

Abstract
A thickened, white patch — leukoplakia — in the oral cavity is usually benign, but sometimes (in ~9% of individuals) it progresses to malignant tumour. Because the genomic basis of this progression is poorly understood, we undertook this study and collected samples of four tissues — leukoplakia, tumour, adjacent normal, and blood — from each of 28 patients suffering from gingivobuccal oral cancer. We performed multiomics analysis of the 112 collected tissues (four tissues per patient from 28 patients) and integrated information on progressive changes in the mutational and transcriptional profiles of each patient to create this genomic narrative. Additionally, we generated and analysed whole‐exome sequence data from leukoplakia tissues collected from 11 individuals not suffering from oral cancer. Nonsynonymous somatic mutations in the CASP8 gene were identified as the likely events to initiate malignant transformation, since these were frequently shared between tumour and co‐occurring leukoplakia. CASP8 alterations were also shown to enhance expressions of genes that favour lateral spread of mutant cells. During malignant transformation, additional pathogenic mutations are acquired in key genes (TP53, NOTCH1, HRAS) (41% of patients); chromosomal‐instability (arm‐level deletions of 19p and q, focal‐deletion of DNA‐repair pathway genes and NOTCH1, amplification of EGFR) (77%), and increased APOBEC‐activity (23%) are also observed. These additional alterations were present singly (18% of patients) or in combination (68%). Some of these alterations likely impact immune‐dynamics of the evolving transformed tissue; progression to malignancy is associated with immune suppression through infiltration of regulatory T‐cells (56%), depletion of cytotoxic T‐cells (68%), and antigen‐presenting dendritic cells (72%), with a concomitant increase in inflammation (92%). Patients can be grouped into three clusters by the estimated time to development of cancer from precancer by acquiring additional mutations (range: 4–10 years). Our findings provide deep molecular insights into the evolutionary processes and trajectories of oral cancer initiation and progression. © 2022 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of The Pathological Society of Great Britain and Ireland.
Keywords: leukoplakia, oral cancer, CASP8, pathogenic mutation, progression, immune suppression
Introduction
Globally, oral squamous cell carcinoma (OSCC) is the 16th most common malignancy and causes high levels of mortality in India; ranking highest among men, and sixth among women [1]. OSCC is highly prevalent in East Asia and the Indian subcontinent, where tobacco chewing is popular [1, 2]. Although early detection and treatment can result in longer disease‐free survival [3], OSCC patients in these regions are usually diagnosed at advanced stages with poor disease‐free prognosis and low overall survival after treatment [2]. Epithelial cancers are known to progress from precancerous lesions [4, 5]. Leukoplakia, mostly dysplastic, white, thick tissue often present in the oral cavity, serves as a precursor to oral cancer [6, 7] in a fraction (~9%) of patients [8]. Prolonged chewing of tobacco damages DNA and results in protumorigenic mutations in oral epithelial cells [9, 10]. We posit that epithelial cells that acquire specific genomic alterations develop tissue dysplasia that sometimes becomes neoplastic with acquisition of additional genomic changes. Among oral cancer patients, recurrent mutations and copy number variations in several cancer driver genes, some with large‐scale genome alterations [2, 11], APOBEC enzymatic activity [12], and immune cell dysregulation [13] are often detected. Only a fraction of individuals with leukoplakia eventually develop oral cancer [8]. Individuals with leukoplakia are usually clinically examined for progression every 6 or 12 months [14]. Although multiple loss of heterozygosity (LOH) loci have been identified [15], biomarkers based on genome‐wide studies have not been identified to predict malignant transformation in an individual with oral premalignant lesions.
We undertook this study to gain insights into the molecular events associated with malignant progression of leukoplakia to cancer (OSCC) that may help identify biomarkers to guide clinical management. We also sought to obtain evidence of a sequential accumulation of genomic alterations during the evolution of normal to malignant tissue in the oral cavity.
Earlier studies on oral cancer with similar objectives have the indicated involvement of TP53 mutation [16, 17, 18], amplification, and deletion of specific chromosomal arms [19, 20]. These studies, however, were based on small sample sizes of independent sets of patients with leukoplakia and cancer, and lacked a genome‐wide approach. The strength of our study is that we recruited oral cancer patients with concomitant leukoplakia and collected and carried out a genome‐wide search for alterations from normal to premalignant and malignant tissues in each patient.
Among OSCC patients in India, the most frequently (~60%) affected site is the gingivobuccal region of the oral cavity, comprising buccal mucosa, retro‐molar trigone, and the lower gum [2]. We therefore focused on this subset (OSCC‐GB) of OSCC patients. We collected multiple tissue samples from tumour, leukoplakia, adjacent normal and blood from OSCC‐GB patients, and performed DNA and RNA sequencing. We adopted a multiomics approach in identifying recurrent somatic genomic alterations associated with progression from leukoplakia to OSCC‐GB, by analysing tissues from only patients with a concomitant presence of leukoplakia and malignant tumour.
Materials and methods
Patient recruitment, characteristics, and sample acquisition
Our study was approved by the Institutional Ethics Committees of all collaborating institutions in India: Dr. R. Ahmed Dental College & Hospital, Kolkata (RADCH); the Indian Statistical Institute (ISI), Kolkata; and the National Institute of Biomedical Genomics (NIBMG), Kalyani. Data and biospecimens were collected with voluntary informed consent, including consent to publish. From RADCH, treatment‐naive OSCC‐GB patients (n = 28; 21 men and 7 women; age range: 32–70 years; median age: 53 years) with the concomitant presence of pathological dysplastic lesion (clinically reported as leukoplakia) and malignant lesion in the gingivobuccal region of the oral cavity were recruited. Self‐reported information on consumption of tobacco (100%) and alcohol (14.3%), and other relevant variables were collected (supplementary material, Table S1). For each patient, the clinician recorded precise locations of sampling of tissue during surgery from the tumour, leukoplakia, and adjacent normal (all tissue samples were collected at the same time for a given patient). Representative fresh tissue sections from tumour core, leukoplakia (whitish keratinised lesion with dysplasia) and adjacent normal (from the opposite cheek) were surgically microdissected, collected in RNAlater (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), and stored as per recommended protocols for DNA and RNA sequencing. Sections of collected tissues were formalin‐fixed and characterised histopathologically after hematoxylin and eosin (H&E) staining to determine grades of dysplasia and invasiveness. DNA and RNA were isolated from the stored blood samples and tissues. Additionally, we recruited 11 patients who were diagnosed with leukoplakia, but without any oral tumour. From each of them, we collected blood samples and leukoplakia tissue. DNA was isolated from these samples.
Whole‐exome sequence data generation and detection of somatic and germline alterations
Whole‐exome sequencing (WES) was performed for blood, leukoplakia, and tumour tissue samples using Illumina HiSeq‐2500 at ≥100× depth of coverage (supplementary material, Table S2) as described in the Supplementary materials and methods. An approach previously published by us [2, 11] was used to identify somatic and germline single nucleotide alterations and somatic copy number alterations (sCNA) from WES data (see Supplementary materials and methods for details). Briefly a suite of packages was used for quality control (QC), alignment, and postalignment processing that includes FastQC [21], BWA‐MEM [22], PICARD [23], and GATK [24] tools. Somatic variant calling was independently performed using four variant callers, i.e. Mutect2 [24], MuSE [25], strelka‐2 [26], and BaseByBase‐variant‐caller [2]; somatic variants detected by at least two of the four callers were accepted for further analysis (details described in Supplementary materials and methods). Variants were filtered for sequencing artefacts (e.g. low‐frequency, strand bias, Oxo‐G, low‐complexity‐regions, etc.) and polymorphic germline loci (in 1000G [27] and Genomeasia100K [28]) to get a high‐confidence somatic variant set. Further somatic mutations in known cancer driver genes were manually verified by IGV [29] and validated from the available RNAseq data. Arm‐level and focal sCNAs were detected using GISTIC [30] and ASCAT [31] algorithms. Mutational signatures were estimated from data on somatic single nucleotide variants of tumour and leukoplakia lesions, by Signal Analyse (v. 2) [32] package (https://signal.mutationalsignatures.com/analyse2) with 100 bootstraps and a p value cutoff of 0.05 for signature fitting. Further, a method previously described by Noë et al [33] was utilised to estimate progression time of leukoplakia to a malignant tumour from information on the number of additional nonsynonymous mutations acquired in tumour than leukoplakia of the same individual. Germline variants for each patient were detected using the GATK HaplotypeCaller joint calling algorithm. Annotation and classification of variants were obtained using ANNOVAR [34] and Oncotator [35], respectively. Methodological details of the above‐mentioned steps (tools, parameters, databases used) are provided in the Supplementary materials and methods.
Whole transcriptome sequence data generation, expression quantification, and identification of differentially expressed genes
Whole transcriptome sequence data on tumour, leukoplakia, and adjacent normal tissue samples were performed to obtain ~100 million reads (supplementary material, Table S3) (see Supplementary materials and methods for details). RNA sequence data were analysed as per GDC recommendations (https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/). Briefly, FastQC, STAR [36] and HTSeq [37] packages were used for QC, alignment, and gene expression quantification. The method encoded in the DESeq2 [38] package was used to detect differentially expressed genes between various comparison groups. Additional details of methodology of analysis are provided in the Supplementary materials and methods.
Deconvolution of immune cell composition from bulk RNA sequence data
The immune cell compositions in normal oral tissue, leukoplakia, and oral tumour were deconvoluted separately for each patient (n = 25, for whom RNAseq data were available) using CIBERSORT [39] with LM22 signature genes (provided with CIBERSORT) and 100 permutations (tool default) for the statistical analysis. Further, findings on the abundance of CD8+ T cell was validated using an orthogonal method encoded in EPIC [40]. The gene expression TPM (transcript per million) values for immune deconvolution analysis was computed using the method included with TPMCalculator [41] package.
Results
Number and characteristics of somatic mutations correlate with distance between tumour and leukoplakia lesion
WES data, at ~100X coverage, of DNA from blood, leukoplakia, and tumour tissues of 28 patients (described in supplementary material, Table S1) were generated and analysed (Figure 1A). Histopathological characteristics of representative tissue sections are described in Figure 1B. A total of 2,355 somatic protein‐coding mutations were identified in tumour tissues and 1,237 in leukoplakia tissues (supplementary material, Tables S4 and S5). The mean number of somatic mutations in tumour (84.1; range: 10–248) was significantly higher (p < 0.01, t‐test for equality of means) than in leukoplakia tissue of the same patient (mean = 44.2; range: 3–164) (Figure 1C). Somatic mutational landscapes of tumour and leukoplakia are depicted in Figure 2A–C. Among somatic alterations, frameshift deletion was significantly more frequent (p < 0.02, Kolmogorov–Smirnov test) in tumour (range: 0–10.53%, mean = 3.13%) than in leukoplakia (0–5.26%, mean = 1.28%) (Figure 1D). Overall, a significantly higher (p < 0.0004, t‐test for equality of means) somatic mutation rate per Mb was observed in tumour (0.14–8.84/Mb, mean = 2.88/Mb) than in leukoplakia (0.17–5.06/Mb, mean = 1.21/Mb); the rate was even lower (0.03–0.89/Mb, mean = 0.39/Mb) in leukoplakia without the presence of tumour in the oral cavity (n = 11, additionally recruited patients; supplementary material, Table S6).
Figure 1.
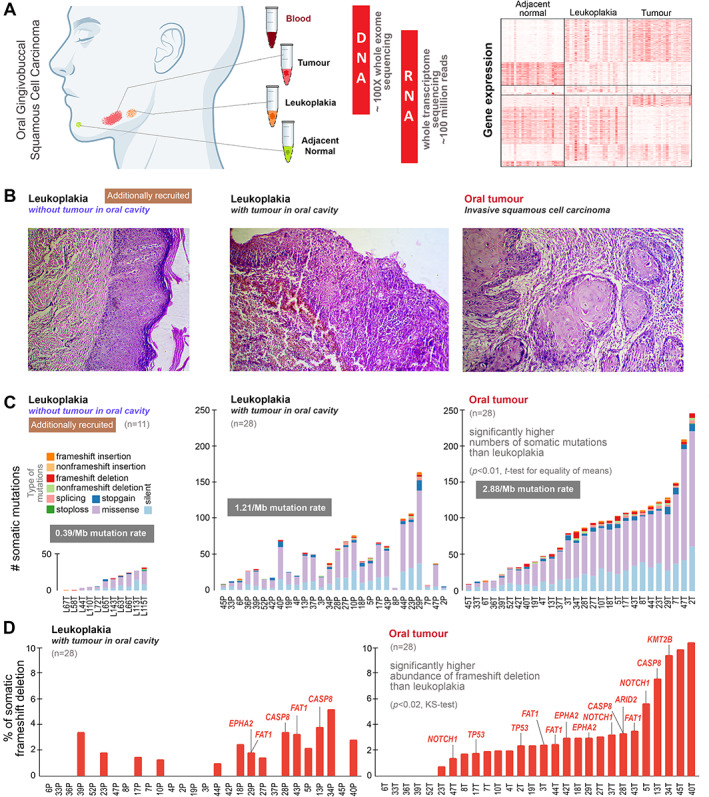
(A) To understand the genomic underpinnings of malignant progression of oral potentially malignant lesion (leukoplakia), oral cancer patients were recruited in whom there was the presence of leukoplakia with cancer. From each patient, tissue samples from tumour, leukoplakia, adjacent normal, and blood were collected. Good‐quality DNA was extracted from tumour, leukoplakia, and blood and sequenced for whole‐exome at about 100X depth; RNA was extracted from tumour, leukoplakia, and adjacent normal tissue and whole transcriptome sequence data were generated (the right‐hand portion represents mRNA expression patterns of differentially expressed genes in leukoplakia and tumour with respect to adjacent normal tissue, described in the supplementary information). (B) Additionally, we recruited 11 individuals diagnosed having oral leukoplakia without the presence of oral tumour. The left‐hand‐side H&E‐stained image showing a leukoplakia lesion in squamous epithelium with features of dysplasia in patients without the presence of tumour in the oral cavity. The middle image represents the histopathology of leukoplakia with the concomitant presence of tumour within the oral cavity, revealing progression from mildly dysplastic epithelium to invasive squamous cell carcinoma with infiltrating dyscohesive neoplastic epithelial cells. And the right‐hand‐side image shows the histopathological features of invasive squamous cell carcinoma with invading neoplastic squamous cells islands in connective tissue stroma. (C) The numbers of somatic mutations in leukoplakia of 11 individuals without tumour in the oral cavity, and in both leukoplakia and tumour in 28 patients with concomitant leukoplakia and cancer are provided. The somatic mutation rate in oral tumours (2.88/Mb) was higher than in leukoplakia (1.21/Mb). The mean number of somatic mutations in tumours (84) was significantly higher than in leukoplakia lesions (44) of the same patient. Additionally, we generated whole‐exome sequence data from the oral leukoplakia tissue of 11 individuals without oral cancer, among whom the mean number of somatic mutations (14) was smaller. (D) The proportion of frameshift deletions in tumours was significantly higher than in leukoplakia tissue. Frameshift deletions in known oral cancer driver genes are provided for each patient.
Figure 2.
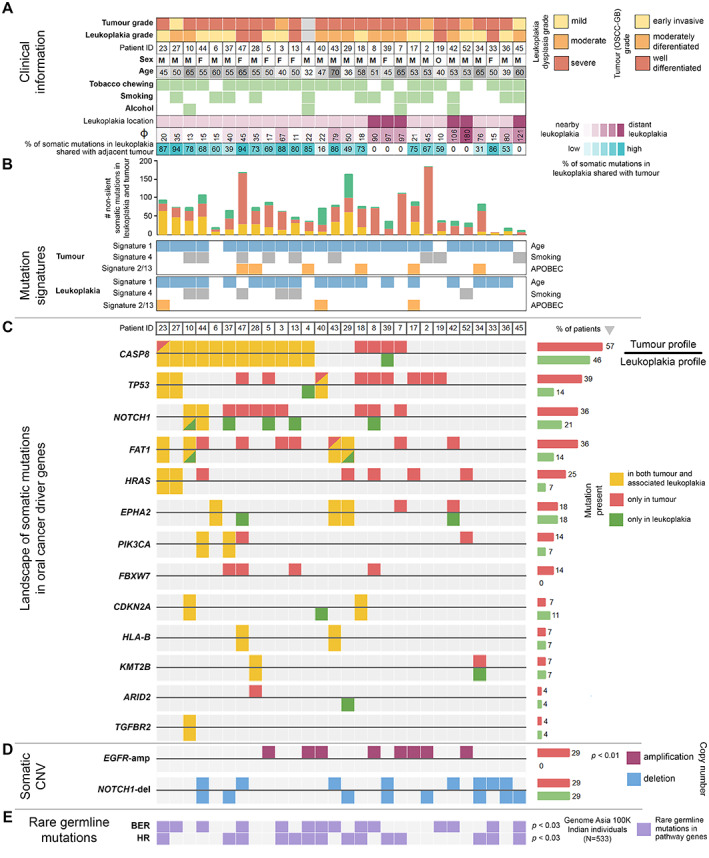
Mutational landscapes of OSCC‐GB patients with the concomitant presence of oral potentially malignant lesions. (A) Histopathological grade of tumour and leukoplakia, clinical information, and location of leukoplakia. (B) Somatic mutations identified in leukoplakia lesion and tumour were grouped as: (1) present in both leukoplakia and tumour, (2) present only in tumour, and (3) present only in leukoplakia. Although none of the mutation signatures associated with tumour age, tobacco smoking, or APOBEC activity showed a statistically significant difference between the leukoplakia lesion and tumour; for the APOBEC mutational signature the difference was high between the leukoplakia lesion (11%) and tumour (21%). The proportion of mutations in a leukoplakia lesion that was shared with the paired tumour was negatively correlated with the angular distance between leukoplakia and tumour (φ). Distant (φ > 80°) leukoplakia lesions (n = 6) did not share any somatic mutation with paired tumours. (C) Nonsynonymous somatic mutations were found in the known driver genes of oral cancer in tumour and leukoplakia. Recurrent somatic mutations in CASP8 were found to be shared among 43% of patients. (D) Amplification of EGFR was found only in the tumour tissue (29%), but not in the leukoplakia lesion. NOTCH1 was deleted in both tumours (29%) and the leukoplakia state (29%). (E) Rare germline mutations in DNA repair pathways —BER (Base Excision Repair) and HR (Homologous Recombination) — in these patients were significantly higher than in the normal healthy Indian population (GenomeAsia100K data).
From the diagrammatic records kept by the clinician on the physical locations of leukoplakia and tumour within the oral cavity, we measured the angular distance between tumour and leukoplakia as described in supplementary material, Figure S1A. Based on the angular distance (φ), which ranged from 10°–180° (median = 37.5°, supplementary material, Table S1), we categorised the patients into the following groups: (1) leukoplakia patch in proximity (n = 22, having φ ≤ 80° [i.e. below the upper quartile of φ distribution]; median = 22°; mean = 31.75°), and (2) leukoplakia patch at a distance (n = 6, remaining patients; median = 101.5°; mean = 115°). The mean φ for group‐2 was significantly larger (p < 0.001, t‐test for equality of means) than for group‐1. The proportion of mutations shared between leukoplakia and tumour decreased with an increase in the distance (φ) between their locations (Pearson's correlation coefficient, r = −0.72; p < 0.0001) (supplementary material, Table S7 and Figure S1B). The mean number of somatic mutations in the leukoplakia tissue was significantly higher (p < 0.001, t‐test for equality of means) if it was located in proximity to a tumour than if it was distantly located, although the mean number of mutations in the tumour was the same (p > 0.2, t‐test for equality of means) irrespective of the distance of the leukoplakia patch from the tumour.
Of the somatic mutations present in the leukoplakia of a patient with a tumour in proximity, a high proportion (median 73.3%; range 16.4–94.1% across patients) was shared with somatic mutations present in the tumour. No somatic mutation was shared between leukoplakia and tumour if these were distantly located (φ > 80°). The results of sharing of mutations are indicative of a proximal leukoplakia being a potentially malignant lesion that progressed to cancer. Each somatic mutation can be placed in one of three distinct classes: (1) shared between paired leukoplakia and tumour, (2) unique to tumour, and (3) unique to leukoplakia (Figure 2B). A somatic mutation that is shared between the leukoplakia and tumour is indicative of having arisen in the nonmalignant lesion and possibly early in tumorigenesis; unshared mutations are likely to have arisen independently and later. Variant allele frequency (VAF) can be used to assess the time of occurrence of somatic mutations in tumour evolution; the expectation is that a mutation that arose early will have a higher VAF and will be present in a larger number of cells than one that arose later [42, 43, 44]. The median VAFs of shared somatic mutations between leukoplakia and tumour located in proximity were similar or higher compared to mutations that were unique to the tumour in 91% (20 of 22) of patients, consistent with the expectation of early origins for shared mutations. It may be argued that somatic mutations are shared between the proximal leukoplakia and tumour tissues of a patient because the tissue surgically resected from the leukoplakia also contained some tumour tissue and is therefore ‘contaminated’. Using an approach adopted in a recent study [33] on malignant progression in neoplastic pancreatic cysts, we systematically investigated and excluded this possibility, as described in the Supplementary materials and methods. Further, analysis of available mRNA expression data also showed distinct gene expression patterns even in leukoplakia and tumour located in proximity within the same oral cavity (Figure 1A, supplementary material, Figures S2 and S3).
Mutations in the CASP8 gene are early events in oral cancer evolution
Nonsynonymous somatic mutations in all protein coding genes (~20,000 genes) were identified from WES data of tumour and leukoplakia tissues (28 patients). The mutational landscapes of oral cancer driver genes [2, 45, 46, 47] and additionally in the head and neck cancer driver genes [48] (FBXW7, TGFBR2) are described in supplementary material, Table S8 and Figure 2C. These mutations were validated using the RNA sequence data from tumour and leukoplakia tissue that were available for a subset of patients (n = 22). Somatic mutations identified from tumour and leukoplakia tissues were not detected from RNAseq data available for paired adjacent normal tissues. TP53, CASP8, FAT1, PIK3CA, NOTCH1, KMT2B, HRAS, ARID2, EPHA2, HLA‐B, and TGFBR2 genes were found mutated in both tumour and leukoplakia, while FBXW7 mutations were only detected in tumour samples (Figure 2C). The vast majority (71% on average) of somatic mutations present in driver genes in the leukoplakia tissue of a patient were also present in their tumour. We found CASP8 to be the most frequently mutated gene in these OSCC‐GB tumours. A high level of co‐occurrence of mutations, along with those in CASP8, were found in NOTCH1 (62.5%), TP53 (43.7%), and FAT1 (43.7%), while the level of co‐occurrence was moderate for HRAS (25%) and FBXW7 (25%). The same somatic mutation was shared between leukoplakia and tumour most frequently for the CASP8 gene (in 43% of patients; 55% among patients in whom the leukoplakia lesion was in proximity to the tumour), but not for the most recurrently mutated gene—TP53—in OSCC‐GB, for which the frequency of such sharing was only 14%. The presence of these CASP8 mutations in both tumour and leukoplakia is indicative of their early origin in leukoplakia, likely providing it the initial drive to malignancy. We also note that for 81.25% (n = 13 of 16) patients harbouring CASP8 mutations in their tumour, the VAF of the CASP8 mutation was greater than or equal to the median VAF of all somatic mutations (supplementary material, Table S9), providing further supportive evidence for their early origin during tumorigenesis. Most of the shared CASP8 mutations (83%) were highly deleterious (stopgain/indel/hotspot) and the remaining were predicted to be deleterious by multiple prediction algorithms. Most (81.25%) CASP8 mutations were found on the conserved protease domain (supplementary material, Figure S4). We confirmed that the evidence of most CASP8 mutations being confined to the protease domain was not a chance finding (see details in the Supplementary materials and methods). We reanalysed the data from multiple biopsies taken from an OSCC patient by Tabatabaeifar et al [49] (supplementary material, Table S10) and found high VAF (0.26; higher than 95% of all somatic mutations of the tumour core) of CASP8 protease domain stopgain mutations to be present in multiple tissue locations, including the tumour core. This is consistent with our finding of an early occurrence of deleterious mutations in CASP8 during oral cancer evolution.
These findings prompted us to further investigate the downstream transcriptional perturbations that are specifically associated with CASP8 mutation. Genes associated with cell survival, migration, and invasion (e.g. FABP4 [50], TNFRSF17 [51], FMOD [52], CALB2 [53], etc.) and with cellular stemness (e.g. ALDH1L2 [54], WNT10A [55], etc.) were significantly upregulated compared with adjacent normal tissue in both tumour and leukoplakia with mutated CASP8, but not in those without any CASP8 mutation (supplementary material, Table S11). These results, with supportive evidence from other studies [56, 57], indicate that deleterious CASP8 mutations in oral squamous cells may provide increased survival potential to cells by inactivating apoptosis. In turn, this may induce intraepithelial cell migration and facilitate the lateral spread of a protumorigenic layer of cells as an initiator event in oral tumour evolution, possibly inducing field cancerization.
APOBEC activity contributes to elevate the mutation rate, thereby promoting tumour development
An increase in the mutation rate in a nonmalignant tissue can promote the development of cancer. The cytidine deaminases of the APOBEC family deaminate cytosines in single‐stranded DNA and cause mutations in human cancers [58]. In 21.4% of the OSCC‐GB patients recruited by us, the APOBEC mutational signature (RefSig signatures 2 and 13) [32] was present in the tumour (Figure 2B). However, the presence of this signature was less frequent in leukoplakia (10.7%); Figure 2B. The mutation rate (number of somatic mutations per Mb of genome) was at least 2‐fold higher in tumour than in leukoplakia (n = 12), of which 42% (n = 5) had a tumour‐specific APOBEC mutational signature (supplementary material, Table S12). In the remaining patients (n = 10) with leukoplakia lesion in proximity to tumour, the mutation rate in tumour was not elevated compared to the leukoplakia; none of these patients possessed a tumour‐specific APOBEC mutation signature. The enzymatic activity of APOBEC3A and APOBEC3B has been shown to be responsible for inducing this specific type of mutational signature in multiple cancer types [32, 59, 60]. Compared to the normal tissue, both APOBEC3A (log2 fold‐change = 3.67, p false discovery rate [FDR] < 0.0001) and APOBEC3B (log2 fold‐change = 1.87, p[FDR] < 0.0001) genes were significantly overexpressed in tumours but not in leukoplakia tissues (log2 fold‐change = 0.92 and 0.63, p[FDR] > 0.1). Our in vivo results provide support for a recent in vitro study that provided evidence of APOBEC‐induced mutagenesis in head and neck squamous cell carcinoma (HNSCC) [12].
After initiation of tumorigenesis, pathogenic mutations are required for development of tumour
Among 72% of patients (n = 16 of 22), oral cancer driver gene mutations were shared between tumours and paired leukoplakia located in proximity (φ < 80°). However, all patients possessed additional mutations in one or more driver genes in their tumours, indicating that multiple driver mutations are required for eventual transformation to malignant tumour (Figures 2C and 3A). We have reconstructed the most probable trajectory of mutations in driver genes for development of a potentially malignant state and cancer in each patient (n = 28) and the proportional contributions of these genes to cellular transformation (Figure 3A), as estimated from VAFs of driver mutations. These trajectories showed complex patterns and histories of mutations in leukoplakia and tumour tissues in the patients. Among patients in whom the leukoplakia was located in proximity, 41% (n = 9) harboured at least two additional oral driver gene mutations in their tumours. Among the shared driver gene mutations, ~83% had higher VAF in tumours than in leukoplakia (Figure 3C), consistent with cells with these mutations clonally propagating rapidly to tumour (Figure 3A,C,D). Most pathogenic mutations (frameshift indels, stopgain, and hotspot mutations) in frequently mutated oral cancer driver genes, TP53, NOTCH1, and HRAS, were present almost exclusively in tumours but not in leukoplakia (Figure 3B); mutations in these genes in leukoplakia were predominantly less pathogenic (nonhotspot missense only) (Figure 3B).
Figure 3.
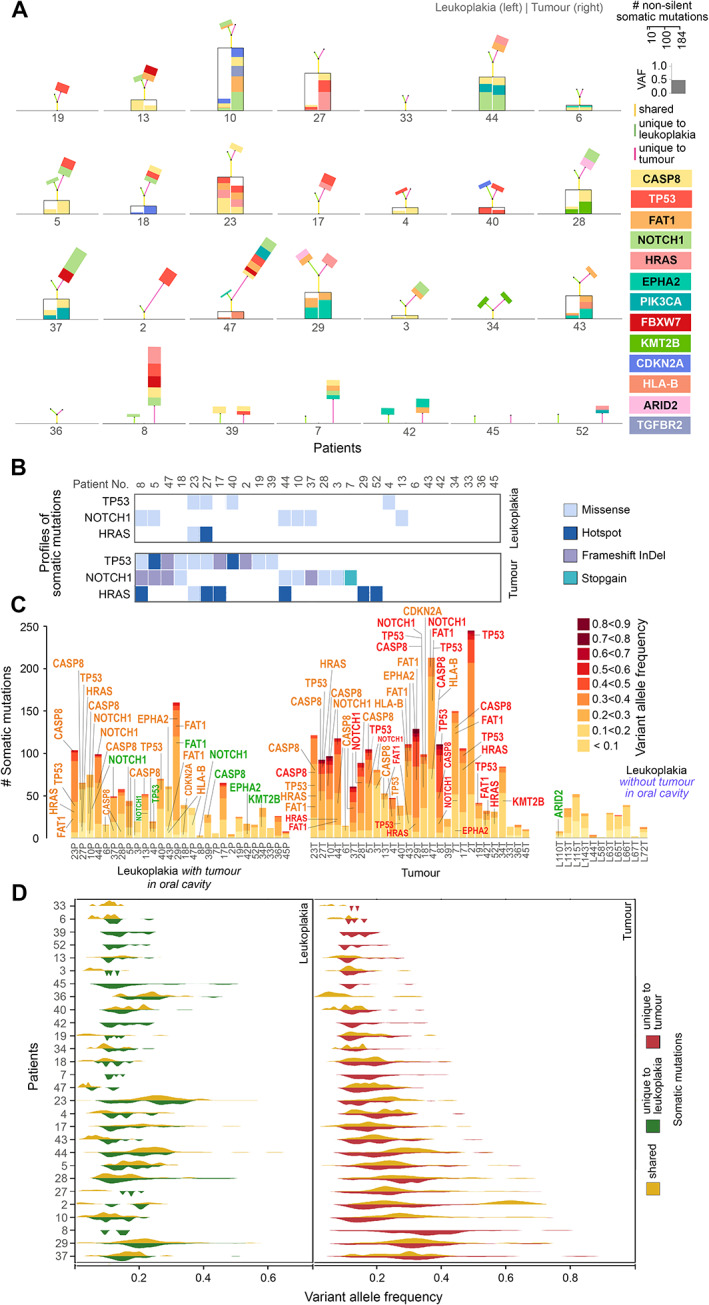
(A) Landscape of shared and unique somatic mutations in oral cancer driver genes for each patient. The genes are colour‐coded as indicated. The height of the box representing a driver gene mutation is scaled in proportion to its variant allele frequency. Mutations in genes that are shared between tumour (right) and leukoplakia (left) tissues are placed within the black box. Additional unique mutations in the tumour (right) and/or leukoplakia (left) are placed on the top of each box. The heights of the colour‐coded lines (yellow: shared, red: unique to tumour; green: unique to leukoplakia) are proportional to the number of mutations. The angle between the two lines representing the number of unique mutations in leukoplakia (green) and tumour (red) is proportional to the angular distance between them. (B) The number of pathogenic somatic mutations (hotspot, frameshift InDel, and stopgain) in oral cancer driver genes is higher in tumours than in leukoplakia lesions. (C) Numbers of somatic mutations in oral cancer driver genes in various allele frequency classes in (1) leukoplakia lesions, (2) adjacent tumours, and (3) leukoplakia lesions without tumour in the oral cavity. Genes are coloured based on their presence: only in tumour (red), only in leukoplakia lesion (green), and in both leukoplakia lesion and tumour (orange). (D) Allele frequencies (VAFs) of variants in tumours were significantly higher than in leukoplakia tissues, indicative of larger clonal fractions with variants in tumours. The allele frequencies of shared somatic variants were significantly higher than tumour‐ or leukoplakia‐specific variants indicating that those shared mutations originate early during tumorigenesis.
The most frequently mutated gene in many other cancers, TP53, was found to be mutated in 39% (n = 11) of OSCC‐GB tumours, significantly higher (Fisher's exact test, p < 0.04) than in leukoplakia tissues (14%, n = 4). Among the tumour‐specific mutations in TP53, 56% were hotspot or frameshift; hotspot mutations (oncogenic p.R273H, p.C176G) and were only detected in tumours but not in leukoplakia tissues. One patient (patient 40) harboured two TP53 mutations: (1) a missense mutation (p.E258Q) that was shared with the leukoplakia tissue, and (2) a hotspot mutation (p.C176G) that was present only in the tumour. Among tumour‐specific NOTCH1 mutations, 62.5% were stopgain or frameshift, whereas all mutations in the leukoplakia tissue were missense. Oncogenic mutations in HRAS (p.G12D, p.G12S, p.G13D) were found in 21.4% (n = 6) of tumours, but in only one (3.5%) leukoplakia tissue, with a very small cellular fraction (VAF < 0.05) that was shared with the tumour (VAF > 0.6 in tumour); details in supplementary material, Table S8. The overall picture that emerges is that malignant transformation finally requires some aggressive mutations in cancer driver genes in some cells of the leukoplakia tissue that results in clonal propagation of these cells to form a malignant tumour.
Chromosome‐level somatic structural alterations are late events in malignant transformation
The mean number of somatic copy number alterations (sCNA)—in particular, chromosomal arm‐level amplifications and deletions—identified from high coverage exome sequence data, was significantly greater (p < 0.001, t‐test for equality of means) in a tumour compared to the leukoplakia lesion of the patient (Figure 4A,B). Among patients (n = 22) in whom there was the presence of leukoplakia in proximity, the median proportion of chromosomal arm‐level alterations that were unique to tumour (that is, unshared with leukoplakia tissue) was 80%, compared to a significantly lower proportion (Kolmogorov–Smirnov test, p < 0.01) of 56.3% for single nucleotide variants (SNVs) (supplementary material, Table S13). This indicates that sCNA events occur later and facilitate the transition from potentially malignant lesion to cancer. The frequently amplified oncogenic oral cancer driver, EGFR, was found to be amplified in 29% (n = 8) of tumours, but completely absent even in a leukoplakia tissue in proximity (Figure 2D). NOTCH1, which is frequently mutated in OSCC‐GB, was found to be deleted (29%) in both tumour and leukoplakia. Among all chromosomal arms, seven arms (7p, 7q, 8p, 10p, 19p, 19q, and 20q) were amplified/deleted in tumours in higher proportions compared to leukoplakia tissues (Fisher's exact test, p < 0.05; Figure 4A,B). Notable increases from leukoplakia to tumour were on 8p, from 25 to 46%; 19p, from 14 to 61%; and 19q, from 14 to 50%. We found that 13 DNA‐repair pathway genes (nucleotide excision repair, eight genes; base excision repair, four genes; mismatch repair, three genes; Fanconi anaemia, three genes; homologous recombination, two genes) were deleted in tumour significantly more often (p < 0.02, Fisher's exact test) compared to leukoplakia (supplementary material, Table S14). In sum, arm‐level sCNA events along with amplification of EGFR and deletion of DNA‐repair genes were more frequent in tumours compared to leukoplakia tissues in proximity and are likely late, but essential, events for transformation to cancer.
Figure 4.
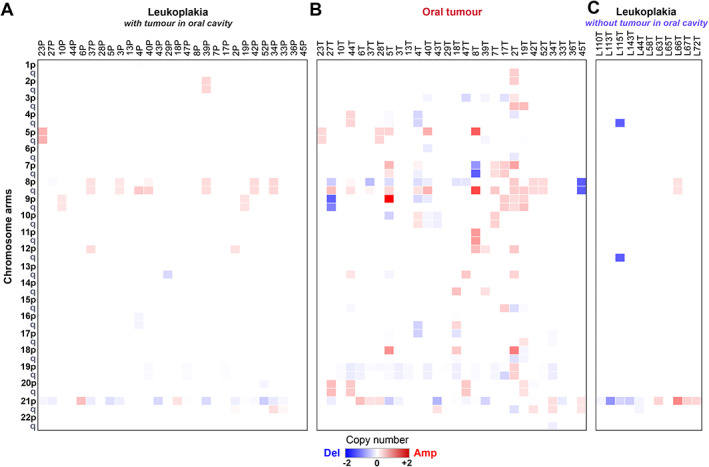
Profiles of somatic copy number alterations (sCNA) in (A) tumour and (B) leukoplakia showed a higher proportion of chromosomal arm‐level alterations in tumours. Seven chromosomal arms (7p and q, 8p, 10p, 19p and q and 20q) were amplified/deleted in a significantly higher proportion in tumours than in the potentially malignant lesions. (C) Among individuals with leukoplakia but without cancer, the proportion of chromosomal arm‐level alterations in the leukoplakia tissue was less. Our analysis showed that sCNAs occur late during the development of malignancy.
Oral tumour progression is associated with depletion of CD8+ T cells
Immune cell composition plays a pivotal role at various stages of tumour development and modulation of the tumour microenvironment [61]. The relative proportion of immune cells in leukoplakia, tumour, and adjacent normal tissues were estimated using CIBERSORT [38] from bulk RNAseq data (supplementary material, Table S15, Figure 5 and supplementary material, Figure S5). We have detected significantly higher abundance of CD4+ memory activated T cells (p < 0.004, Kolmogorov–Smirnov test) and M1 macrophages (p < 0.04, Kolmogorov–Smirnov test) in both tumours and leukoplakia tissues than adjacent normal tissues. This observation is also supported by transcriptomic data; CTLA4 [62] (expressed by the subset of CD4+ T cells called Treg cells) was found upregulated in both tumour (log2 fold‐change = 2.83, p[FDR] < 0.0001) and leukoplakia tissues (log2 fold‐change = 2.44, p[FDR] < 0.0001) compared to adjacent normal, which provided further supportive evidence for Treg‐mediated immunoregulatory activity [62] in potentially malignant leukoplakia and malignant tumour. The relative abundance of CD4+ memory activated T cells was at least 2‐fold higher in tumours of 56.52% patients compared to their adjacent leukoplakia tissues (Figure 5). On the other hand, the abundance of CD8+ T cells (cytotoxic T cells: TC) was significantly lower (p < 0.02, Kolmogorov–Smirnov test) in tumour than in the leukoplakia, in 68% of patients (Figure 5 and supplementary material, Figure S6). This possibly indicates the inability of cytotoxic T cells to enter into the tumour microenvironment or to survive there after entry, suggestive of an immunosuppression program during tumour progression. In support of this interpretation, we found that the immune regulatory gene IDO1, which is known to suppress TC proliferation [63, 64], was significantly upregulated in tumour tissue than in both leukoplakia lesion (log2 fold‐change = 1.37, p[FDR] < 0.01) and adjacent normal tissue (log2 fold‐change = 4.8, p[FDR] < 0.0001). Depletion of TC cells in the tumour microenvironment was been noted earlier [65]. Additionally, we observed a significant sequential depletion in abundance of dendritic cells (resting) from normal to leukoplakia (p < 0.0004, Kolmogorov–Smirnov test) to tumour (p < 0.04, Kolmogorov–Smirnov test), which is known to be associated with immune suppression by adversely affecting the degree of antigen presentation [66] besides depletion of TC in the tumour microenvironment (Figure 5). The abundances of M0 macrophages and activated mast cells were significantly higher (p < 0.004, Kolmogorov–Smirnov test) in tumours, compared to both leukoplakia lesions and adjacent normal tissues, possibly contributing to inflammation in tumours [13]. A recent study on early lung cancer patients also found similarly evolving patterns of composition of immune cells from early to late‐stage [67].
Figure 5.
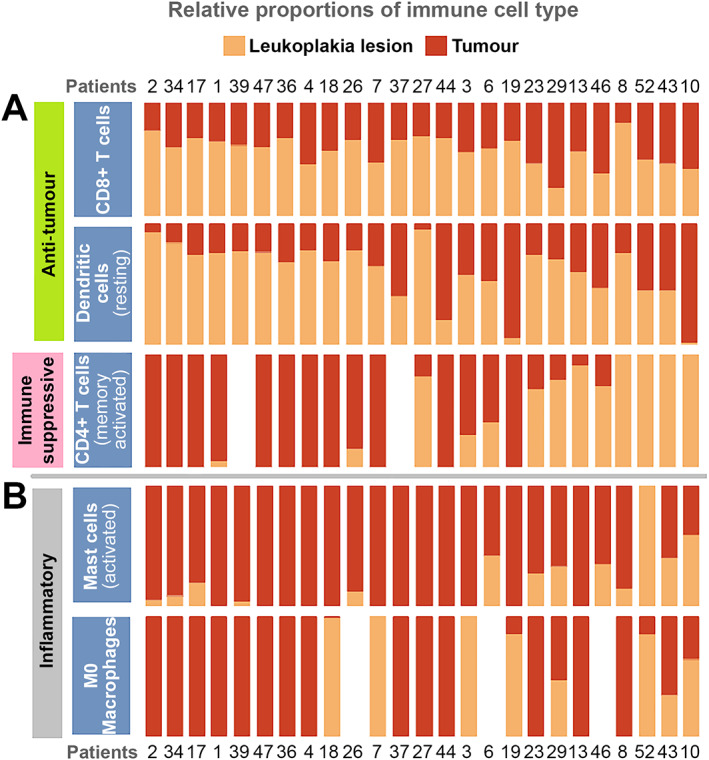
Immune deconvolution analysis from a bulk RNA sequence data showed: (A) depletion of relative abundance of (1) cytotoxic CD8+ T cells; and (2) antigen‐presenting dendritic cells; and (3) increased infiltration of immune regulatory CD4+ memory activated T cells in tumours compared to nonmalignant leukoplakia lesions that provided evidence for immune suppression in oral tumour progression. (B) The higher abundance of inflammatory mast cells and M0 macrophages in tumours indicates increased inflammation in tumours promoting tumorigenesis.
The time to malignant transformation of a potentially malignant leukoplakia ranges from 4 to 10 years
We estimated (supplementary material, Table S16) the time required for a leukoplakia tissue to transform to a malignant tumour, using a previously described method [33] and a range of somatic mutation rates (1–10 mutations per year). Among these patients, 27% had a APOBEC mutagenesis signature in tumour, which is known to elevate the mutation rate. In our data, a >2‐fold elevated mutation rate was observed in tumour compared to leukoplakia, likely due to APOBEC activity; thus, we have taken the possible range of the somatic mutation rate to be 6–10 mutations per year for patients with an APOBEC mutational signature in tumours. The average time to malignant transformation of leukoplakia located in proximity to tumour was estimated to be ~7 years (95% confidence interval [CI]: 3.8–9.8 years). Three broad groups were observed on the basis of the time to malignant transformation: Group I (11 patients), with an average time of ~3 years (95% CI: 1.8–4.0 years) (average number of additional mutations during transformation ~18); Group II (nine patients) with an average time of ~7 years (95% CI: 6.2–8.5 years) (average number of additional mutations ~45); and Group III (two patients), patients 2 and 47 (with 184 and 140 additional mutations in tumours), with estimated times as 34 and 18 years. For more than 80% patients, the time taken to progress to malignancy after a potentially malignant condition (leukoplakia) had arisen was over 3 years, providing a significant window of opportunity for observation and clinical management, consistent with findings of some previous studies [68, 69].
Discussion
In this study we sought to identify the genomic changes that occur during the evolution of a normal tissue in the oral cavity to a potentially malignant lesion (leukoplakia), and finally to a malignant tumour. In particular, we investigated whether there is consistent sequential accumulation of genomic alterations during this evolution. Our analysis provides evidence for (1) early genomic alterations in leukoplakia, and (2) the accumulation of additional alterations along with a change in the immune dynamics that promote progression to malignancy.
Most patients in our cohort were habitual tobacco chewers. Tobacco is a major source of ROS (reactive oxygen species) that increases oxidative damage to DNA [70]. DNA changes that are induced by oxidation are primarily repaired by base excision repair (BER)‐mediated mechanisms [71]. We have found a significantly higher (Fisher's exact test, p < 0.03) number of rare germline alterations in genes of the DNA repair pathway — BER and homologous recombination (HR) — in patients compared to normal healthy individuals (GenomeAsia100k‐Indian cohort; n = 533); details are in supplementary material, Table S17 and Figure 2E. Rare germline alterations in DNA repair pathway genes likely predispose individuals to develop lesions [72, 73] on habitual exposure to tobacco.
We observed that (1) there is considerable sharing of genomic variants between a leukoplakia and a tumour in the same oral cavity (79%, n = 22), and (2) the number of shared variants between the two tissues decreases with an increase of the distance between them. The presence of shared somatic mutations in two physically distinct locations, i.e. tumour and leukoplakia in proximity and their higher cellular fractions in tumours further confirmed their early occurrence [42, 74]. Somatic mutations that were specific to leukoplakia and tumour likely arose after the committed transformation to malignancy.
CASP8 appears to be a major driver to initiate malignant transformation in a benign leukoplakia lesion. The proportion of shared somatic mutations in CASP8 between leukoplakia and cancer was significantly higher (Fisher's exact test, p < 0.02) than for any other gene. Among OSCC‐GB patients, we found that most CASP8 mutations are in the most conserved protease domain [which is evolutionarily more conserved (Bit score: 341.12) than the known conserved DED domain (Bit score: 132.23); https://www.ncbi.nlm.nih.gov/cdd/], which is required for cleavage and activation of downstream caspases to trigger apoptosis [75]. Most (83%) mutations in CASP8 shared between leukoplakia and tumour were highly deleterious: stopgain, frameshift, or hotspot [R292Q (CASP8:NM_001080125) or R233Q (CASP8:NM_033355)]; the remaining observed missense mutations were also predicted to be highly deleterious by at least two of three mutation functional effect prediction algorithms (MutationAccessor, SIFT, and PolyPhen‐2). CASP8 is a key regulator of the apoptosis pathway, inactivation of which may result in cellular immortality [76]. Genes linked to cellular invasion, migration, and stemness were upregulated in CASP8‐mutated leukoplakia and tumours. Loss of function of CASP8 has been previously shown in head and neck cancer [77], neuroblastoma [78], lung cancer cell lines [79], and model systems [56] to be associated with cell survival, migration, and invasive capacities, as well as immune escape [80], especially in the case of head and neck tumours. We infer from our data and supportive evidence from multiple functional studies that CASP8 mutations are early initiators of oral cancer facilitating survival advantage and lateral spread of mutant cells.
Besides acquisition of additional pathogenic mutations and CNAs, the immune contexture of the tumours was found to be protumorigenic, i.e. abundance of regulatory T cells (Treg) and depletion of cytotoxic T cells (TC) and antigen‐presenting dendritic cells. We also analysed gene expression data from 43 paired tumour‐normal samples from the TCGA‐HNSC cohort and found that 55.8 and 65.1% of patients had at least a 2‐fold greater abundance, respectively, of TC and resting dendritic cells in adjacent normal than in their tumours, adding further support to our observations on Indian oral cancer patients. These findings indicate that immunotherapy can be used to stabilize cytotoxic T‐cell and antigen‐presenting dendritic cells to retard malignant transformation of oral, potentially malignant, disorders. An increase of inflammatory immune cell infiltration was observed from normal oral tissue to leukoplakia to tumour. The expression of proinflammatory cytokines and interleukins, e.g. CXCL8 [81], CXCL11 [82], IL24 [83], IL1A [84, 85], and IL11 [86] were significantly upregulated in tumours as compared to both adjacent normal and leukoplakia tissues. Our gene expression data showed tumour‐specific upregulation of proangiogenic factor VEGFC, due possibly to inflammation in the tumour microenvironment.
Our multiomics characterisation of oral tumours and associated potentially malignant lesions helped delineate the sequence of genomic events during malignant transformation and progression. We identified early events (CASP8 and NOTCH1 alterations), intermediate events (APOBEC activity and tumour‐only additional pathogenic mutations), late events (EGFR amplification, chromosome arm‐level instability), and deregulation of immune contexture (depletion of antitumour immune cells and increased inflammation) during the course of oral tumour initiation and progression. Our findings also open up possibilities for incorporation of: (1) therapy to inhibit APOBEC activity [87] activity, and (2) immunotherapy in stimulating cytotoxic T cells [88] and dendritic cells [89], to prevent malignant transformation of oral, potentially malignant, conditions.
Author contributions statement
NKB was responsible for conceiving the study. SG and BR were responsible for coordinating patient recruitment and clinical assessments. AM was responsible for guiding and coordinating massively parallel sequencing. PPM was responsible for providing statistical guidance. AG, CD and NKB carried out statistical and bioinformatics analysis of whole‐exome and transcriptomic data. AG, CD, PPM and NKB wrote the article.
Supporting information
Supplementary materials and methods
Figure S1. Relationship between the proximity of lesions and the sharing of somatic mutations
Figure S2. Differential gene expression from mRNA sequencing data showed distinct patterns in oral tumour and leukoplakia lesions
Figure S3. The predominantly‐overexpressed genes of oral hairy leukoplasias and oral tumours
Figure S4. CASP8 mutations in this study, the ICGC‐India study, and the TCGA‐HNSC collection
Figure S5. Relative proportions of anti‐tumour (CD8+ cytotoxic T cells, dendritic cells), immune suppressive (CD4+ memory activated T cells) and inflammatory (activated mast cells and M0 macrophages) cell types in normal tissue, leukoplakia and tumour from the same patients
Figure S6. The abundance of CD8+ T cells estimated in each tissue sample from tumour and leukoplakia using an orthogonal algorithm encoded in EPIC to validate the findings from CIBERSORT results
Figure S7. Distributions of variant allele frequencies (VAF) of somatic mutations in leukoplakia that are unique to leukoplakia (green) and shared with nearby tumours (yellow)
Figure S8. Patient‐wise variant allele frequencies (VAF) of somatic mutations in tumours that are unique to tumours (red) and shared with adjacent leukoplakia lesions (yellow)
Table S1. Summary of demographic, exposure, clinical and other variables of the recruited cohort of patients
Table S2. Whole‐exome sequencing data generation
Table S3. Whole transcriptome sequencing data generation
Table S4. Distributions of types of somatic SNVs in tumour and leukoplakia
Table S5. Summary of different types of somatic mutations in tumour and leukoplakia
Table S6. Somatic mutations in exonic regions found in leukoplakia of 11 individuals without presence of tumour in the oral cavity
Table S7. In the cohort of patients with concomitant presence of tumour and leukoplakia, the numbers and proportions of mutations that are unshared and shared between tumour and leukoplakia.
Table S8. Nonsynonymous protein coding somatic mutations in oral cancer driver genes in oral primary tumour (OPT) and oral leukoplakia (OLK)
Table S9. Median VAF of all somatic mutations and VAF of CASP8 mutations in tumours (n = 16; patients with CASP8 mutation)
Table S10. Somatic mutations from multiple biopsies from an OSCC patient having evidence of field cancerization by Tabatabaeifar et al [49].
Table S11. Upregulated genes that are associated with cellular migration and invasion and cellular stemness in CASP8 mutated tumour (OPT) and leukoplakia (OLK)
Table S12. Mutation rate (number of mutations per Mb of genome) in paired leukoplakia and tumour in OSCC patients with concomitant presence of leukoplakia in proximity and presence of APOBEC signature in the tumour and leukoplakia lesion
Table S13. Numbers of somatic SNVs and chromosomal arm‐level alterations in tumours that are shared with leukoplakia and are specific to tumours
Table S14. Number of patients with copy number deletion in DNA repair pathway genes in tumour and leukoplakia lesions
Table S15. Difference in abundance of various types of immune cells present in tumour, leukoplakia, and normal tissue
Table S16. Estimation of time window for malignant progression of leukoplakia adjacent to oral tumour
Table S17. Numbers of individuals with rare germline mutations in genes belonging to base excision repair (BER) and homologous recombination (HR) pathways drawn randomly from populations, from OSCC patients with or without concomitant presence of leukoplakia lesion and from cohorts of patients with other rare diseases
Table S18. Estimation of sample contamination with respect to paired blood from WES data
Acknowledgements
PPM acknowledges financial support from his National Science Chair grant from SERB, Government of India. We acknowledge the guidance provided by Mr. Subrata Das for analysis of sequence data and Mr. Animesh K. Singh for providing help in data visualization. The software pipelines development part of this project was supported by an NSM project grant, MeitY. We also thank Dr. Analabha Basu for providing critical comments on an early draft of the article.
This study was funded by the Department of Biotechnology, Government of India through the SyMeC project.
No conflicts of interest were declared.
Data availability statement
Whole‐exome and transcriptome sequence CRAM files for each sample are deposited in the European Nucleotide Archive under study accession PRJEB42969.
References
References 90–98 are cited only in the supplementary material.
- 1. Sung H, Ferlay J, Siegel RL, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021; 71: 209–249. [DOI] [PubMed] [Google Scholar]
- 2. India Project Team of the International Cancer Genome Consortium . Mutational landscape of gingivo‐buccal oral squamous cell carcinoma reveals new recurrently‐mutated genes and molecular subgroups. Nat Commun 2013; 4: 2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tiziani S, Lopes V, Günther UL. Early stage diagnosis of oral cancer using 1H NMR‐based metabolomics. Neoplasia 2009; 11: 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hu B, Castillo E, Harewood L, et al. Multifocal epithelial tumors and field cancerization from loss of mesenchymal CSL signaling. Cell 2012; 149: 1207–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Curtius K, Wright NA, Graham TA. An evolutionary perspective on field cancerization. Nat Rev Cancer 2018; 18: 19–32. [DOI] [PubMed] [Google Scholar]
- 6. Feller LL, Khammissa RR, Kramer BB, et al. Oral squamous cell carcinoma in relation to field precancerisation: pathobiology. Cancer Cell Int 2013; 13: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gangadharan P, Paymaster JC. Leukoplakia–an epidemiologic study of 1504 cases observed at the Tata Memorial Hospital, Bombay, India. Br J Cancer 1971; 25: 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Iocca O, Sollecito TP, Alawi F, et al. Potentially malignant disorders of the oral cavity and oral dysplasia: a systematic review and meta‐analysis of malignant transformation rate by subtype. Head Neck 2020; 42: 539–555. [DOI] [PubMed] [Google Scholar]
- 9. Mohan M, Jagannathan N. Oral field cancerization: an update on current concepts. Oncol Rev 2014; 8: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wiencke JK. DNA adduct burden and tobacco carcinogenesis. Oncogene 2002; 21: 7376–7391. [DOI] [PubMed] [Google Scholar]
- 11. Biswas NK, Das C, Das S, et al. Lymph node metastasis in oral cancer is strongly associated with chromosomal instability and DNA repair defects. Int J Cancer 2019; 145: 2568–2579. [DOI] [PubMed] [Google Scholar]
- 12. Cannataro VL, Gaffney SG, Sasaki T, et al. APOBEC‐induced mutations and their cancer effect size in head and neck squamous cell carcinoma. Oncogene 2019; 38: 3475–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hadler‐Olsen E, Wirsing AM. Tissue‐infiltrating immune cells as prognostic markers in oral squamous cell carcinoma: a systematic review and meta‐analysis. Br J Cancer 2019; 120: 714–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kumar A, Cascarini L, McCaul JA, et al. How should we manage oral leukoplakia? Br J Oral Maxillofac Surg 2013; 51: 377–383. [DOI] [PubMed] [Google Scholar]
- 15. William WN Jr, Papadimitrakopoulou V, Lee JJ, et al. Erlotinib and the risk of oral cancer: the erlotinib prevention of oral cancer (EPOC) randomized clinical trial. JAMA Oncol 2016; 2: 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsuda H, Konishi N, Hiasa Y, et al. Alterations of p16/CDKN2, p53 and ras genes in oral squamous cell carcinomas and premalignant lesions. J Oral Pathol Med 1996; 25: 232–238. [DOI] [PubMed] [Google Scholar]
- 17. Boyle JO, Hakim J, Koch W, et al. The incidence of p53 mutations increases with progression of head and neck cancer. Cancer Res 1993; 53: 4477–4480. [PubMed] [Google Scholar]
- 18. Saranath D, Tandle AT, Teni TR, et al. p53 inactivation in chewing tobacco‐induced oral cancers and leukoplakias from India. Oral Oncol 1999; 35: 242–250. [PubMed] [Google Scholar]
- 19. Partridge M, Emilion G, Pateromichelakis S, et al. Allelic imbalance at chromosomal loci implicated in the pathogenesis of oral precancer, cumulative loss and its relationship with progression to cancer. Oral Oncol 1998; 34: 77–83. [DOI] [PubMed] [Google Scholar]
- 20. Graveland AP, Bremmer JF, de Maaker M, et al. Molecular screening of oral precancer. Oral Oncol 2013; 49: 1129–1135. [DOI] [PubMed] [Google Scholar]
- 21. Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010; [Accessed 16th April 2022]. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
- 22. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM; 2013. arXiv:1303.3997 [Not peer reviewed].
- 23. Broad Institute . Picard Tools. version 2.18.2Broad Institute, GitHub Repos; 2018.
- 24. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010; 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fan Y, Xi L, Hughes DST, et al. MuSE: accounting for tumor heterogeneity using a sample‐specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol 2016; 17: 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim S, Scheffler K, Halpern AL, et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 2018; 15: 591–594. [DOI] [PubMed] [Google Scholar]
- 27. 1000 Genomes Project Consortium , Auton A, Abecasis GR, et al. A global reference for human genetic variation. Nature 2015; 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. GenomeAsia100K Consortium . The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature 2019; 576: 106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 2013; 14: 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mermel CH, Schumacher SE, Hill B, et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy‐number alteration in human cancers. Genome Biol 2011; 12: R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Loo P, Nordgard SH, Lingjærde OC, et al. Allele‐specific copy number analysis of tumors. Proc Natl Acad Sci U S A 2010; 107: 16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Degasperi A, Amarante TD, Czarnecki J, et al. A practical framework and online tool for mutational signature analyses show inter‐tissue variation and driver dependencies. Nat Cancer 2020; 1: 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Noë M, Niknafs N, Fischer CG, et al. Genomic characterization of malignant progression in neoplastic pancreatic cysts. Nat Commun 2020; 11: 4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010; 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ramos AH, Lichtenstein L, Gupta M, et al. Oncotator: cancer variant annotation tool. Hum Mutat 2015; 36: E2423–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dobin A, Davis CA, Schlesinger F, et al. STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high‐throughput sequencing data. Bioinformatics 2015; 31: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015; 12: 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Racle J, de Jonge K, Baumgaertner P, et al. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. Elife 2017; 6: e26476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vera Alvarez R, Pongor LS, Mariño‐Ramírez L, et al. TPMCalculator: one‐step software to quantify mRNA abundance of genomic features. Bioinformatics 2019; 35: 1960–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jolly C, Van Loo P. Timing somatic events in the evolution of cancer. Genome Biol 2018; 19: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Faltas BM, Prandi D, Tagawa ST, et al. Clonal evolution of chemotherapy‐resistant urothelial carcinoma. Nat Genet 2016; 48: 1490–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McGranahan N, Favero F, de Bruin EC, et al. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med 2015; 7: 283ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Singh R, Das S, Datta S, et al. Study of Caspase 8 mutation in oral cancer and adjacent precancer tissues and implication in progression. PLoS One 2020; 15: e0233058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011; 333: 1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Murugan AK, Munirajan AK, Tsuchida N. Genetic deregulation of the PIK3CA oncogene in oral cancer. Cancer Lett 2013; 338: 193–203. [DOI] [PubMed] [Google Scholar]
- 48. Cancer Genome Atlas Network . Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015; 517: 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tabatabaeifar S, Larsen MJ, Larsen SR, et al. Investigating a case of possible field cancerization in oral squamous cell carcinoma by the use of next‐generation sequencing. Oral Oncol 2017; 68: 74–80. [DOI] [PubMed] [Google Scholar]
- 50. Gharpure KM, Pradeep S, Sans M, et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat Commun 2018; 9: 2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pelekanou V, Notas G, Athanasouli P, et al. BCMA (TNFRSF17) induces APRIL and BAFF mediated breast cancer cell stemness. Front Oncol 2018; 8: 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mondal B, Patil V, Shwetha SD, et al. Integrative functional genomic analysis identifies epigenetically regulated fibromodulin as an essential gene for glioma cell migration. Oncogene 2017; 36: 71–83. [DOI] [PubMed] [Google Scholar]
- 53. Landemaine T, Jackson A, Bellahcène A, et al. A six‐gene signature predicting breast cancer lung metastasis. Cancer Res 2008; 68: 6092–6099. [DOI] [PubMed] [Google Scholar]
- 54. Rodriguez‐Torres M, Allan AL. Aldehyde dehydrogenase as a marker and functional mediator of metastasis in solid tumors. Clin Exp Metastasis 2016; 33: 97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Long A, Giroux V, Whelan KA, et al. WNT10A promotes an invasive and self‐renewing phenotype in esophageal squamous cell carcinoma. Carcinogenesis 2015; 36: 598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gorelick‐Ashkenazi A, Weiss R, Sapozhnikov L, et al. Caspases maintain tissue integrity by an apoptosis‐independent inhibition of cell migration and invasion. Nat Commun 2018; 9: 2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Keller N, Ozmadenci D, Ichim G, et al. Caspase‐8 function, and phosphorylation, in cell migration. Semin Cell Dev Biol 2018; 82: 105–117. [DOI] [PubMed] [Google Scholar]
- 58. Roberts SA, Lawrence MS, Klimczak LJ, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet 2013; 45: 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Burns MB, Lackey L, Carpenter MA, et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013; 494: 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Langenbucher A, Bowen D, Sakhtemani R, et al. An extended APOBEC3A mutation signature in cancer. Nat Commun 2021; 12: 1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Galon J, Bruni D. Tumor immunology and tumor evolution: intertwined histories. Immunity 2020; 52: 55–81. [DOI] [PubMed] [Google Scholar]
- 62. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression ‐ implications for anticancer therapy. Nat Rev Clin Oncol 2019; 16: 356–371. [DOI] [PubMed] [Google Scholar]
- 63. Löb S, Königsrainer A, Rammensee HG, et al. Inhibitors of indoleamine‐2,3‐dioxygenase for cancer therapy: can we see the wood for the trees? Nat Rev Cancer 2009; 9: 445–452. [DOI] [PubMed] [Google Scholar]
- 64. Munn DH, Sharma MD, Lee JR, et al. Potential regulatory function of human dendritic cells expressing indoleamine 2,3‐dioxygenase. Science 2002; 297: 1867–1870. [DOI] [PubMed] [Google Scholar]
- 65. Zhou X, Zhao S, He Y, et al. Precise spatiotemporal interruption of regulatory T‐cell‐mediated CD8+ T‐cell suppression leads to tumor immunity. Cancer Res 2019; 79: 585–597. [DOI] [PubMed] [Google Scholar]
- 66. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12: 265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mascaux C, Angelova M, Vasaturo A, et al. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature 2019; 571: 570–575. [DOI] [PubMed] [Google Scholar]
- 68. Silverman S Jr, Gorsky M. Proliferative verrucous leukoplakia: a follow‐up study of 54 cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 1997; 84: 154–157. [DOI] [PubMed] [Google Scholar]
- 69. Silverman S Jr, Gorsky M, Lozada F. Oral leukoplakia and malignant transformation. A follow‐up study of 257 patients. Cancer 1984; 53: 563–568. [DOI] [PubMed] [Google Scholar]
- 70. Loft S, Poulsen HE. Cancer risk and oxidative DNA damage in man. J Mol Med (Berl) 1996; 74: 297–312. [DOI] [PubMed] [Google Scholar]
- 71. Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutat Res Rev Mutat Res 2015; 763: 212–245. [DOI] [PubMed] [Google Scholar]
- 72. Lu C, Xie M, Wendl MC, et al. Patterns and functional implications of rare germline variants across 12 cancer types. Nat Commun 2015; 6: 10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mijuskovic M, Saunders EJ, Leongamornlert DA, et al. Rare germline variants in DNA repair genes and the angiogenesis pathway predispose prostate cancer patients to develop metastatic disease. Br J Cancer 2018; 119: 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Salichos L, Meyerson W, Warrell J, et al. Estimating growth patterns and driver effects in tumor evolution from individual samples. Nat Commun 2020; 11: 732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stennicke HR, Jürgensmeier JM, Shin H, et al. Pro‐caspase‐3 is a major physiologic target of caspase‐8. J Biol Chem 1998; 273: 27084–27090. [DOI] [PubMed] [Google Scholar]
- 76. Fritsch M, Günther SD, Schwarzer R, et al. Caspase‐8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019; 575: 683–687. [DOI] [PubMed] [Google Scholar]
- 77. Li C, Egloff AM, Sen M, et al. Caspase‐8 mutations in head and neck cancer confer resistance to death receptor‐mediated apoptosis and enhance migration, invasion, and tumor growth. Mol Oncol 2014; 8: 1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Stupack DG, Teitz T, Potter MD, et al. Potentiation of neuroblastoma metastasis by loss of caspase‐8. Nature 2006; 439: 95–99. [DOI] [PubMed] [Google Scholar]
- 79. Barbero S, Mielgo A, Torres V, et al. Caspase‐8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res 2009; 69: 3755–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rooney MS, Shukla SA, Wu CJ, et al. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015; 160: 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Karin M. Inflammation and cancer: the long reach of Ras. Nat Med 2005; 11: 20–21. [DOI] [PubMed] [Google Scholar]
- 82. Qin S, Alcorn JF, Craigo JK, et al. Epigallocatechin‐3‐gallate reduces airway inflammation in mice through binding to proinflammatory chemokines and inhibiting inflammatory cell recruitment. J Immunol 2011; 186: 3693–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sauane M, Su ZZ, Gupta P, et al. Autocrine regulation of mda‐7/IL‐24 mediates cancer‐specific apoptosis. Proc Natl Acad Sci U S A 2008; 105: 9763–9768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Liu S, Lee JS, Jie C, et al. HER2 overexpression triggers an IL1α proinflammatory circuit to drive tumorigenesis and promote chemotherapy resistance. Cancer Res 2018; 78: 2040–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Suwara MI, Green NJ, Borthwick LA, et al. IL‐1α released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol 2014; 7: 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Putoczki TL, Thiem S, Loving A, et al. Interleukin‐11 is the dominant Il‐6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013; 24: 257–271. [DOI] [PubMed] [Google Scholar]
- 87. Barzak FM, Harjes S, Kvach MV, et al. Selective inhibition of APOBEC3 enzymes by single‐stranded DNAs containing 2’‐deoxyzebularine. Org Biomol Chem 2019; 17: 9435–9441. [DOI] [PubMed] [Google Scholar]
- 88. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 2020; 20: 651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Wculek SK, Cueto FJ, Mujal AM, et al. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 2020; 20: 7–24. [DOI] [PubMed] [Google Scholar]
- 90. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. García‐Alcalde F, Okonechnikov K, Carbonell J, et al. Qualimap: evaluating next‐generation sequencing alignment data. Bioinformatics 2012; 28: 2678–2679. [DOI] [PubMed] [Google Scholar]
- 93. Costello M, Pugh TJ, Fennell TJ, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res 2013; 41: e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol 1990; 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 95. Bailey MH, Tokheim C, Porta‐Pardo E, et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018; 173: 371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Lawrence MS, Stojanov P, Mermel CH, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014; 505: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016; 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kanehisa M, Furumichi M, Tanabe M, et al. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res 2017; 45: D353–D361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Figure S1. Relationship between the proximity of lesions and the sharing of somatic mutations
Figure S2. Differential gene expression from mRNA sequencing data showed distinct patterns in oral tumour and leukoplakia lesions
Figure S3. The predominantly‐overexpressed genes of oral hairy leukoplasias and oral tumours
Figure S4. CASP8 mutations in this study, the ICGC‐India study, and the TCGA‐HNSC collection
Figure S5. Relative proportions of anti‐tumour (CD8+ cytotoxic T cells, dendritic cells), immune suppressive (CD4+ memory activated T cells) and inflammatory (activated mast cells and M0 macrophages) cell types in normal tissue, leukoplakia and tumour from the same patients
Figure S6. The abundance of CD8+ T cells estimated in each tissue sample from tumour and leukoplakia using an orthogonal algorithm encoded in EPIC to validate the findings from CIBERSORT results
Figure S7. Distributions of variant allele frequencies (VAF) of somatic mutations in leukoplakia that are unique to leukoplakia (green) and shared with nearby tumours (yellow)
Figure S8. Patient‐wise variant allele frequencies (VAF) of somatic mutations in tumours that are unique to tumours (red) and shared with adjacent leukoplakia lesions (yellow)
Table S1. Summary of demographic, exposure, clinical and other variables of the recruited cohort of patients
Table S2. Whole‐exome sequencing data generation
Table S3. Whole transcriptome sequencing data generation
Table S4. Distributions of types of somatic SNVs in tumour and leukoplakia
Table S5. Summary of different types of somatic mutations in tumour and leukoplakia
Table S6. Somatic mutations in exonic regions found in leukoplakia of 11 individuals without presence of tumour in the oral cavity
Table S7. In the cohort of patients with concomitant presence of tumour and leukoplakia, the numbers and proportions of mutations that are unshared and shared between tumour and leukoplakia.
Table S8. Nonsynonymous protein coding somatic mutations in oral cancer driver genes in oral primary tumour (OPT) and oral leukoplakia (OLK)
Table S9. Median VAF of all somatic mutations and VAF of CASP8 mutations in tumours (n = 16; patients with CASP8 mutation)
Table S10. Somatic mutations from multiple biopsies from an OSCC patient having evidence of field cancerization by Tabatabaeifar et al [49].
Table S11. Upregulated genes that are associated with cellular migration and invasion and cellular stemness in CASP8 mutated tumour (OPT) and leukoplakia (OLK)
Table S12. Mutation rate (number of mutations per Mb of genome) in paired leukoplakia and tumour in OSCC patients with concomitant presence of leukoplakia in proximity and presence of APOBEC signature in the tumour and leukoplakia lesion
Table S13. Numbers of somatic SNVs and chromosomal arm‐level alterations in tumours that are shared with leukoplakia and are specific to tumours
Table S14. Number of patients with copy number deletion in DNA repair pathway genes in tumour and leukoplakia lesions
Table S15. Difference in abundance of various types of immune cells present in tumour, leukoplakia, and normal tissue
Table S16. Estimation of time window for malignant progression of leukoplakia adjacent to oral tumour
Table S17. Numbers of individuals with rare germline mutations in genes belonging to base excision repair (BER) and homologous recombination (HR) pathways drawn randomly from populations, from OSCC patients with or without concomitant presence of leukoplakia lesion and from cohorts of patients with other rare diseases
Table S18. Estimation of sample contamination with respect to paired blood from WES data
Data Availability Statement
Whole‐exome and transcriptome sequence CRAM files for each sample are deposited in the European Nucleotide Archive under study accession PRJEB42969.