0 | 1 (12.5) | 2 (25.0) | 3 (12.5) |
| Injection site erythema | 0 | 0 | 2 (25.0) | 2 (8.3) |
| Injection site inflammation | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Injection site pruritus | 0 | 0 | 2 (25.0) | 2 (8.3) |
| Injection site reaction | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Injection site swelling | 0 | 0 | 1 (12.5) | 1 (4.2) |
| Laboratory investigations b | 1 (12.5) | 0 | 0 | 1 (4.2) |
| Fibrin D‐dimer increased | 1 (12.5) | 0 | 0 | 1 (4.2) |
| Nervous system disorders | 0 | 3 (37.50) | 0 | 3 (12.5) |
| Dizziness | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Headache | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Ischaemic stroke | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Transverse sinus thrombosis | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Skin and subcutaneous tissue disorders | 0 | 0 | 1 (12.5) | 1 (4.2) |
| Erythema | 0 | 0 | 1 (12.5) | 1 (4.2) |
| AESI | 0 | 2 (25.0) | 1 (12.5) | 3 (12.5) |
| Ischemic stroke | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Retinal artery thrombosis | 0 | 0 | 1 (12.5) | 1 (4.2) |
| Transverse sinus thrombosis | 0 | 1 (12.5) | 0 | 1 (4.2) |
| Any TEAE leading to discontinuation | 1 (12.5) | 2 (25.0) | 0 | 3 (12.5) |
| Any serious TEAE | 1 (12.5) | 2 (25.0) | 1 (12.5) | 4 (16.7) |
| Any study drug‐related serious TEAE | 0 | 2 (25.0) | 1 (12.5) | 3 (12.5) |
| Any serious TEAE leading to discontinuation | 1 (12.5) c | 2 (25.0) | 1 (12.5) d | 3 (12.5) |
Data are shown for each participant based on their initial dose cohort.
Some participants experienced ≥1 TEAE;
Increased levels of fibrin D‐dimer.
Serious TEAE of nasal bleeding and paranasal sinus tumour, considered unrelated to study drug.
Study drug was reported to be interrupted for serious TEAE of retinal artery thrombosis; however, the study drug was subsequently discontinued in all subjects and the study was terminated.
AESI, adverse event of special interest; TEAE, treatment‐emergent adverse event. TEAE grades are listed according to the Common Terminology Criteria for Adverse Events, version 5.0 : 36
Grade 1, Mild; asymptomatic or mild symptoms; clinical or diagnostic observations only; intervention not indicated. Grade 2, Moderate; minimal, local, or non‐invasive intervention indicated; limiting age‐appropriate instrumental activities of daily living (ADL). Grade 3, Severe or medically significant but not immediately life‐threatening; hospitalisation or prolongation of hospitalisation indicated; disabling; limiting self‐care ADL. Grade 4, Life‐threatening consequences; urgent intervention indicated. Grade 5, Death related to AE.
FIGURE 3.
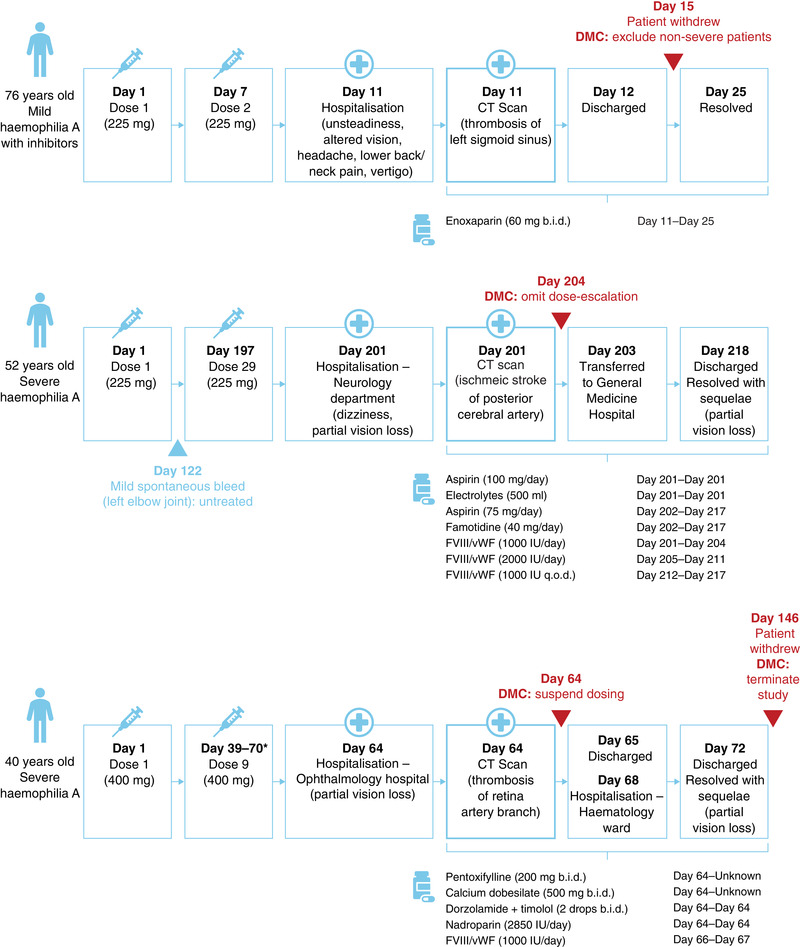
Timelines for the three serious adverse events recorded during the study. *Exact date of final dose unknown. b.i.d., twice daily; CT, computed tomography; DMC, Data Monitoring Committee; FVIII/vWF, factor VIII and von Willebrand factor complex; IU, international units; q.o.d., every other day
The first SAE occurred in January 2019 in a 76‐year‐old participant with mild haemophilia A and a recent diagnosis of inhibitors. In August 2018, 5 months prior to the first study dose, this participant underwent surgery to remove skin cancer and received treatment with recombinant FVIII for 4 days to treat post‐surgical bleeding. Following slow and incomplete resolution of bleeding, the presence of FVIII inhibitors (10.2 BU/ml) were confirmed in September 2018. The participant was subsequently enrolled in the intermediate‐dose cohort and received two 225 mg doses of befovacimab, administered every 7 days. At screening (December 2019), the patient's FVIII level was 10%, with FVIII inhibitors of 13 BU. Four days after the second dose, the participant presented with a two‐day history of unsteadiness, impaired vision, headache, lower back/neck pain and vertigo, and was hospitalised. A computed tomography (CT) scan identified thrombosis of the left sigmoid venous sinus. Treatment with study drug was interrupted as the participant was initiated on enoxaparin and discharged from the hospital on the following day. The thrombosis resolved within 2 weeks and the participant recovered without sequelae. FVIII level and inhibitor titre were not recorded in the time period immediately before/after the SAE. Following this first SAE, DMC recommendations were that the study continued, but excluded participants with non‐severe haemophilia. A protocol amendment to exclude patients with mild haemophilia was subsequently implemented.
The second SAE occurred in July 2019 in a 52‐year‐old participant with severe haemophilia A and no history of inhibitors. This participant had received previous on‐demand treatment with FVIII/von Willebrand factor complex and experienced 26 bleeding episodes in the six months prior to the study. The participant was enrolled in intermediate‐dose cohort (225 mg), and reported one mild spontaneous bleed after 17 weeks, which affected the left elbow joint and was not treated with any medication. Following 29 weekly doses of befovacimab, the participant presented with a history of dizziness and partial loss‐of‐vision and was admitted to the hospital. A CT scan confirmed an ischaemic stroke of the posterior cerebral artery, which resolved after 17 days. During this time, the participant received treatment with aspirin, electrolytes, and famotidine, and was also restarted on FVIII/von Willebrand factor complex. The participant was subsequently discharged and continued to experience partial vision loss. At this point, DMC recommendations were to continue the study, but not to further escalate participants to the higher 400 mg dose until further evaluation.
The third SAE occurred in July 2019 in a 40‐year‐old male with severe haemophilia A without inhibitors, who had previously been treated on‐demand with FVIII/von Willebrand factor complex. This participant had experienced 18 bleeds in the six months prior to the study and was enrolled in the high‐dose (400 mg) cohort. After nine weekly doses of befovacimab treatment and zero bleeds, he presented with a history of partial loss‐of‐vision in the right eye and was hospitalised. The participant spent two days in a specialised ophthalmology hospital, where he was diagnosed with thrombosis of the retinal artery branch, and a further five days in a haematology ward, before being fully discharged. Treatment during this time included pentoxifylline, calcium dobesilate, nadroparin and FVIII/von Willebrand factor complex. The participant's thrombosis resolved, but his partial loss‐of‐vision continued. Dosing was immediately suspended in all participants upon the report of the third SAE, in accordance with the safety rules of the protocol. Upon further evaluation of all data, and considering the safety and the efficacy results, the trial was prematurely terminated.
Laboratory analyses in these participants did not show any significant findings with regards to platelet consumption, haemolysis or microangiopathy, platelet counts, levels of fibrinogen, factor V and antithrombin (data not shown). In addition, comparison of participants who did and did not experience SAEs indicated that there was no correlation between SAE occurrence and PK/PD parameters, including plasma befovacimab concentration or level of TFPI suppression (Figure 4). Total TFPI levels showed a high level of inter‐participant variability throughout the study duration and were below the lower limit of quantification (LLOQ) in the majority of participants who reported SAEs.
FIGURE 4.
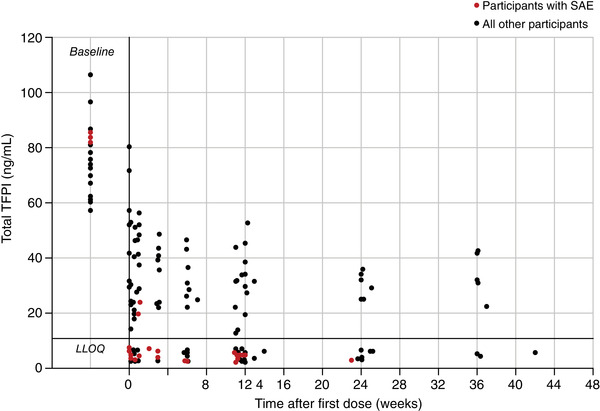
Summary of total TFPI levels over the duration of the study for participants who did and did not experience serious adverse events. LLOQ, lower limit of quantification; SAE, serious adverse event; TFPI, tissue factor pathway inhibitor
3.2. Efficacy/dose finding
A total of 17/24 participants (70.8%) experienced spontaneous bleeding events during the study (Table 4). Of the 128 spontaneous bleeds recorded, the majority (71.9%) were classed as mild and were treated (78.9%). The majority of bleeds occurred within the low‐ (100 mg) and intermediate‐ (225 mg) dose cohorts.
TABLE 4.
Spontaneous bleeds during the study (safety population)
| Spontaneous bleeds | Cohort 1 (100 mg) (n = 8) | Cohort 2 (225 mg) (n = 8) | Cohort 3 (400 mg) (n = 8) | Total (N = 24) |
|---|---|---|---|---|
| Participants with spontaneous bleeds, n (%) | 7 (87.5) | 7 (87.5) a | 3 (37.5) | 17 (70.8) |
| Number of spontaneous bleeds, n (%) | 79 | 37 | 12 | 128 |
| Mild | 69 (87.3) | 16 (43.2) | 7 (58.3) | 92 (71.9) |
| Moderate | 10 (12.7) | 20 (54.1) | 5 (41.7) | 35 (38.0) |
| Severe | 0 | 1 (2.7) | 0 | 1 (2.9) |
| Treated | 65 (82.3) | 25 (67.6) | 11 (91.7) | 101 (78.9) |
| Untreated | 14 (17.7) | 12 (32.4) | 1 (8.3) | 27 (21.1) |
Data are shown for each participant based on their initial dose cohort.
Number of bleeds recorded for n = 7 participants in Cohort 2.
Available data indicated that bleeding control improved in both the low‐ (100 mg) and intermediate‐ (225 mg) befovacimab dose cohorts, compared with pre‐study bleeding rates, with a dose‐dependent effect (Figure 5). Median ABR decreased from 19.6 during the 6 months prior to the study to 13.0 after 3 to 6 months treatment in the low‐dose participants (a decrease of 33.7%), and from 23.9 to 8.7 in the medium‐dose participants (a decrease of 63.6%).
FIGURE 5.
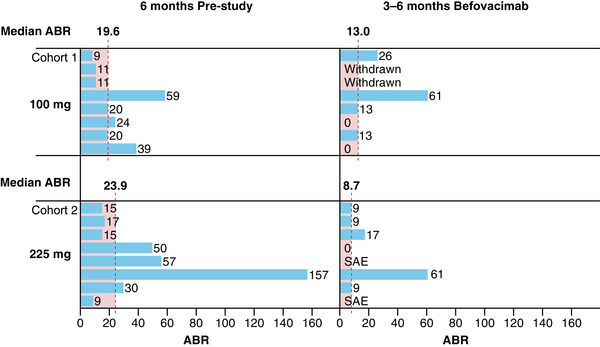
Median ABR in the 6 months prior to study entry and after 3 to 6 months of befovacimab treatment for participants in the low‐ (100 mg; n = 8) and intermediate‐ (225 mg; n = 8) dose cohorts. Data are shown for each participant based on their initial dose cohort. ABR, annualised bleeding rate
4. DISCUSSION
This open‐label, dose‐escalation Phase 2 study did not confirm a positive safety profile for befovacimab in patients with severe haemophilia A.
Although dose‐dependent improvements in bleeding control were observed between the low‐ (100 mg) and intermediate‐ (225 mg) befovacimab dose cohorts median ABR remained relatively high following three to six months of treatment (13.0 and 8.7 in the 100 and 225 mg cohorts, respectively), suggesting a higher dose would be needed for optimal bleeding control. However, the occurrence of two of the three SAEs/AESIs in the 225 mg dose cohort suggests further dose increases would not be advisable. Taken together, these data raise concerns around the therapeutic window of befovacimab (i.e., the dose range at which a drug is both effective and safe), and based on these data, there is no rationale to investigate a higher nor lower dose.
We did not observe any evidence of platelet consumption, haemolysis or microangiopathy in participants who experienced SAEs and no clinically relevant laboratory abnormalities were identified. Furthermore, befovacimab has been shown to bind TFPI with high affinity and high specificity in preclinical investigations, 18 , 19 , 20 and no SAEs or off‐target effects on fibrinolysis were observed during the initial Phase 1 study. While no clinically relevant laboratory abnormalities were identified in the present study, previous safety results were not confirmed. In addition, comparison of circulating befovacimab and TFPI levels between participants who did and did not experience SAEs failed to identify any differentiating PK/PD characteristics related to SAE occurrence. The lack of correlation between the level of TFPI suppression and the occurrence of SAEs contrasts with previous results identifying low levels of free/total TFPI as a risk factor for deep‐vein thrombosis. Together with the high level of inter‐participant variability observed in TFPI concentration, these findings raise concerns about the predictability of thrombosis during anti‐TFPI treatment.
The safety concerns observed in this study should be considered together with existing evidence of both thrombotic and bleeding events with other anti‐TFPI therapies; including the BAX499 aptamer and the concizumab antibody. 23 , 24 , 25 The Phase 1 trial of BAX499 (NCT01191372) was prematurely suspended 23 , 24 , 25 , 26 , 27 , 28 and the Phase 2 and 3 Explorer trials of concizumab (NCT03196297, NCT04082429 and NCT04083781) were put transiently on hold. However, following implementation of a risk mitigation strategy (involving the use of the lowest approved dose of factor/bypassing agents and restrictions on dose interval) the Explorer trials were resumed in September 2020. 29
Whether the mechanism of action of befovacimab can explain the observed thrombotic events is a research question beyond the scope of the study. However, interestingly, these anti‐TFPI therapies target distinct Kunitz‐type domains of the TFPI protein. In fact, concizumab binds K2, 16 , 30 , 31 BAX499 binds K1 and K3, 32 while befovacimab binds K1 and K2. Moreover, the biological aspect that renders difficult the correlation between the PK/PD effect of the drug and the thrombotic events is the target mediated drug disposition which characterises the binding of anti‐TFPI agents in the body. Indeed, TFPI exists in several compartments in the body including the vascular endothelium 14 ; however, what is measurable is the amount of free TFPI in plasma after the binding to anti‐TFPI which does not reflect exactly the inhibitory effect of the drug in the body. These observations suggest a more complex role for TFPI than is currently understood and the effect of anti‐TFPI inhibitors in the endothelial compartment could be related to the occurrence of thrombotic events in certain body districts, such as the central nervous system.
Taken together, these findings suggest that, irrespective of the specific binding targets of the therapeutic agent, caution is needed when TFPI is considered as target for medical intervention. In addition to FVIIa and FXa, TFPI also alters the function of FVa and inhibits prothrombinase assembly. 17 , 33 The coagulation cascade exists in a delicate state of balance that tips towards either bleeding or thrombosis depending on a multitude of internal and external factors; any intervention in this process needs to be very carefully assessed. The results of this study showing thrombosis in the absence of cardiovascular risk factors further reiterate this. Furthermore, thrombotic events have been reported with non‐TFPI, non‐replacement therapies that target the haemostatic pathway, 1 , 34 , 35 confirming the need to further explore and understand the mode of action and the predictability of efficacy and safety of new therapeutic interventions to treat haemophilia.
This study has several limitations; the early suspension of the study prevented analysis of escalating and/or high‐ (400 mg) doses of befovacimab, as originally planned. In addition, the short study duration, exacerbated by its premature termination, prevented analysis of the long‐term effects of befovacimab treatment. The open‐label design and relatively small participant population are also potential limitations, although are broadly consistent with previous clinical trials for other anti‐TFPI therapies and haemophilia studies. 24 , 26 , 27 , 28 , 31
5. CONCLUSION
The occurrence of the three thrombotic events observed in this study highlights the unpredictability of the occurrence of thrombosis when intervening on the haemostatic balance. These events, along with suboptimal bleeding protection, led to suspension of study dosing and early termination of this Phase 2 study with befovacimab. The results presented here will hopefully help to build our understanding of the utility of anti‐TFPI agents as a therapeutic option.
CONFLICT OF INTEREST
MEM has acted as a paid consultant, advisor and/or speaker for Bayer, BioMarin, CSL Behring, Grifols, Kedrion, LFB, Novo Nordisk, Octapharma, Pfizer, Roche, Sanofi, Sobi, Spark Therapeutics, Takeda and UniQure; SI and MK are internal Bayer employees.
AUTHOR CONTRIBUTIONS
All authors participated equally in the preparation of this manuscript (including study design, data acquisition/analysis/interpretation, and drafting/revising for important intellectual content). All authors approved the final version for publication.
Supporting information
Supporting Information
ACKNOWLEDGEMENTS
All authors contributed equally to the development of this manuscript. The authors would like to thank the participants and their families, and the investigators, pharmacists, nurses, and trial staff at each centre for participating in the trial. The authors would also like to thank Frank Driessler (Bayer AG) for his scientific advice and review of the manuscript. This study was supported by Bayer. Medical writing assistance was provided by Jason Foreman of Fishawack Communications, Ltd, part of Fishawack Health, and was fully funded by Bayer.
Mancuso ME, Ingham SJM, Kunze M. Befovacimab, an anti‐tissue factor pathway inhibitor antibody: Early termination of the multiple‐dose, dose‐escalating Phase 2 study due to thrombosis. Haemophilia. 2022;28:702–712. 10.1111/hae.14595
At time of study accomplishment Dr Mancuso was a hematologist at IRCCS Fondazione Ca’ Granda, Ospedale Maggiore Policlinico, Milan, Italy.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from Bayer. Restrictions apply to the availability of these data, which were used under license for this study. Availability of the data underlying this publication will be determined according to Bayer's commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing.” This pertains to scope, time point, and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers participant‐level clinical trial data, study‐level clinical trial data, and protocols from clinical trials in participants for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymised participant‐level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve participant care. Information on the Bayer criteria for listing studies and other relevant information is provided in the Study sponsors section of the portal. Data access will be granted to anonymised participant‐level data, protocols, and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that participant privacy is safeguarded.
REFERENCES
- 1. Arruda VR, Doshi BS, Samelson‐Jones BJ. Emerging therapies for hemophilia: controversies and unanswered questions. F1000Res. 2018:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thornburg C, Duncan N. Treatment adherence in hemophilia. Patient Prefer Adherence. 2017;11:1677‐1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Knobe K, Berntorp E. Haemophilia and joint disease: pathophysiology, evaluation, and management. J Comorb. 2011;1:51‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Franchini M, Mannucci PM. Non‐factor replacement therapy for haemophilia: a current update. Blood Transfus. 2018;16(5):457‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Key NS. Inhibitors in congenital coagulation disorders. Br J Haematol. 2004;127(4):379‐391. [DOI] [PubMed] [Google Scholar]
- 6. Abdi A, Bordbar MR, Hassan S, et al. Prevalence and incidence of non‐neutralizing antibodies in congenital hemophilia A – a systematic review and meta‐analysis. Front Immunol. 2020;11:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Male C, Andersson NG, Rafowicz A, et al. Inhibitor incidence in an unselected cohort of previously untreated patients with severe haemophilia B: a PedNet study. Haematologica. 2021;106(1):123‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kizilocak H, Young G. Diagnosis and treatment of hemophilia. Clin Adv Hematol Oncol. 2019;17(6):344‐351. [PubMed] [Google Scholar]
- 9. Mackman N. The role of tissue factor and factor VIIa in hemostasis. Anesth Analg. 2009;108(5):1447‐1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hackeng TM, Seré KM, Tans G, Rosing J. Protein S stimulates inhibition of the tissue factor pathway by tissue factor pathway inhibitor. Proc Natl Acad Sci USA. 2006;103(9):3106‐3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mast AE. Tissue factor pathway inhibitor: multiple anticoagulant activities for a single protein. Arterioscler Thromb Vasc Biol. 2016;36(1):9‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peterson JA, Maroney SA, Mast AE. Targeting TFPI for hemophilia treatment. Thromb Res. 2016;141(Suppl 2):S28‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wun TC, Kretzmer KK, Girard TJ, Miletich JP, Broze GJ. Cloning and characterization of a cDNA coding for the lipoprotein‐associated coagulation inhibitor shows that it consists of three tandem Kunitz‐type inhibitory domains. J Biol Chem. 1988;263(13):6001‐6004. [PubMed] [Google Scholar]
- 14. Broze Jr GJ. Tissue factor pathway inhibitor: structure‐function. Front Biosci (Landmark Ed). 2012;17:262‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maroney SA, Mast AE. New insights into the biology of tissue factor pathway inhibitor. J Thromb Haemost. 2015;13(Suppl 1):S200‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chowdary P. Inhibition of tissue factor pathway inhibitor (TFPI) as a treatment for haemophilia: rationale with focus on concizumab. Drugs. 2018;78(9):881‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wood JP, Bunce MW, Maroney SA, Tracy PB, Camire RM, Mast AE. Tissue factor pathway inhibitor‐alpha inhibits prothrombinase during the initiation of blood coagulation. Proc Natl Acad Sci USA. 2013;110(44):17838‐17843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gu J‐M, Zhao X‐Y, Schwarz T, et al. Mechanistic modeling of the pharmacodynamic and pharmacokinetic relationship of tissue factor pathway inhibitor‐neutralizing antibody (BAY 1093884) in cynomolgus monkeys. AAPS J. 2017;19(4):1186‐1195. [DOI] [PubMed] [Google Scholar]
- 19. Yegneswaran S, Marquardt T, Evans V, et al. BAY 1093884 binds to the Kunitz 1 and 2 domain interface of tissue factor pathway inhibitor and inhibits its function (PB 892). International Society on Thrombosis and Haemostasis ‐ 26th Congress. Research and Practice in Thrombosis and Haemostasis: Berlin, Germany; 2017. [Google Scholar]
- 20. Yegneswaran S, Kollnberger M, Evans V, Patel C. In vitro mechanism of BAY 1093884 inhibition of tissue factor pathway inhibitor. International Society on Thrombosis and Haemostasis ‐ 25th Congress. Journal of Thrombosis and Haemostasis: Toronto, Canada; 2015. [Google Scholar]
- 21. Bauzon M, Liu B, Grudzinska‐Goebel J, et al. Characterization of a high‐affinity fully human IgG2 antibody against tissue factor pathway inhibitor as a bypass agent for the treatment of hemophilia. Blood. 2015;126(23):3513‐3513. [Google Scholar]
- 22. Chowdary P, Lissitchkov T, Willmann S, et al. Pharmacodynamics, pharmacokinetics and safety of BAY 1093884, an antibody directed against human TFPI, in patients with factor VIII or IX deficiency (with and without inhibitors): a Phase 1 study. 60th American Society of Hematology Annual Meeting. Blood: San Diego, California, USA; 2018. [Google Scholar]
- 23. Dockal M, Pachlinger R, Hartmann R, et al. Biological explanation of clinically observed elevation of TFPI plasma levels after treatment with TFPI‐antagonistic aptamer BAX 499. 54th American Society of Hematology Annual Meeting. Blood: Atlanta, Georgia, USA; 2012. [Google Scholar]
- 24. ClinicalTrials.gov . First‐in‐human and proof‐of‐mechanism study of ARC19499 administered to hemophilia patients (NCT01191372). April 2020; https://clinicaltrials.gov/ct2/show/NCT01191372
- 25. Nordisk N. Novo Nordisk pauses the clinical trials investigating concizumab (anti‐TFPI mAB) in haemophilia A and B with or without inhibitors. March 2020; https://www.novonordisk.com/media/news‐details.2265611.html
- 26. ClinicalTrials.gov . A trial evaluating efficacy and safety of prophylactic administration of concizumab in patients with severe haemophilia A without inhibitors (explorer™5) (NCT03196297). April 2020; https://clinicaltrials.gov/ct2/show/NCT03196297
- 27. ClinicalTrials.gov . Research study to look at how well the drug concizumab works in your body if you have haemophilia without inhibitors (explorer8) (NCT04082429). April 2020; https://clinicaltrials.gov/ct2/show/NCT04082429
- 28. ClinicalTrials.gov . Research study to look at how well the drug concizumab works in your body if you have haemophilia with inhibitors (explorer7) (NCT04083781). April 2020; https://clinicaltrials.gov/ct2/show/NCT04083781
- 29. Abstract #ABS 188: Seremetis S , et al. Presented at EAHAD 2021.
- 30. Hilden I, Lauritzen B, Sørensen BB, et al. Hemostatic effect of a monoclonal antibody mAb 2021 blocking the interaction between FXa and TFPI in a rabbit hemophilia model. Blood. 2012;119(24):5871‐5878. [DOI] [PubMed] [Google Scholar]
- 31. Chowdary P, Lethagen S, Friedrich U, et al. Safety and pharmacokinetics of anti‐TFPI antibody (concizumab) in healthy volunteers and patients with hemophilia: a randomized first human dose trial. J Thromb Haemost. 2015;13(5):743‐754. [DOI] [PubMed] [Google Scholar]
- 32. Waters EK, Genga RM, Thomson HA, et al. Aptamer BAX 499 mediates inhibition of tissue factor pathway inhibitor via interaction with multiple domains of the protein. J Thromb Haemost. 2013;11(6):1137‐1145. [DOI] [PubMed] [Google Scholar]
- 33. Camire RM. Rethinking events in the haemostatic process: role of factor V and TFPI. Haemophilia. 2016;22(Suppl 5):3‐8. [DOI] [PubMed] [Google Scholar]
- 34. Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med. 2018;9:135‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809‐818. [DOI] [PubMed] [Google Scholar]
- 36. Common Terminology Criteria for Adverse Events (CTCAE) v5.0. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm#ctc_50 [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from Bayer. Restrictions apply to the availability of these data, which were used under license for this study. Availability of the data underlying this publication will be determined according to Bayer's commitment to the EFPIA/PhRMA “Principles for responsible clinical trial data sharing.” This pertains to scope, time point, and process of data access. As such, Bayer commits to sharing upon request from qualified scientific and medical researchers participant‐level clinical trial data, study‐level clinical trial data, and protocols from clinical trials in participants for medicines and indications approved in the United States (US) and European Union (EU) as necessary for conducting legitimate research. This applies to data on new medicines and indications that have been approved by the EU and US regulatory agencies on or after January 01, 2014. Interested researchers can use www.clinicalstudydatarequest.com to request access to anonymised participant‐level data and supporting documents from clinical studies to conduct further research that can help advance medical science or improve participant care. Information on the Bayer criteria for listing studies and other relevant information is provided in the Study sponsors section of the portal. Data access will be granted to anonymised participant‐level data, protocols, and clinical study reports after approval by an independent scientific review panel. Bayer is not involved in the decisions made by the independent review panel. Bayer will take all necessary measures to ensure that participant privacy is safeguarded.