

SUMMARY
To unveil the evolution of mitochondrial RNA editing in gymnosperms, we characterized mitochondrial genomes (mitogenomes), plastid genomes, RNA editing sites, and pentatricopeptide repeat (PPR) proteins from 10 key taxa representing four of the five extant gymnosperm clades. The assembled mitogenomes vary in gene content due to massive gene losses in Gnetum and Conifer II clades. Mitochondrial gene expression levels also vary according to protein function, with the most highly expressed genes involved in the respiratory complex. We identified 9132 mitochondrial C‐to‐U editing sites, as well as 2846 P‐class and 8530 PLS‐class PPR proteins. Regains of editing sites were demonstrated in Conifer II rps3 transcripts whose corresponding mitogenomic sequences lack introns due to retroprocessing. Our analyses reveal that non‐synonymous editing is efficient and results in more codons encoding hydrophobic amino acids. In contrast, synonymous editing, although performed with variable efficiency, can increase the number of U‐ending codons that are preferentially utilized in gymnosperm mitochondria. The inferred loss‐to‐gain ratio of mitochondrial editing sites in gymnosperms is 2.1:1, of which losses of non‐synonymous editing are mainly due to genomic C‐to‐T substitutions. However, such substitutions only explain a small fraction of synonymous editing site losses, indicating distinct evolutionary mechanisms. We show that gymnosperms have experienced multiple lineage‐specific duplications in PLS‐class PPR proteins. These duplications likely contribute to accumulated RNA editing sites, as a mechanistic correlation between RNA editing and PLS‐class PPR proteins is statistically supported.
Keywords: gymnosperms, mitochondria, mitogenomes, RNA editing, PPR proteins
Significance Statement
We conducted a systematic and comprehensive investigation on RNA editing in extant gymnosperm clades. In total, 9132 mitochondrial RNA editing sites and 11 376 PPR proteins were identified. Our results unveil the difference in functional roles between non‐synonymous and synonymous editing, regains of editing sites, evolution of losses or gains of editing events, multiple duplications in PLS‐class PPR proteins, and the positive association between PLS‐class PPR protein diversity and RNA editing site abundance in gymnosperms.
INTRODUCTION
RNA editing, a widespread post‐transcriptional process, alters RNA molecules to carry genetic information differing from their genomic templates. In plants, RNA editing is confined to terrestrial species, leading to the hypothesis that RNA editing might have evolved to facilitate adaptation to land environments where plants face increased UV damage compared to water habitats (Fujii & Small, 2011). On the contrary, constructive neutral evolution posits that existence of editing activities predates emergence of RNA editing sites and such activities are derived from pre‐existing enzymes serving other functions (Gray, 2012).
Numerous C‐to‐U editing sites were previously documented in land plant mitochondria (Ichinose & Sugita, 2016). In contrast, U‐to‐C editing sites were not found in seed plants (Gagliardi & Gualberto, 2004; Takenaka et al., 2013), although they were frequently detected in other vascular plants, such as lycophytes (Grewe et al., 2011) and ferns (Knie et al., 2016). Plant mitochondrial RNA editing sites generally occur in protein‐coding regions, concentrated at first and second codon positions, and involve restoration of evolutionarily conserved codons (Edera et al., 2018). Therefore, RNA editing is thought to be a repair system vital to mitochondrial biogenesis and function (Li et al., 2019). RNA editing is also detectable at third codon positions, where a C‐to‐U change has no effect on the encoded amino acids. These synonymous editing events usually are performed with low efficiency as they occur only in a subset of transcripts (Edera et al., 2018; Wu et al., 2015). Partially edited transcripts were previously considered to be intermediates generated from RNA processing (Verbitskiy et al., 2006), while synonymous editing was hypothesized to occur accidentally (Bentolila et al., 2013; Picardi et al., 2010; Wu et al., 2015).
Nuclear‐encoded pentatricopeptide repeat (PPR) proteins are characterized by tandem arrays of a degenerated PPR motif containing about 35 amino acids. They comprise one of the largest protein families in plants and are composed of two major classes: P and PLS (Cheng et al., 2016). P‐class PPR proteins only have canonical P motifs, while PLS‐class PPR proteins harbor P, L, and S motifs that form tandemly repeated PLS triplets (Small & Peeters, 2000). PLS‐class PPR proteins are further divided into subclasses PLS, E+, E1, E2, and DYW based on their C‐terminal domains (Cheng et al., 2016; Manna, 2015; Wang et al., 2021). It has been known that PLS‐class PPR proteins and other trans‐acting factors constitute multi‐protein complexes, termed editosomes, which can recognize cis‐elements surrounding target sites to carry out RNA editing (Small et al., 2020; Sun et al., 2016). Moreover, positive correlations between PLS‐class PPR protein diversity and RNA editing site abundance were reported in some plant lineages (Dong et al., 2019; Fujii & Small, 2011). Interestingly, recognition flexibility enables a single editosome to be responsible for multiple editing sites (Shikanai, 2015). Unspecific binding of cis‐elements also suggests a mechanism for low editing efficiencies usually observed at synonymous sites (Bentolila et al., 2013; Picardi et al., 2010; Sun et al., 2016).
Investigations of RNA editing sites rely on comparing genomic sequences with their RNA counterparts. Advances in next‐generation sequencing (NGS) technologies have made significant improvements in exploration of RNA editing sites (Hao et al., 2021), especially of partially edited sites that can be hidden in Sanger sequencing. Extant gymnosperms, a seed plant group sister to angiosperms, comprise five major clades. Their mitochondrial genomes (mitogenomes) are highly variable in size, gene content, and intron number due to extensive rearrangements (Guo et al., 2020; Kan et al., 2021). To date, investigations of gymnosperm mitochondrial RNA editing sites based on NGS data have been only conducted for a few taxa such as Ginkgo, Welwitschia (Fan et al., 2019), and Taxus (Kan et al., 2020). A systematic study and a broader taxon sampling are required to better understand the evolution and patterns of mitochondrial RNA editing in gymnosperms.
In this study, plastid genomes (plastomes), mitogenomes, and transcriptomes were assembled from 10 diverse taxa representing four of the five extant gymnosperm clades: cycads, gnetophytes, Conifer I, and Conifer II. RNA editing sites were identified in plastid and mitochondrial protein‐coding transcripts. We elucidated their variation, conservation, editing efficiency, bias occurrence, loss, gain, association with gene expression, and effects on alternations of amino acid products. In addition, PPR protein sequences were identified and retrieved from the assembled transcriptomes and their associations with RNA editing sites were examined across the sampled gymnosperm taxa. We also discuss the possible mechanisms underlying gains of RNA editing sites.
RESULTS
Gene content varies among gymnosperm mitogenomes
We obtained 11 to 43 mitochondrial scaffolds from the 10 sequenced gymnosperm representatives. Their average k‐mer coverage ranges from 4.3× to 46.1×, while GC content is between 43.4% and 51.7% (Table S2). The total length of the assembled mitochondrial scaffolds ranges from 201.2 to 3000 kb, indicating that gymnosperm mitogenomes vary drastically in size.
Using Cycas (NC010303) and Ginkgo (NC027976) as the references, 41 mitochondrial protein‐coding genes were identified in the two cycads, Dioon and Zamia, and Conifer I taxa, Pseudotsuga and Keteleeria, except for rps7, which was missing in Pseudotsuga (Figure 1). Several genes were not detected in other gymnosperms. For example, (i) seven genes, rpl2, rps1, rps2, rps7, rps10, rps11, and rps14, were found neither in Gnetum nor in any sampled Conifer II taxa (i.e., cupressophytes), including Nageia, Agathis, Sciadopitys, Cephalotaxus, and Cunninghamia; (ii) four genes, rpl5, rpl16, rps13, and rps19, were uniquely absent from Gnetum; (iii) sdh3 was not detected in Gnetum, Sciadopitys, Cephalotaxus, and Cunninghamia; and (iv) rpl10 was lost from all cupressophytes except Sciadopitys. Collectively, we found 41 mitochondrial protein‐coding genes in cycads, 40–41 in Conifer I, 29 in Gnetum, and 32–33 in Conifer II, and 28 are shared among all examined gymnosperms. Our data also indicate that genes involved in respiratory complexes and cytochromes are mostly retained, while those associated with ribosomal subunits are frequently absent, leading to highly variable gene content among the gymnosperm mitogenomes (Figure 1).
Figure 1.
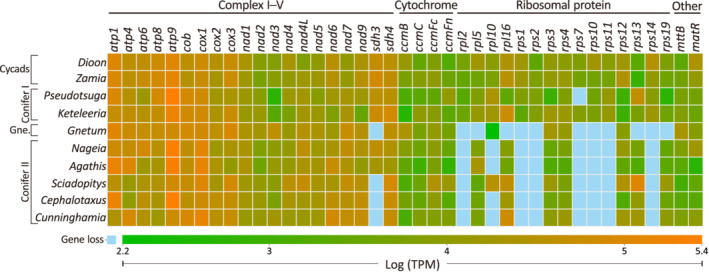
Comparisons of mitochondrial protein‐coding genes and their expression levels among the 10 studied gymnosperms. Gene expression levels are normalized to transcripts per million (TPM) values and represented by a color gradient from green (lowest) to orange (highest). Light‐blue squares specify losses of genes. Gne., gnetophytes.
The complete sequences of plastomes were also recovered from Dioon, Gnetum, Pseudotsuga, and Zamia. Their circular molecules vary from 114.8 to 165.3 kb in size (Table S2). Plastomes from the other six taxa have been assembled and elucidated in our earlier study (Wu et al., 2021).
Mitochondrial gene expression levels are functionally dependent
Approximately 20 million RNA‐seq read pairs per taxon were generated from young leaves under controlled light conditions (see Experimental Procedures). These reads were then mapped to the corresponding scaffolds. To evaluate gene expression levels in mitochondria, reads that matched identified genes were counted and normalized to transcripts per million (TPM) values. Our analyses revealed that gene expression levels in mitochondria are dependent more on function than on phylogenetic relatedness. For example, respiration‐associated genes, such as atp, cob, cox, and sdh, are in general expressed at higher levels than other functional genes (Figure 1). These results are in good agreement with the fact that mitochondria are responsible for aerobic respiration. Despite being highly expressed, sdh3 is missing in some taxa as mentioned above. This suggests that retention of mitochondrial genes is likely independent of their gene expression levels despite frequent losses of low‐expressed ribosomal genes during gymnosperm mitogenome evolution (Figure 1).
Characteristics of organelle RNA editing sites in gymnosperms
We counted nucleotide mismatches between protein‐coding genes and their associated RNA‐seq reads. In total, 9132 mismatches were identified as C‐to‐U RNA editing events across the 10 gymnosperm mitochondria (Figure 2; Table S3). We did not find U‐to‐C editing sites because none of them passed the identification threshold. The C‐to‐U editing events mainly occurred at the second codon position (42.2–62.6%), followed by the first (24.2–34.1%) and third (8.9–23.7%) codon positions (Figure S1). As a result, the majority (75.7–87.8%) of them involve amino acid changes or creation of initial/terminal codons, the so‐called non‐synonymous editing. Synonymous editing, which does not cause amino acid changes, accounts for only 12.2–24.3% of the editing sites (Figure 2). In addition, approximately 30% of the non‐synonymous editing sites occur at the first codon position, while approximately 70% occur at the second codon position (Figure S1). By contrast, approximately 5% of the synonymous editing sites occur at the first codon position and the remaining approximately 95% occur at the third codon position (Figure S1).
Figure 2.
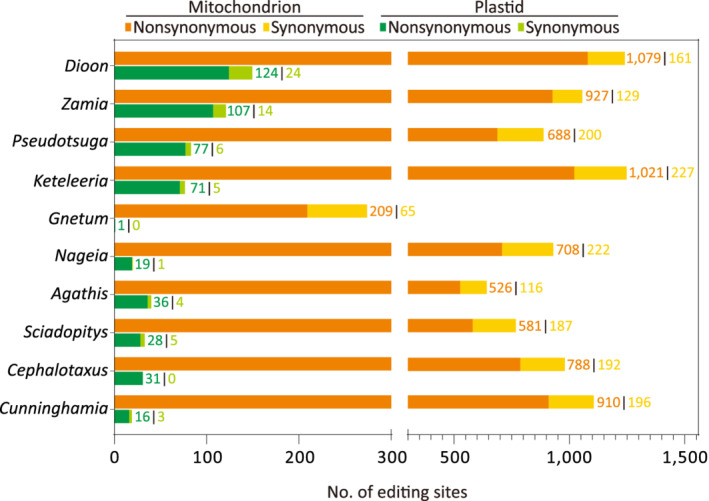
Variations in plastid and mitochondrial RNA editing sites across the 10 surveyed gymnosperms.
Notably, numbers of mitochondrial RNA editing sites exhibit striking variation among the gymnosperms. Gnetum contains the fewest (274), about only 22.1% of those in Dioon (1240; Figure 2). Overall, mitochondrial RNA editing occurs more frequently at non‐synonymous sites than at synonymous sites in all sampled gymnosperms (paired t‐test, P < 0.01 after Bonferroni correction).
We also identified 148 plastid C‐to‐U editing sites in Dioon, 121 in Zamia, 83 in Pseudotsuga, and one in Gnetum (Figure 2; Table S3). Plastid RNA editing sites of the other six taxa were based on our earlier study without counting those at antisense transcripts (Wu et al., 2021). The ratio of plastid non‐synonymous to synonymous editing sites is between 4.2:1 and 19:1 across the gymnosperms except for Cephalotaxus and Gnetum, whose plastid RNA does not undergo synonymous editing (Figure 2).
To assess if RNA editing affects biochemical properties of mitochondrial proteins, we compared amino acids encoded by the codons with and without modifications from non‐synonymous editing. Codons specific to hydrophobic amino acids (i.e., hydrophobic codons) increase drastically from 44.7% to 86.9% in the edited codons. In contrast, hydrophilic codon prevalence drops from 55.3% to 13.1% (Figure S2). Therefore, RNA editing apparently leads to increased hydrophobicity of mitochondrial proteins.
Editing efficiency differs remarkably between non‐synonymous and synonymous sites
We measured editing efficiency by estimating the frequency of C‐to‐U changes (or G‐to‐A changes in the reverse complementary strand) in mapped mitochondrial RNA reads. Editing efficiency ranges between 10 and 100% (the minimal threshold for editing detection was set to 10%; Figure 3). Average editing efficiency at non‐synonymous sites is 77.3–85.1%, about two‐fold higher than that at synonymous sites (34.0–50.9%). Editing efficiency at non‐synonymous sites significantly differs from that at synonymous sites in all examined taxa (two‐tailed t‐test, all P < 0.01).
Figure 3.
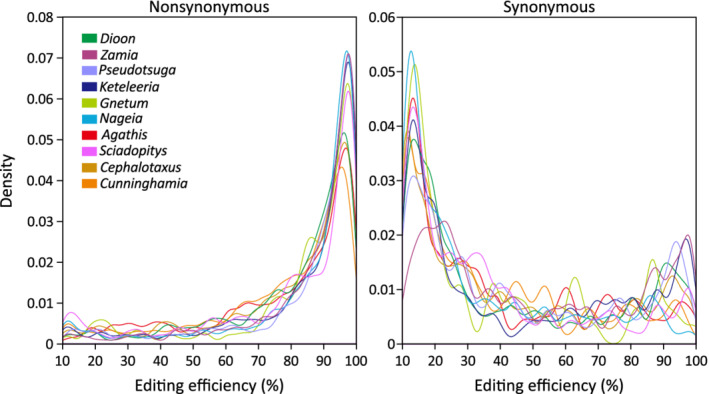
Comparisons of editing efficiency between non‐synonymous (right panel) and synonymous (left panel) sites using kernel density plots.
As expected, non‐synonymous editing sites in all taxa exhibit a unimodal distribution, with 70.9–85.1% of the sites exhibiting >70% editing efficiency (Figure 3; Figure S3). However, high editing efficiency was also detected at some synonymous sites. For example, 14.9–32.6% of these sites are edited with efficiency greater than 70%, resulting in a clearly bimodal distribution observed in Dioon, Zamia, Pseudotsuga, and Keteleeria (Figure 3).
We further estimated the relationship between editing conservation and efficiency based on editing sites in the 28 shared mitochondrial genes. Our results show that there is no significant correlation at non‐synonymous edited sites (Pearson's Rho = 0.473, P = 0.167; Figure S4), suggesting that most such positions are efficiently edited, irrespective of their conservation status among the gymnosperms. By contrast, strongly conserved synonymous sites appear to be edited relatively efficiently as compared to weakly conserved ones, as evidenced by a positive correlation between editing conservation and efficiency at these sites (Pearson's Rho = 0.884, P = 0.0016; Figure S4).
Losses/gains of RNA editing sites during gymnosperm mitochondrial evolution
We inferred gains and losses of mitochondrial RNA editing sites across the 10 sampled gymnosperms using Dollo parsimony and ‘gnepines’ topology (Chaw et al., 2000; Wu et al., 2011). To avoid potential effects from missing data (i.e., losses of editing sites due to gene losses), we only analyzed 8348 editing sites detected in the 28 shared mitochondrial genes. In total, 4040 losses and 1945 gains were inferred across the tree where excesses of losses over gains appear on all branches, with a few exceptions, such as those leading to Dioon, to Conifer I, and to the Cephalotaxus–Cunninghamia clade (Figure 4a). Overall, the loss‐to‐gain ratios across the gymnosperm tree, on average, are 2.1:1 at all editing sites, 2.7:1 at non‐synonymous editing sites, and 1.1:1 at synonymous editing sites (Figure 4b).
Figure 4.
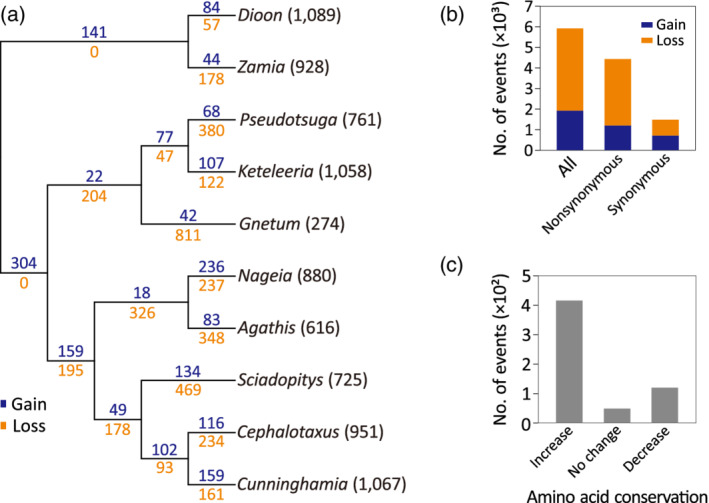
Evolution of mitochondrial RNA editing sites in gymnosperms. (a) Gains (blue) and losses (orange) of RNA editing sites across the evolution of gymnosperms. Values in parentheses behind taxa are the number of editing sites detected in the 28 shared genes. (b) Gains or losses of RNA editing taking place at non‐synonymous or synonymous sites. (c) Changes in amino acid conservation due to gains of editing sites on the terminal branches.
The inferred gains of non‐synonymous editing on the terminal branches allow us to investigate the effects of RNA editing on amino acid sequences. We found that the majority (71.1%) of these gains resulted in increased amino acid identities among the gymnosperms (Figure 4c), suggesting a fundamental role of RNA editing in maintaining amino acid sequence conservation. To examine if genomic C‐to‐T substitutions are responsible for the loss of mitochondrial RNA editing, we retrieved genomic sequences at sites whose RNA editing capability was inferred to be lost on the terminal branches. Our data indicate that 72.1% of these formerly non‐synonymous editing sites are thymines (Figure S5), suggesting that genomic C‐to‐T substitutions primarily drive the loss of non‐synonymous editing. However, at formerly synonymous editing sites, most genomic nucleotides are cytosines (79.8%) rather than thymines (15.2%). This suggests that the evolution of non‐synonymous and synonymous editing sites is shaped by different mechanisms in the gymnosperm mitochondria.
Regains of editing sites in transcripts of intron‐less rps3 genes
We compared frequencies of editing sites, i.e., the number of such sites per kb, among genes (Figure S6). Remarkable variation among genes is observed within lineages, with the most extreme in Nageia: its ccmB gene contains the highest frequency of editing sites (87.6 sites/kb) but its rps3 gene contains the lowest frequency (4.3 sites/kb). Despite high variation among genes, ccmB and mttB are generally the two most heavily edited genes in the examined taxa. Previously, Fan et al. (2019) detected a negative correlation between gene expression levels and editing site frequencies, and they concluded that retroprocessing drives the loss of RNA editing sites because highly transcribed genes are more prone to such conversions. However, such correlations are not significant in our set of taxa except for Sciadopitys (Figure S6). These insignificant results are expected because both losses and gains occurred during evolution (Figure 4a), but retroprocessing accounts only for losses.
A retroprocessing event was previously reported to account for the loss of rps3 introns from the common ancestor of Conifer II taxa (Ran et al., 2010). Indeed, all sampled Conifer II taxa lack introns in their rps3 gene (Figure 5), making it an ideal target for investigating the evolution of RNA editing after intron losses via retroprocessing. Two competing scenarios are possible. Scenario I assumes that a large‐scale retroprocessing event occurred across the entire rps3 gene, resulting in the intron losses. In contrast, scenario II hypothesizes that multiple rounds of retroprocessing took place across only the introns and their adjacent regions. In scenario I, we would expect no editing sites to be shared between Conifer II and other gymnosperms because ancient editing sites were purged by retroprocessing in the Conifer II common ancestor. We found between two and nine editing sites shared between Conifer II taxa and other gymnosperms (Figure 5). Therefore, the large‐scale retroprocessing scenario is unlikely. However, we also discovered a few shared editing sites next to putative splicing sites that are lost in Agathis (one site), Cephalotaxus (two sites), and Cunninghamia (two sites; Figure 5). This result argues against scenario II, where ancient editing sites adjacent to splicing sites would be removed by retroprocessing during the intron losses. Therefore, we propose that either these shared editing sites have been regained in the transcripts of the intron‐less rps3 genes, or neither of the scenarios we proposed can account for the rps3 intron losses.
Figure 5.
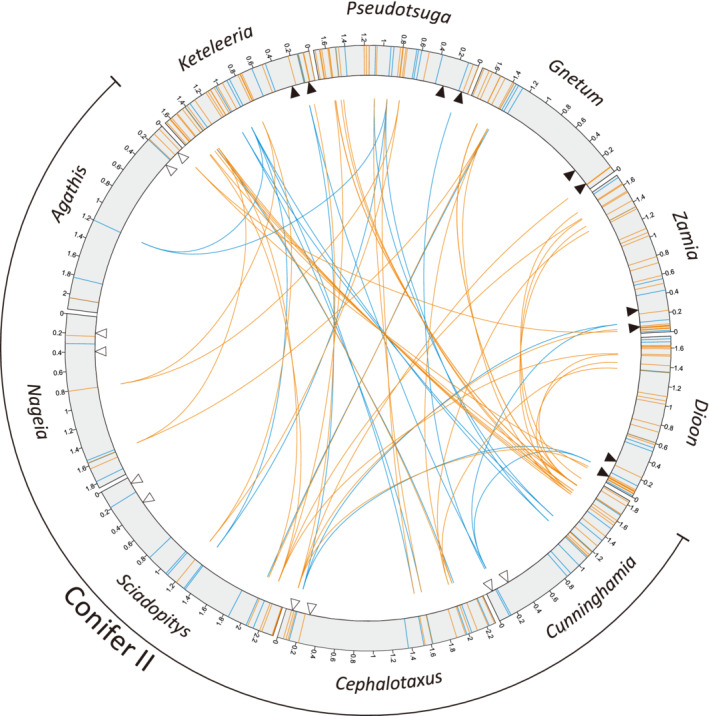
RNA editing sites detected in gymnosperm mitochondrial rps3 transcripts. The rps3 gene is illustrated as a gray box where non‐synonymous and synonymous editing sites are designated as orange and blue lines, respectively. Editing sites shared between Conifer II and other gymnosperm taxa are linked with the inner lines. Black triangles indicate splicing sites, while white ones denote putative splicing sites that were lost during Conifer II evolution.
PPR proteins and their association with RNA editing sites
We identified 11 376 PPR protein sequences, including 2846 P‐class and 8530 PLS‐class proteins, in our assembled transcriptomes (Figure 6; Table S4). PPR protein copy numbers range from 509 in Gnetum to 2131 in Zamia, with an average of 1138 per taxon. Copy number variation among taxa is relatively low in P‐class PPR proteins (232–316) compared to PLS‐class PPR proteins (277–1822). The PLS subclass contributes to 46.9–71.6% of the PPR protein diversity. The other four subclasses (E+, E1, E2, and DYW) contribute 28.4–58.1% to the diversity. To trace the evolutionary trajectory of the PPR proteins, we constructed phylogenetic trees of subclasses E+, E1, E2, and DYW (Figures [Link], [Link], [Link], and S10). We did not conduct phylogenetic analyses for the PLS subclass because their sequences are too divergent to be reliably aligned. We found several gene duplication events, for example E+ in cycads, Conifer I, and Conifer II (Figure S7), E1 in cycads and Conifer II (Figure S8), E2 in Gnetum and Conifer II (Figure S9), and DYW in all four sampled gymnosperm clades (Figure S10). These lineage‐specific duplications have expanded and diversified the PPR proteins in gymnosperms. We did not detect any DYW:KP domains (Gutmann et al., 2020), in agreement with the absence of U‐to‐C editing sites in our assembled transcripts.
Figure 6.
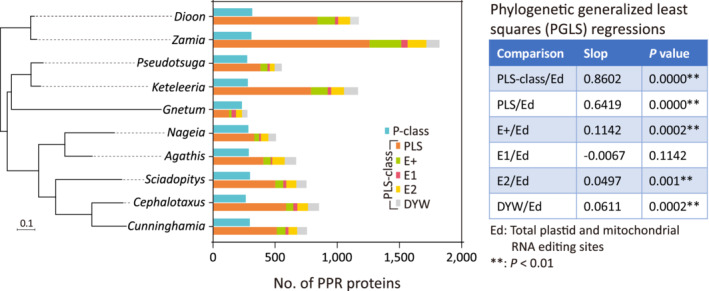
PPR protein diversity in gymnosperms. The stacked bar charts (central panel) represent the diversity of identified P‐class and PLS‐class PPR proteins, the latter including five subclasses. Correlations between the diversity of PLS‐class proteins and the abundance of RNA editing sites were examined using PGLS regression (left panel) with incorporation of a tree (right panel) inferred from the concatenation of shared mitochondrial and plastid genes.
We also examined the association between PLS‐class protein diversity and the RNA editing site abundance using phylogenetic generalized least squares (PGLS) regression. PGLS regression takes into account non‐independent residuals that arise from shared phylogenetic history (Grafen, 1989). Our results reveal that the diversity of not only the PLS‐class group but also its subclasses (except for E1) is positively correlated with the number of editing sites identified in both plastids and mitochondria (Figure 6). These positive correlations highlight a mechanistic link between RNA editing and PLS‐class PPR proteins in gymnosperms.
DISCUSSION
Gymnosperm and angiosperm RNA editing machineries have a common origin
Our assemblies show losses of many mitochondrial genes in Gnetum and Conifer II but not in cycads or Conifer I (Figure 1). These results support recent studies on gymnosperm mitogenomes (Guo et al., 2020; Kan et al., 2020, 2021), which indicated that gene losses have shaped gymnosperm mitogenome evolution. Because polyA tails act as degradation signals for organellar RNAs (Chang & Tong, 2012; Hirayama et al., 2013), we adopted Rib‐Zero and random hexamer methods rather than oligo dT priming to deplete rRNAs and synthesize cDNAs. We also constructed strand‐specific libraries that enable clear separation of sense and antisense transcripts, making RNA‐seq reads more specific (Levin et al., 2010). Collectively, our data are consistent with the general review that respiratory genes are more abundantly expressed than other genes in plant mitochondria (Stone & Storchova, 2015).
We identified 9132 mitochondrial RNA editing sites with a stringent threshold (editing efficiency ≥ 10%) to minimize false positives. Because of the incomplete mitogenome assemblies, we did not search for editing sites in non‐coding transcripts, although a few such sites were documented in the intronic sequences of Welwitschia (Fan et al., 2019) and some angiosperms, such as grape (Vitis vinifera) (Picardi et al., 2010), tobacco (Nicotiana tabacum) (Grimes et al., 2014), and moth orchid (Phalaenopsis aphrodite subsp. formosana) (Chen et al., 2020). This implies that the real prevalence of RNA editing sites in the examined mitochondria might have been underestimated.
Due to different methods for constructing RNA libraries and detecting editing sites, our comparisons did not include other gymnosperms whose mitochondrial RNA editing sites were previously reported. Nevertheless, the set of editing sites we identified is the most up‐to‐date and comprehensive. Although gymnosperms and angiosperms have diverged for 3 million years, our data show that they are highly similar in several aspects of RNA editing, including absence of U‐to‐C editing, an overwhelming prevalence of editing at non‐synonymous sites, an increase in codons encoding hydrophobic amino acids, high conservation of non‐synonymous editing sites, and low efficiency at synonymous sites (Edera et al., 2018; Giegé & Brennicke, 1999). These observations suggest that the RNA editing machineries of seed plants have a common origin and that their editing sites may play similar roles in mitochondrial biogenesis and function.
The evolutionary role of synonymous editing in gymnosperm mitochondria
Approximately one tenth to one fourth of the total RNA editing sites in the sampled gymnosperms were detected as synonymous. Most of them occur at third codon positions with lower editing efficiency than non‐synonymous sites (Figure 3). This low efficiency likely results from non‐specific binding of editing factors since editosomes can recognize cis‐elements with a few base changes (Picardi et al., 2010; Sun et al., 2016). In contrast, high editing efficiency at most non‐synonymous sites implicates strong selection constraints on both editosomes and cis‐elements to maintain their binding affinities. This reasonably explains our observation that losses of non‐synonymous editing sites are mainly due to genomic C‐to‐T substitutions (Figure S5). Conversely, relaxation of selection constraints on editosomes, cis‐elements, or both accounts for losses of most synonymous editing sites where the corresponding genomic bases are still cytosines (Figure S5).
However, the recognition flexibility of editosomes cannot explain the existence of synonymous sites that are edited with >70% efficiency (Figure 3; Figure S3). Moreover, we observed that the more conserved a synonymous editing site is, the more efficiently it is edited (Figure S4). These highly conserved and efficiently edited synonymous sites likely play an unknown but important role, such as stabilizing RNA molecules, as proposed by Edera et al. (2018) and Small et al. (2020). In addition, C‐to‐U editing changes synonymous codon types. Therefore, we wonder whether synonymous editing has effects on translation efficiency. Our estimates of the relative synonymous codon usage reveal a strong bias towards U‐ending codons (Figure S11), suggesting that gymnosperm mitochondria preferentially utilize U‐ending codons for translation. Hence, it is logical for us to associate synonymous editing with translation efficiency, and the former likely regulates the latter because synonymous sites are edited with variable efficiencies among lineages, individuals, tissue types, developmental stages, and growth conditions (Bentolila et al., 2008; Ichinose & Sugita, 2016; Zehrmann et al., 2008).
Possible mechanisms underlying regains of RNA editing sites
In the rps3 transcripts of Conifer II taxa, we identified several editing sites being recruited after the gene had experienced retroprocessing and lost its two introns (Figure 5). Interestingly, some of these edited loci are shared with other gymnosperms, indicating regains of the editing capability at these specific loci in Conifer II. Point mutations and retroprocessing are two proposed mechanisms causing losses of RNA editing sites (Edera et al., 2018; Fan et al., 2019; Grewe et al., 2014; Mower, 2008; Sloan et al., 2010). Modeling tests have suggested that replacements of edited sites with genomic thymidines happen more frequently than degradation of cis‐elements (or motifs) that are recognized by editosomes (Edera et al., 2018). Many PPR proteins can recognize several cis‐elements, resulting in multiple remote sites edited by a single editosome (Fujii & Small, 2011; Ichinose & Sugita, 2016; Shikanai, 2015). This capability of editing multiple sites might permit retention of specific editosomes when one of their targets was removed by genomic point mutations or retroprocessing. Therefore, regains of particular RNA editing sites can be achieved if the associated editosomes and cis‐elements still persist in the mitochondria. This point of view is in good agreement with the concept of constructive neutral evolution whereby pre‐existence of RNA editing systems rescues the mutational effects at the RNA level, thus allowing genomic T‐to‐C mutations to be fixed at editable positions (Covello & Gray, 1993; Gray, 2012).
Expansion of PLS‐class PPR proteins accounts for gains of RNA editing sites in gymnosperms
Loss‐to‐gain ratios of mitochondrial RNA editing sites were estimated to be 14:1 at non‐synonymous and 2:1 at synonymous sites in angiosperms (Richardson et al., 2013). Using the same method and criteria, we inferred the ratio at non‐synonymous and synonymous sites to be 2.7:1 and 1.1:1 in gymnosperms. Apparently, loss‐to‐gain ratios are relatively low in gymnosperms. The lower ratio may have resulted from reduced loss rates or increased gain rates during RNA editing site evolution. We favor the latter scenario because we occasionally observe excesses of gains over losses on some branches of the gymnosperm tree (Figure 4a). This is in contrast to the angiosperm evolution, where losses always overwhelmingly exceed gains (Richardson et al., 2013).
It is well known that PLS‐class PPR proteins are responsible for RNA editing (reviewed in Small et al., 2020). There are several ways to gain novel editing sites, for instance, proliferation of PLS‐class PPR proteins that target different RNA sequences and reassembly of E‐ and DYW‐subclass proteins to generate new editosome complexes that increase the number of accessible sites (Takenaka et al., 2018). PPR proteins in angiosperms typically comprise 400–600 members (Fujii & Small, 2011), fewer than in gymnosperms (509–2131). In gymnosperms, PLS‐class PPR proteins contribute most to PPR protein diversity (Figure 6) and have experienced several rounds of lineage‐specific duplications (Figures [Link], [Link], [Link], and S10). These duplications not only expand PLS‐class PPR proteins but also add new substrates to recombine editosomes that permit gains of novel editing sites. Consequently, we observed that the PLS‐class PPR protein diversity is positively correlated with the editing site abundance in gymnosperms (Figure 6).
In conclusion, we analyzed plastid and mitochondrial genomic and transcriptomic data from 10 taxa representing four of the five extant gymnosperm clades. Our results indicate that Gnetum and Conifer II have undergone massive losses of ribosomal genes, resulting in the highly variable gene content currently observed among gymnosperm mitogenomes. In addition, the RNA editing site prevalence and PLS‐class PPR protein diversity are statistically correlated, highlighting a mechanistic link between them in gymnosperms. Although gymnosperms and angiosperms have similar RNA editing characteristics, the gain‐to‐loss ratios of their mitochondrial RNA editing sites are strikingly different, reinforcing distinctive trends in evolution of their PPR proteins. Previous experiments have demonstrated that mitochondrial hydrophobic membrane proteins, if synthesized in the cytosol, are misdirected into the endoplasmic reticulum (Björkholm et al., 2015). This supports the hypothesis that some genes are retained in organelles because their highly hydrophobic products fail to be transported from the cytosol to organelles, thus limiting their intracellular transfer from organelles to the nucleus (Daley & Whelan, 2005; von Heijne, 1986). Recently, large‐scale studies also proposed that high GC content and protein hydrophobicity facilitate retention of genes in mitochondria (Johnston & Williams, 2016; Kan et al., 2021). Notably, C‐to‐U editing not only results in increased protein hydrophobicity (Giegé & Brennicke, 1999; Edera et al., 2018; this study), but also eliminates deleterious effects of genomic T‐to‐C substitutions that ultimately elevate whole‐genome GC content. Whether C‐to‐U editing plays an important role in retention of mitochondrial genes is worthy of investigation in the future.
EXPERIMENTAL PROCEDURES
Plant material collection, DNA and RNA extraction, and sequencing
Young shoots bearing leaves were collected from individuals of the 10 studied gymnosperms. Voucher information is shown in Table S1. To reduce variability across growth conditions, these shoots were grown hydroponically in a growth chamber (GC‐550R, Yihder Company, New Taipei City) at 25°C with a light intensity of 100 μmol m−2 sec−1 for 24 h. Fresh leaves on shoots were then excised for DNA and RNA extraction using the methods described in Stewart and Via (1993 ) and Kolosova et al. (2004), respectively. The obtained DNA was sequenced on an Illumina HiSeq 4000 instrument at Genomics BioSci & Tech (New Taipei City, Taiwan) to generate 150‐bp paired end DNA‐seq reads. The same sequencing platform was used to obtain strand‐specific RNA‐seq reads after DNase I (Invitrogen, Carlsbad, CA, USA.) treatment, rRNA depletion (Illumina Ribo‐Zero rRNA Removal kits, Plant Leaf version), and cDNA library construction with dUTP and random hexamers. NGS data generated in this study and their SRA accession numbers are detailed in Table S2.
Sequence assembly, mitochondrial scaffold identification, and annotation
After quality trimming by Trimmomatic 0.38 (Bolger et al., 2014), DNA‐seq reads were de novo assembled using SPAdes 3.13 (Bankevich et al., 2012) with the option of ‘careful’ and the k‐mer size increasing from 21 to 33, 55, 77, and 99. To facilitate blast searches, we discarded scaffolds that were shorter than 500 bp. Mitochondrial scaffolds were identified by blast searches against protein‐coding genes retrieved from Cycas (NC010303) and Ginkgo (NC027976) mitogenomes with an expected threshold of 10−10, followed by manual inspections. We used Pilon 1.24 (Walker et al., 2014) to do base‐scale corrections on the identified scaffolds. Mitochondrial genes were annotated using Cycas and Ginkgo mitogenomes as the references in Geneious Prime 2021.2 (https://www.geneious.com). For the assembly of the Dioon, Zamia, Pseudotsuga, and Gnetum plastomes, we used GetOrganelle v1.7.5.3 (Jin et al., 2020) with default parameters. Table S2 details GenBank accession numbers for the plastomes and identified mitochondrial scaffolds.
RNA mapping and RNA editing site exploration
Twenty million read pairs per taxon were retrieved from raw read pools using Seqtk (https://github.com/lh3/seqtk) after quality trimming. These strand‐specific RNA‐seq reads were mapped to our assembled mitochondrial scaffolds and plastomes using TopHat 2.1.1 (Kim et al., 2013) with the following parameters: library‐type = fr‐firststrand, read‐mismatches = 15, read‐gap‐length = 0, and read‐edit‐dist = 15. Mapped reads were sorted, filtered, and combined to generate BAM files using Samtools 1.9 (Li et al., 2009). These BAM files and their corresponding DNA scaffolds were imported together into Geneious Prime to calculate read counts for estimating expression levels and other downstream analyses.
The ‘Find Variations’ function implemented in Geneious Prime was employed to search RNA editing sites on the protein‐coding transcripts with the following thresholds: minimum coverage = 50, minimum variant frequency = 0.1, maximum variant P‐value = 10−6, and minimum strand‐bias P‐value = 10−5 when exceeding 65% bias. Indel variants were not considered.
Gains and losses of RNA editing sites
Sequences of the 28 shared mitochondrial genes containing editing sites were aligned using MUSCLE 3.8.425 (Edgar, 2004) implemented in Geneious Prime. Positions of editing sites in alignments were exported for constructing a presence/absence matrix, in which sites with and without editing were coded as ‘1’ and ‘0’, respectively. This matrix was then used for calculating the degree of editing conservation and inferring gains and losses of editing sites across the gymnosperm evolution using the Count software (Csűös, 2010).
RNA‐seq assembly, PPR protein identification, and phylogenetic inference
De novo assembly of RNA‐seq data was conducted using Trinity 2.14.0 (Grabherr et al., 2011), with library type = RF. Open reading frames were extracted from the assembly using TransDecoder 5.5.0 (https://github.com/TransDecoder), followed by removal of redundancies using CD‐HIT 4.8.1 (Fu et al., 2012) under the identity threshold of 0.9. Unique sequences were then searched for P‐ and PLS‐class PPR proteins in the PPR database (https://ppr.plantenergy.uwa.edu.au/; access date: March, 2022). The five subclasses of PLS‐class proteins were identified based on their specific C‐terminal motifs described in Cheng et al. (2016). Sequences of E+‐, E1‐, E2‐, and DYW‐subclass proteins were aligned using Clustal Omega 1.2.2 (Sievers et al., 2011), with refinement iterations = 2. We used FastTree 2.1.11 (Price et al., 2010) and the Jones–Taylor–Thornton model to construct phylogenetic trees for each PPR subclass.
PGLS regression analysis
Correlations between RNA editing sites and PLS‐class PPR proteins were assessed using PGLS regression implemented in the R packages ape and nlme. The incorporated tree inferred from the concatenation of mitochondrial and plastid genes was built using RAxML 8.2.4 (Stamatakis, 2014), with a GTRGAMMA model and a constrained monophyletic group comprising Pseudotsuga, Keteleeria, and Gnetum. The covariance structure between examined variables was set to be a Brownian motion process across the tree.
ACCESSION NUMBERS
Mitochondrial scaffolds, plastomes, and DNA‐seq and RNA‐seq data are deposited in GenBank and SRA databases under the following accession numbers: LC712386–LC712389, LC649831–LC649858, LC649586–LC649611, LC650159–LC650199, LC651066–LC651106, LC650058–LC650085, LC649230–LC649233, LC649399–LC649405, LC649584, LC649585, LC649896–LC649917, LC650964–LC651026, SRR15616203–SRR15616212, and SRR15647520–SRR15647529.
Supporting information
Figure S1. Stacked bars depicting the proportion of mitochondrial RNA editing at the first, second, and third codon positions. (A) All editing sites. (B) Non‐synonymous editing sites. (C) Synonymous editing sites.
Figure S2. Bar charts comparing the biochemical nature of amino acids encoded by edited codons before and after C‐to‐U editing. Overall increases of protein hydrophobicity in the gymnosperm mitochondria are supported by decreased hydrophilic codons (from 55.3% to 13.1%) but increased hydrophobic codons (from 44.7% to 86.9%) after RNA editing.
Figure S3. Bar charts illustrating proportions of the editing sites classified into nine bins of editing efficiency. Note that several synonymous sites are edited with more than 70% efficiency, resulting in a clear bimodal distribution in Dioon, Zamia, Pseudotsuga, Keteleeria, and Cephalotaxus.
Figure S4. Scatter plots depicting editing efficiency changes across 10 editing conservation levels. The editing efficiency data for 100% conserved synonymous editing sites are missing because none of the synonymous editing sites is shared among all studied gymnosperm taxa.
Figure S5. Genomic nucleotide types at the sites whose C‐to‐U RNA editing capability was lost along the tree branches leading to the 10 sampled gymnosperm taxa.
Figure S6. Scatter plots showing editing efficiency (y‐axis) versus expression level (x‐axis) in gymnosperm mitochondrial genes. Genes are labeled with their names if their editing efficiency is in the top three. Black lines denote regression between editing efficiency and gene expression level.
Figure S7. Phylogenetic placements and diversification of 899 E+‐subclass PPR proteins from 10 sampled gymnosperm taxa. Lineage‐specific duplications mentioned in the main text are highlighted with colored background.
Figure S8. Phylogenetic placements and diversification of 264 E1‐subclass PPR proteins from 10 sampled gymnosperm taxa. Lineage‐specific duplications mentioned in the main text are highlighted with colored background.
Figure S9. Phylogenetic placements and diversification of 835 E2‐subclass PPR proteins from 10 sampled gymnosperm taxa. Lineage‐specific duplications mentioned in the main text are highlighted with colored background.
Figure S10. Phylogenetic placements and diversification of 794 DYW‐subclass PPR proteins from 10 sampled gymnosperm taxa. Lineage‐specific duplications mentioned in the main text are highlighted with colored background.
Figure S11. Mitochondrial RSCU values showing a highly significant difference between U‐ and C‐ending codons in all sampled gymnosperm taxa. Horizontal lines within boxes represent medians. ‘**’ indicates a highly significant difference (P < 0.01). RSCU, relative synonymous codon usage.
Table S1. Sequenced taxa and their voucher information.
Table S2. Characteristics of plastomes and mitogenomic scaffolds elucidated in this study.
Table S3. Mitochondrial and plastid C‐to‐U RNA editing sites detected in the examined gymnosperm taxa.
Table S4. PPR proteins identified in this study.
ACKNOWLEDGMENTS
We thank Taipei Botanical Garden for providing plant materials. This study was supported by research grants from the Ministry of Science and Technology, Taiwan (106‐2311‐B‐001‐005) and Biodiversity Research Center, Academia Sinica, Taiwan. We are indebted to the editor and the two anonymous reviewers for helpful and suggestive comments.
REFERENCES
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A.A. , Dvorkin, M. , Kulikov, A.S. et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19, 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentolila, S. , Elliott, L.E. & Hanson, M.R. (2008) Genetic architecture of mitochondrial editing in Arabidopsis thaliana. Genetics, 178, 1693–1708. 10.1534/genetics.107.073585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentolila, S. , Oh, J. , Hanson, M.R. & Bukowski, R. (2013) Comprehensive high‐resolution analysis of the role of an Arabidopsis gene family in RNA editing. PLoS Genetics, 9, e1003584. 10.1371/journal.pgen.1003584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkholm, P. , Harish, A. , Hagström, E. , Ernst, A.M. & Andersson, S.G. (2015) Mitochondrial genomes are retained by selective constraints on protein targeting. Proceedings of the National Academy of Sciences of the United States of America, 112, 10154–10161. 10.1073/pnas.1421372112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. & Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, J.‐H. & Tong, L. (2012) Mitochondrial poly(a) polymerase and polyadenylation. Biochimica et Biophysica Acta, 1819, 992–997. 10.1016/j.bbagrm.2011.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaw, S.M. , Parkinson, C.L. , Cheng, Y. , Vincent, T.M. & Palmer, J.D. (2000) Seed plant phylogeny inferred from all three plant genomes: monophyly of extant gymnosperms and origin of Gnetales from conifers. Proceedings of the National Academy of Sciences of the United States of America, 97, 4086–4091. 10.1073/pnas.97.8.4086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T.C. , Su, Y.Y. , Wu, C.H. , Liu, Y.C. , Huang, C.H. & Chang, C.C. (2020) Analysis of mitochondrial genomics and transcriptomics reveal abundant RNA edits and differential editing status in moth orchid, Phalaenopsis aphrodite subsp. formosana . Scientia Horticulturae, 267, 109304. 10.1016/j.scienta.2020.109304 [DOI] [Google Scholar]
- Cheng, S. , Gutmann, B. , Zhong, X. , Ye, Y. , Fisher, M.F. , Bai, F. et al. (2016) Redefining the structural motifs that determine RNA binding and RNA editing by pentatricopeptide repeat proteins in land plants. The Plant Journal, 85, 532–547. 10.1111/tpj.13121 [DOI] [PubMed] [Google Scholar]
- Covello, P.S. & Gray, M.W. (1993) On the evolution of RNA editing. Trends in Genetics, 9, 265–268. 10.1016/0168-9525(93)90011-6 [DOI] [PubMed] [Google Scholar]
- Csűös, M. (2010) Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics, 26, 1910–1912. 10.1093/bioinformatics/btq315 [DOI] [PubMed] [Google Scholar]
- Daley, D.O. & Whelan, J. (2005) Why genes persist in organelle genomes. Genome Biology, 6, 110. 10.1186/gb-2005-6-5-110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, S. , Zhao, C. , Zhang, S. , Wu, H. , Mu, W. , Wei, T. et al. (2019) The amount of RNA edited sites in liverwort organellar genes is correlated with GC content and nuclear PPR protein diversity. Genome Biology and Evolution, 11, 3233–3239. 10.1093/gbe/evz232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edera, A.A. , Gandini, C.L. & Sanchez‐Puerta, M.V. (2018) Towards a comprehensive picture of C‐to‐U RNA editing sites in angiosperm mitochondria. Plant Molecular Biology, 97, 215–231. 10.1007/s11103-018-0734-9 [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 19, 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, W. , Guo, W. , Funk, L. , Mower, J.P. & Zhu, A. (2019) Complete loss of RNA editing from the plastid genome and most highly expressed mitochondrial genes of Welwitschia mirabilis . Science China. Life Sciences, 62, 498–506. 10.1007/s11427-018-9450-1 [DOI] [PubMed] [Google Scholar]
- Fu, L. , Niu, B. , Zhu, Z. , Wu, S. & Li, W. (2012) CD‐HIT: accelerated for clustering the next‐generation sequencing data. Bioinformatics, 28, 3150–3152. 10.1093/bioinformatics/bts565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii, S. & Small, I. (2011) The evolution of RNA editing and pentatricopeptide repeat genes. The New Phytologist, 191, 37–47. 10.1111/j.1469-8137.2011.03746.x [DOI] [PubMed] [Google Scholar]
- Gagliardi, D. & Gualberto, J.M. (2004) Gene expression in higher plant mitochondria. In: Day, D.A. , Millar, A.H. & Whelan, J. (Eds.) Plant mitochondria: from genome to function. Dordrecht, The Netherlands: Springer Science and Business Media LLC, pp. 55–82. [Google Scholar]
- Giegé, P. & Brennicke, A. (1999) RNA editing in Arabidopsis mitochondria effects 441 C to U changes in ORFs. Proceedings of the National Academy of Sciences of the United States of America, 96, 15324–15329. 10.1073/pnas.96.26.15324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. , Levin, J.Z. , Thompson, D.A. & Amit, I. (2011) Full‐length transcriptome assembly from RNA‐seq data without a reference genome. Nature Biotechnology, 29, 644–652. 10.1038/nbt.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafen, A. (1989) The phylogenetic regression. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 326, 119–157. 10.1098/rstb.1989.0106 [DOI] [PubMed] [Google Scholar]
- Gray, M.W. (2012) Evolutionary origin of RNA editing. Biochemistry, 51, 5235–5242. 10.1021/bi300419r [DOI] [PubMed] [Google Scholar]
- Grewe, F. , Edger, P.P. , Keren, I. , Sultan, L. , Pires, J.C. , Ostersetzer‐Biran, O. et al. (2014) Comparative analysis of 11 Brassicales mitochondrial genomes and the mitochondrial transcriptome of Brassica oleracea. Mitochondrion, 19, 135–143. 10.1016/j.mito.2014.05.008 [DOI] [PubMed] [Google Scholar]
- Grewe, F. , Herres, S. , Viehöver, P. , Polsakiewicz, M. , Weisshaar, B. & Knoop, V. (2011) A unique transcriptome: 1782 positions of RNA editing alter 1406 codon identities in mitochondrial mRNAs of the lycophyte Isoetes engelmannii. Nucleic Acids Research, 39, 2890–2902. 10.1093/nar/gkq1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes, B.T. , Sisay, A.K. , Carroll, H.D. & Cahoon, A.B. (2014) Deep sequencing of the tobacco mitochondrial transcriptome reveals expressed ORFs and numerous editing sites outside coding regions. BMC Genomics, 15, 31. 10.1186/1471-2164-15-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, W. , Zhu, A. , Fan, W. , Adams, R.P. & Mower, J.P. (2020) Extensive shifts from cis‐ to trans‐splicing of gymnosperm mitochondrial introns. Molecular Biology and Evolution, 37, 1615–1620. 10.1093/molbev/msaa029 [DOI] [PubMed] [Google Scholar]
- Gutmann, B. , Royan, S. , Schallenberg‐Rüdinger, M. , Lenz, H. , Castleden, I.R. , McDowell, R. et al. (2020) The expansion and diversification of pentatricopeptide repeat RNA‐editing factors in plants. Molecular Plant, 13, 215–230. 10.1016/j.molp.2019.11.002 [DOI] [PubMed] [Google Scholar]
- Hao, W. , Liu, G. , Wang, W. , Shen, W. , Zhao, Y. , Sun, J. et al. (2021) RNA editing and its roles in plant organelles. Frontiers in Genetics, 12, 757109. 10.3389/fgene.2021.757109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama, T. , Matsuura, T. , Ushiyama, S. , Narusaka, M. , Kurihara, Y. , Yasuda, M. et al. (2013) A poly(a)‐specific ribonuclease directly regulates the poly(a) status of mitochondrial mRNA in Arabidopsis . Nature Communications, 4, 2247. 10.1038/ncomms3247 [DOI] [PubMed] [Google Scholar]
- Ichinose, M. & Sugita, M. (2016) RNA editing and its molecular mechanism in plant organelles. Genes, 8, 5. 10.3390/genes8010005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, J.J. , Yu, W.B. , Yang, J.B. , Song, Y. , de Pamphilis, C.W. , Yi, T.S. et al. (2020) GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biology, 21, 241. 10.1186/s13059-020-02154-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, I.G. & Williams, B.P. (2016) Evolutionary inference across eukaryotes identifies specific pressures favoring mitochondrial gene retention. Cell Systems, 2, 101–111. 10.1016/j.cels.2016.01.013 [DOI] [PubMed] [Google Scholar]
- Kan, S.‐L. , Shen, T.‐T. , Gong, P. , Ran, J.‐H. & Wang, X.‐Q. (2020) The complete mitochondrial genome of Taxus cuspidata (Taxaceae): eight protein‐coding genes have transferred to the nuclear genome. BMC Evolutionary Biology, 20, 10. 10.1186/s12862-020-1582-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan, S.‐L. , Shen, T.‐T. , Ran, J.‐H. & Wang, X.‐Q. (2021) Both conifer II and Gnetales are characterized by a high frequency of ancient mitochondrial gene transfer to the nuclear genome. BMC Biology, 19, 146. 10.1186/s12915-021-01096-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. & Salzberg, S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology, 14, R36. 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knie, N. , Grewe, F. , Fischer, S. & Knoop, V. (2016) Reverse U‐to‐C editing exceeds C‐to‐U RNA editing in some ferns ‐ a monilophyte‐wide comparison of chloroplast and mitochondrial RNA editing suggests independent evolution of the two processes in both organelles. BMC Evolutionary Biology, 16, 134. 10.1186/s12862-016-0707-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolosova, N. , Miller, B. , Ralph, S. , Ellis, B.E. , Douglas, C. , Ritland, K. et al. (2004) Isolation of high‐quality RNA from gymnosperm and angiosperm trees. BioTechniques, 36, 821–824. 10.2144/04365ST06 [DOI] [PubMed] [Google Scholar]
- Levin, J.Z. , Yassour, M. , Adiconis, X. , Nusbaum, C. , Thompson, D.A. , Friedman, N. et al. (2010) Comprehensive comparative analysis of strand‐specific RNA sequencing methods. Nature Methods, 7, 709–715. 10.1038/s41592-018-0014-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Xia, L. , Zhang, Y. , Niu, G. , Li, M. , Wang, P. et al. (2019) Plant editosome database: a curated database of RNA editosome in plants. Nucleic Acids Research, 47, D170–D174. 10.1093/nar/gky1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna, S. (2015) An overview of pentatricopeptide repeat proteins and their applications. Biochimie, 113, 93–99. 10.1016/j.biochi.2015.04.004 [DOI] [PubMed] [Google Scholar]
- Mower, J.P. (2008) Modeling sites of RNA editing as a fifth nucleotide state reveals progressive loss of edited sites from angiosperm mitochondria. Molecular Biology and Evolution, 25, 52–61. 10.1093/molbev/msm226 [DOI] [PubMed] [Google Scholar]
- Picardi, E. , Horner, D.S. , Chiara, M. , Schiavon, R. , Valle, G. & Pesole, G. (2010) Large‐scale detection and analysis of RNA editing in grape mtDNA by RNA deep‐sequencing. Nucleic Acids Research, 38, 4755–4767. 10.1093/nar/gkq202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, M.N. , Dehal, P.S. & Arkin, A.P. (2010) FastTree 2‐‐approximately maximum‐likelihood trees for large alignments. PLoS One, 5, e9490. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, J.H. , Gao, H. & Wang, X.Q. (2010) Fast evolution of the retroprocessed mitochondrial rps3 gene in conifer II and further evidence for the phylogeny of gymnosperms. Molecular Phylogenetics and Evolution, 54, 136–149. 10.1016/j.ympev.2009.09.011 [DOI] [PubMed] [Google Scholar]
- Richardson, A.O. , Rice, D.W. , Young, G.J. , Alverson, A.J. & Palmer, J.D. (2013) The "fossilized" mitochondrial genome of Liriodendron tulipifera: ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biology, 11, 29. 10.1186/1741-7007-11-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikanai, T. (2015) RNA editing in plants: machinery and flexibility of site recognition. Biochimica et Biophysica Acta, 1847, 779–785. 10.1016/j.bbabio.2014.12.010 [DOI] [PubMed] [Google Scholar]
- Sievers, F. , Wilm, A. , Dineen, D. , Gibson, T.J. , Karplus, K. , Li, W. et al. (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal omega. Molecular Systems Biology, 7, 539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan, D.B. , MacQueen, A.H. , Alverson, A.J. , Palmer, J.D. & Taylor, D.R. (2010) Extensive loss of RNA editing sites in rapidly evolving Silene mitochondrial genomes: selection vs. retroprocessing as the driving force. Genetics, 185, 1369–1380. 10.1534/genetics.110.118000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, I.D. & Peeters, N. (2000) The PPR motif—a TPR‐related motif prevalent in plant organellar proteins. Trends in Biochemical Sciences, 25, 45–47. 10.1016/s0968-0004(99)01520-0 [DOI] [PubMed] [Google Scholar]
- Small, I.D. , Schallenberg‐Rüdinger, M. , Takenaka, M. , Mireau, H. & Ostersetzer‐Biran, O. (2020) Plant organellar RNA editing: what 30 years of research has revealed. The Plant Journal, 101, 1040–1056. 10.1111/tpj.14578 [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, C.N. & Via, L.E. (1993) A rapid CTAB DNA isolation technique useful for RAPD fingerprinting and other PCR applications. BioTechniques, 14, 748–750. [PubMed] [Google Scholar]
- Stone, J.D. & Storchova, H. (2015) The application of RNA‐seq to the comprehensive analysis of plant mitochondrial transcriptomes. Molecular Genetics and Genomics, 290, 1–9. 10.1007/s00438-014-0905-6 [DOI] [PubMed] [Google Scholar]
- Sun, T. , Bentolila, S. & Hanson, M.R. (2016) The unexpected diversity of plant organelle RNA editosomes. Trends in Plant Science, 21, 962–973. 10.1016/j.tplants.2016.07.005 [DOI] [PubMed] [Google Scholar]
- Takenaka, M. , Jörg, A. , Burger, M. , Haag, S. et al. (2018) Requirement of various protein combinations for each C‐to‐U RNA editosome in plant organelles. In: Cruz‐Reyes, J. & Gray, M. (Eds.) RNA Metabolism in Mitochondria, Nucleic Acids and Molecular Biology 34. Cham, Switzerland: Springer International Publishing AG, pp. 223–249. [Google Scholar]
- Takenaka, M. , Zehrmann, A. , Verbitskiy, D. , Härtel, B. & Brennicke, A. (2013) RNA editing in plants and its evolution. Annual Review of Genetics, 47, 335–352. 10.1146/annurev-genet-111212-133519 [DOI] [PubMed] [Google Scholar]
- Verbitskiy, D. , Takenaka, M. , Neuwirt, J. , van der Merwe, J.A. & Brennicke, A. (2006) Partially edited RNAs are intermediates of RNA editing in plant mitochondria. The Plant Journal, 47, 408–416. 10.1111/j.1365-313X.2006.02794.x [DOI] [PubMed] [Google Scholar]
- von Heijne, G. (1986) Why mitochondria need a genome. FEBS Letters, 198, 1–4. 10.1016/0014-5793(86)81172-3 [DOI] [PubMed] [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. et al. (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One, 9, e112963. 10.1371/journal.pone.0112963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , An, Y. , Xu, P. & Xiao, J. (2021) Functioning of PPR proteins in organelle RNA metabolism and chloroplast biogenesis. Frontiers in Plant Science, 12, 627501. 10.3389/fpls.2021.627501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C.S. , Sudianto, E. & Chaw, S.M. (2021) Tight association of genome rearrangements with gene expression in conifer plastomes. BMC Plant Biology, 21, 33. 10.1186/s12870-020-02809-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, C.S. , Wang, Y.N. , Hsu, C.Y. , Lin, C.P. & Chaw, S.M. (2011) Loss of different inverted repeat copies from the chloroplast genomes of Pinaceae and cupressophytes and influence of heterotachy on the evaluation of gymnosperm phylogeny. Genome Biology and Evolution, 3, 1284–1295. 10.1093/gbe/evr095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, Z. , Stone, J.D. , Štorchová, H. & Sloan, D.B. (2015) High transcript abundance, RNA editing, and small RNAs in intergenic regions within the massive mitochondrial genome of the angiosperm Silene noctiflora. BMC Genomics, 16, 938. 10.1186/s12864-015-2155-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehrmann, A. , van der Merwe, J.A. , Verbitskiy, D. , Brennicke, A. & Takenaka, M. (2008) Seven large variations in the extent of RNA editing in plant mitochondria between three ecotypes of Arabidopsis thaliana. Mitochondrion, 8, 319–327. 10.1016/j.mito.2008.07.003 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Stacked bars depicting the proportion of mitochondrial RNA editing at the first, second, and third codon positions. (A) All editing sites. (B) Non‐synonymous editing sites. (C) Synonymous editing sites.
Figure S2. Bar charts comparing the biochemical nature of amino acids encoded by edited codons before and after C‐to‐U editing. Overall increases of protein hydrophobicity in the gymnosperm mitochondria are supported by decreased hydrophilic codons (from 55.3% to 13.1%) but increased hydrophobic codons (from 44.7% to 86.9%) after RNA editing.
Figure S3. Bar charts illustrating proportions of the editing sites classified into nine bins of editing efficiency. Note that several synonymous sites are edited with more than 70% efficiency, resulting in a clear bimodal distribution in Dioon, Zamia, Pseudotsuga, Keteleeria, and Cephalotaxus.
Figure S4. Scatter plots depicting editing efficiency changes across 10 editing conservation levels. The editing efficiency data for 100% conserved synonymous editing sites are missing because none of the synonymous editing sites is shared among all studied gymnosperm taxa.
Figure S5. Genomic nucleotide types at the sites whose C‐to‐U RNA editing capability was lost along the tree branches leading to the 10 sampled gymnosperm taxa.
Figure S6. Scatter plots showing editing efficiency (y‐axis) versus expression level (x‐axis) in gymnosperm mitochondrial genes. Genes are labeled with their names if their editing efficiency is in the top three. Black lines denote regression between editing efficiency and gene expression level.
Figure S7. Phylogenetic placements and diversification of 899 E+‐subclass PPR proteins from 10 sampled gymnosperm taxa. Lineage‐specific duplications mentioned in the main text are highlighted with colored background.
Figure S8. Phylogenetic placements and diversification of 264 E1‐subclass PPR proteins from 10 sampled gymnosperm taxa. Lineage‐specific duplications mentioned in the main text are highlighted with colored background.
Figure S9. Phylogenetic placements and diversification of 835 E2‐subclass PPR proteins from 10 sampled gymnosperm taxa. Lineage‐specific duplications mentioned in the main text are highlighted with colored background.
Figure S10. Phylogenetic placements and diversification of 794 DYW‐subclass PPR proteins from 10 sampled gymnosperm taxa. Lineage‐specific duplications mentioned in the main text are highlighted with colored background.
Figure S11. Mitochondrial RSCU values showing a highly significant difference between U‐ and C‐ending codons in all sampled gymnosperm taxa. Horizontal lines within boxes represent medians. ‘**’ indicates a highly significant difference (P < 0.01). RSCU, relative synonymous codon usage.
Table S1. Sequenced taxa and their voucher information.
Table S2. Characteristics of plastomes and mitogenomic scaffolds elucidated in this study.
Table S3. Mitochondrial and plastid C‐to‐U RNA editing sites detected in the examined gymnosperm taxa.
Table S4. PPR proteins identified in this study.