

Abstract
Background
PPKs represent a heterogeneous group of disorders with hyperkeratosis of palmar and/or plantar skin. PPK, hair shaft abnormalities, cardiomyopathy and arrhythmias can be caused by mutations in desmosomal genes, e.g. desmoplakin (DSP). PPK should trigger genetic testing to reveal mutations with possible related cardiac disease.
Objectives
To report a large multigenerational family with a novel DSP mutation associated with early‐onset PPK and adult‐onset cardiomyopathy and arrhythmias.
Methods
A custom‐designed in‐house panel of 35 PPK related genes was used to screen mutations in the index patient with focal PPK. The identified DSP mutation was verified by Sanger sequencing. DNA samples from 20 members of the large multigenerational family were sequenced for the DSP mutation. Medical records were reviewed. Clinical dermatological evaluation was performed, including light microscopy of hair samples. Cardiac evaluation included clinical examination, echocardiography, cardiac magnetic resonance imaging (CMR), electrocardiogram (ECG), Holter monitoring and laboratory tests.
Results
We identified a novel autosomal dominant truncating DSP c.2493delA p.(Glu831Aspfs*33) mutation associated with dilated cardiomyopathy (DCM) with arrhythmia susceptibility and focal PPK as an early cutaneous sign. The mutation was found in nine affected family members, but not in any unaffected members. Onset of dermatological findings preceded cardiac symptoms which were variable and occurred at adult age.
Conclusions
We report a novel truncating DSP mutation causing focal PPK with varying severity and left ventricular dilatation and ventricular extrasystoles. This finding emphasizes the importance of genetic diagnosis in patients with PPK for clinical counselling and management of cardiomyopathies and arrhythmias.
Keywords: palmoplantar keratoderma, dilated cardiomyopathy, genodermatosis, next‐generation sequencing
Introduction
Desmosomes are cell adhesion junctions critical in tissues prone to mechanical stress, and they play an important role in maintaining the integrity of the epidermis and the heart. 1 The desmosomal proteins plakoglobin (PG), plakophilin (PKP, isoforms 1 and 2), desmocollin (DSC, isoforms 1–3), desmoplakin (DSP), desmoglein (DSG, isoforms 1–4) and corneodesmosin (CDSN) are encoded by JUP, PKP1‐2, DSC1‐3, DSP, DSG1‐4, and CDSN, respectively. 2 All these proteins are present in the epidermis while PKP2, DSP, PG, DSC2 and DSG2 are crucial for cardiac desmosomal integrity. 2 Palmoplantar keratodermas (PPK) are a heterogeneous group of rare hereditary skin disorders characterized by variable thickening of the epidermis of palms and soles. PPKs may be associated with several extracutaneous manifestations such as cardiomyopathy and arrhythmias, involvement of other internal organs, deafness and inborn errors of metabolism. 3 , 4 , 5 At least 69 genes have been implicated in PPKs so far. 6 Mutations in DSP, JUP and rarely DSC2 cause syndromes characterized by the triad of PPK, woolly hair and arrhythmogenic cardiomyopathy. 3 Carvajal and Naxos syndromes caused by DSP and JUP mutations, respectively, are potentially life‐threatening conditions due to the cardiac involvement. 2 Among PPK and woolly hair, mutations in DSP, JUP and DSC2 have been linked with arrhythmogenic right ventricular cardiomyopathy (ARVC), and in DSP also with left ventricular and biventricular subtypes. Abnormalities of the teeth, hair and nails may be present. 2
We identified a novel DSP c.2493delA p.(Glu831Aspfs*33) mutation in our index patient with an in‐house PPK gene panel. Further Sanger sequencing in the index patient's large multigenerational family verified that the DSP c.2493delA p.(Glu831Aspfs*33) mutation is associated with PPK, dilated cardiomyopathy (DCM) and arrhythmias.
Materials and methods
Patients
All patients were recruited from the Department of Dermatology, Helsinki University Hospital, where dermatological evaluations were conducted and hair samples were examined with light microscopy. Patients were evaluated clinically by at least one dermatologist (KHJ, VK, LH) and from patient records, or by telephone interviews, photographs and patient records when in‐person evaluation was not possible. Cardiac evaluations were conducted at the Heart and Lung Center in Helsinki University Hospital and all previous hospital records regarding especially cardiac evaluations were examined. Thirteen patients were clinically evaluated by at least one of the authors (TH, KH). All participants gave written informed consent. This study was approved by the Ethical Review Board of the Helsinki and Uusimaa Hospital District, Helsinki, Finland (HUS2004/2018, HUS/3225/2018, HUS/2675/2019) and conducted according to the principles expressed in the Declaration of Helsinki. Permission was granted from Statistics Finland and the Ministry of Social Affairs and Health to obtain clinical data from deceased patients for research purposes. All participants are of Finnish ethnicity.
DNA sequencing
EDTA blood samples were collected from 10 and oral DNA samples from 11 participants by Oracollect‐DNA (ORC‐100, DNAGenotek, Ottawa, ON, Canada). DNA was extracted by standard protocols using Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) or FlexiGene DNA kit (Qiagen). Oral DNA was extracted with ethanol precipitation and prepIT.L2P® (DNAGenotek). The index patient was analysed on a customized in‐house next generation sequencing (NGS) panel designed to target 35 PPK candidate genes and the result was verified by Sanger sequencing. Seventeen family members were Sanger sequenced for the DSP c.2493delA mutation and three family members (III:1, III:2 and II:6) (Fig. 1) were analysed by Blueprint Genetics (Espoo, Finland). DNA samples were not available from deceased family members. Detailed description of sequencing methods is in the (Appendix S1).
Figure 1.
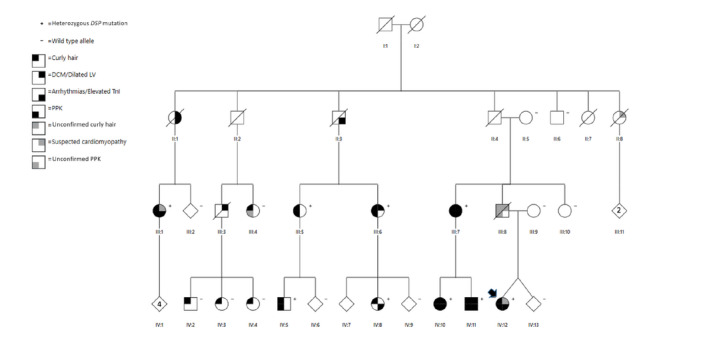
The family pedigree.
The index is marked by an arrow. Genotype: +, heterozygous for DSP c.2493delA p.(Glu831Aspfs*33) mutation; −, wild type allele. Symbols represent the following; right upper quadrant: black for diagnosed DCM or dilated left ventricle, grey for slightly dilated left ventricle or suspected cardiomyopathy; right lower quadrant; black for verified arrhythmias or unexplained elevated TnI concentration, left upper quadrant: black for curly hair, grey for anamnestic reported curly hair that has not been clinically confirmed; left lower quadrant: black for clinically verified PPK, grey for anamnestic reported PPK that has not been clinically confirmed (III:8) or slight hyperkeratosis observed, fitting due to normal friction (III:4). Abbreviations: DCM, dilated cardiomyopathy; TnI, Tropnonin I; PPK, palmoplantar keratoderma.
Cardiac evaluation
The index and 12 available family members underwent cardiac evaluation, including physical examination, echocardiography, 12‐lead electrocardiogram (ECG), appropriate laboratory tests, Holter monitoring and cardiac magnetic resonance imaging (CMR).
DCM was diagnosed using the following criteria; left ventricular systolic dysfunction (LVEF <45%) and left ventricular end‐diastolic diameter (LVEDD) >27 mm/m2 when no abnormal loading conditions or severe coronary artery disease were detected. Patients were considered to be affected when presented with unexplained elevated Troponin I (TnI) concentration or > 100/24 h ventricular extrasystoles (VES) in Holter monitoring, sinus node dysfunction, conduction defects, dilatation (>112% of the predicted LVEDD) or impaired systolic function of the left ventricle (LVEF <45%).
CMR and image analysis are described in detail in the supplemental material (Appendix S1).
CMR diagnostic criteria
Pathologic trabeculation was assessed using the following CMR diagnostic criteria for left‐ventricular non‐compaction cardiomyopathy (LVNC): ratio of non‐compacted to compacted myocardium diameter of ≥2.3 in end‐diastole, mass of non‐compacted myocardium >25%. 7 , 8 LV volumetric was considered abnormal in males and females respectively using the following values: EDV >235 mL and 174 mL, EDV/BSA >112 mL/m2 and 99 mL/m2, SV <66 mL and 61 mL. 9 RV volumetric was considered abnormal in males and females using the following values: EDV >243 mL and 178 mL, EDV/BSA >114 mL/m2 and > 103 mL/m2 and SV <60 mL and 45 mL. 9 Myocardial T1 native relaxation time > 1000 ms was considered increased. 10 , 11
Autopsy material
Autopsy results and death certificates were obtained and examined for eight deceased family members (II:1, II:2, II:3, II:4, II:7, II:8, III:3, III:8) (Fig. 1) in order to find out a possible link to DCM. Additional medical records were available from II:1 and II:3.
Results
Index patient with focal PPK and DSP c.2493delA p.(Glu831Aspfs*33) mutation
The index patient, a 25‐year‐old woman (IV:12) (Fig. 1) was first referred to the Helsinki University Hospital Dermatology clinic because of severe focal PPK on the heels and plantar surface of the toes since the age of one year. The hyperkeratosis showed aquagenic whitening. There was occasional painful fissuring but no blistering, fungal or bacterial skin infections (Table 1, Fig. 2). She had pronounced palmar creasing but no palmar hyperkeratosis and no other skin areas were affected. She had hyperhidrosis of the face and armpits and her hair was slightly curly, but hair microscopy did not show structural abnormalities.
Table 1.
Dermatological manifestations and major cardiac features of the nine patients with DSP c.2493delA p.(Glu831Aspfs*33) mutation
| Patient | Index IV:12 | III:7 | IV:10 | IV:11 | III:5 | III:6 | IV:8 | III:1 | IV:5 |
|---|---|---|---|---|---|---|---|---|---|
| Gender | F | F | F | M | F | F | F | F | M |
| Age at dermatologic evaluation (y) | 24 | 66 | 38 | 32 | 54 | 50 | 23 | 62 | 31 |
| PPK type | Focal | Diffuse | Focal | Focal | Focal | Focal | − | Focal | Focal |
| PPK onset | 1 y | 27 y | Infancy | Puberty | 30 y | Puberty | − | Infancy | Infancy |
| Transgrediens | − | − | + | − | − | − | − | − | − |
| Progrediens | − | + | − | − | − | − | − | − | − |
| Aquagenic whitening | + | − | − | + | + | + | − | + | − |
| Hyperhidrosis | + face, armpits | + feet, hands | NA | − | − | + armpits | + feet, hands, armpits | − | − |
| Curly hair | + | + until early adulthood | +/− wavy and thin | + since prepuberty | + | + | + | + since 30 y | + |
| Hair microscopy | Normal | Normal | Normal | Normal | Normal | Trichodystrophy | Normal | NA | NA |
| Other dermatologic | Atopic eczema | − | Atopic eczema | Atopic eczema | − | − | Atopic eczema | − | − |
| Arrhythmias | + | + | + | + | − | − | − | − | NA |
| DCM | − | + | + | + | − | − | − | − | NA |
| Other cardiac abnormalities | Trabeculation in CMR | Trabeculation in CMR | Trabeculation in CMR | − | − | CMR slightly abnormal | Heart failure | NA |
Transgrediens, PPK contiguously extending on the dorsal surfaces of the feet, hands, Achilles tendons and wrists; progrediens, worsening of PPK with age. Patients III:I and IV:3 were evaluated by telephone interview, photographs and patient records only. Arrhythmias means ventricular extrasystoles, VES, >100/24 h, SVES, supraventricular extrasystoles >100/24 h or NSVT, non‐sustained ventricular tachycardia.
Abbreviations: F, female; M, male, y, years; PPK, palmoplantar keratoderma; NA, not available; DCM, dilated cardiomyopathy; CMR, cardiac magnetic resonanceimaging.
Figure 2.
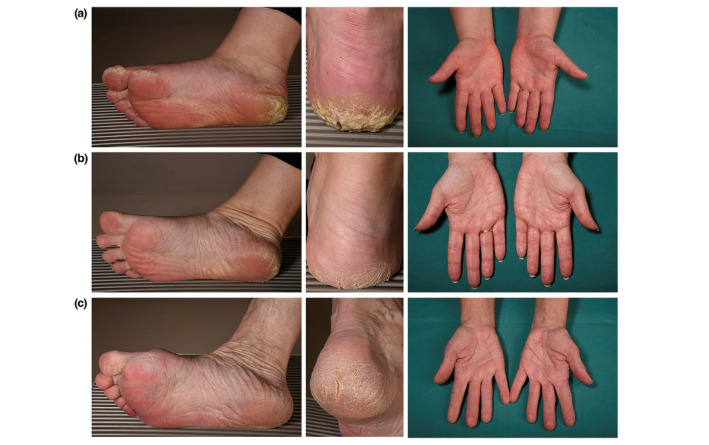
Dermatological findings in patients with DSP c.2493delA p.(Glu831Aspfs*33) mutation.
Variable focal hyperkeratosis of the heels, toes and soles and palmar hyperlinearity (from mild to more prominent) in patients with DSP c.2493delA p.(Glu831Aspfs*33) mutation: (a) index IV:12, (b) III:7, and (c) IV:10.
An in‐house NGS PPK gene panel revealed a heterozygous DSP mutation c.2493delA p.(Glu831Aspfs*33) in exon 18 leading to a frameshift and premature termination of translation. The mutation could not be found in online variant or mutation databases (gnomAD v.2.1.1 / v3.1, LOVD v3.0, ClinVar, HGMD Professional 2020.3). Several pathogenic frameshift, nonsense and splice site mutations in DSP have been described located in the near proximity of the identified deletion, e.g. DSP c.2497C > A (p.Gln833Ter) (ClinVar Variation ID: 572718) and c.2498dup (p.Lys834Glufs*17) (ClinVar Variation ID: 946412). 12 DSP is predicted to be highly loss of function intolerant (pLI = 1.00, gnomAD). 13
The index patient had a history of palpitations during rest and exercise and thorough cardiac evaluation revealed ventricular extrasystoles (VES) 377/24 h in Holter‐monitoring and beta‐blocker treatment was started (Tables 2 and 3). In echocardiography her LVEF was 69% and LVEDD 54 mm, the left ventricle was considered normal (106% of the estimated LVEDD using the Henry's formula), without systolic dysfunction. 14 , 15 Her TnI levels were slightly elevated, 67 ng/L (normal range < 45 ng/L). On CMR, her left ventricular function was otherwise normal but subepicardial inferolateral and inferior late gadolinium enhancement (LGE) were observed. Some edema was observed regionally in the lateral wall. In positron emission tomography‐computed tomography (PET‐CT) no sign of amyloidosis or inflammation was observed.
Table 2.
The cardiac evaluation of thirteen family members with or without DSP c.2493delA p.(Glu831Aspfs*33) mutation
| ID | DSP c.2493delA p.(Glu831Aspfs*33) | Age (M/F) | Arrhythmias | PM, ICD | LVEF (%) | LVEDD (mm) | proBNP (ng/l) | TnI (ng/l) | Phenotype | Age at dg | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Index IV:12 | + | 25F | VES | no | 69 | 54 | 105 | 67 | affected | − | Henry's formula 106% |
| IV:10 | + | 38F | VES, NSVT | no | 40 | 58 | 334 | 76 | DCM | 38 | |
| IV:11 | + | 32 M | VES, SVES, NSVT | ICD | 30 | 75 | 132 | 15 | DCM | 32 | |
| III:7 | + | 66F | VES, SVES, NSVT | ICD | 41 | 62 | 584 | 141 | DCM | 66 | |
| III:6 | + | 50F | no | no | 69 | 54 | 115 | 19 | affected | − | Henry's formula: 119.0% |
| IV:8 | + | 23F | no | no | 55 | 45 | 96 | 437 | affected | − | |
| III:5 | + | 54F | no | no | 68 | 44 | 38 | <3 | unaffected | − | |
| III:1 | + | 62F | no | no | NA | NA | NA | 263 | HF | NA | Aetiology of HF is not known |
| III:10 | − | 59F | no | no | 64 | 48 | 51 | <3 | unaffected | − | |
| IV:3 | − | 32F | WPW | no | 55 | 46 | <35 | <3 | unaffected | − | |
| IV:4 | − | 34F | no | no | 61 | 50 | 137 | 5 | unaffected | − | |
| IV:2 | − | 19 M | no | no | 56 | 59 | 89 | 4 | unaffected | − | Henry's formula: 112%, LV size normal in CMR |
| III:4 | − | 57F | no | no | 83 | 41 | NA | NA | unaffected | − |
Symbols and abbreviations: Genotype: +, heterozygous for DSP c.2493delA p.(Glu831Aspfs*33); −, wild type allele; NA, not available; Age (M/F), Age at evaluation; M, male; F, female; Arrhythmias: VES, ventricular extrasystoles >100/24 h; SVES, supraventricular extrasystoles >100/24 h; NSVT, non‐sustained ventricular tachycardia; WPW, Wolf‐Parkinson‐White syndrome; PM, pacemaker; ICD, implantable cardioverter‐defibrillator; LVEF, left ventricular ejection fraction (%); LVEDD, left ventricular end‐diastolic diameter (mm); ProBNP, pro b‐type natriuretic peptide (ng/L); TnI, Troponin I (ng/L); Age at dg, age at diagnosis of cardiomyopathy; Phenotype, affected if the patient had unexplained elevated (>45 ng/L) TnI concentration, arrhythmias in Holter monitoring, systolic dysfunction (LVEF <45%) or dilated LV (>112% of the predicted LVEDD), unaffected if the previous criteria is not valid.
CMR, cardiac magnetic resonance imaging; DCM, dilated cardiomyopathy; HF, heart failure; Other, other relevant clinical information: Henry's formula: LV size compared to the estimated LVEDD using the Henry's formula.
Table 3.
Cardiac MRI results of eleven family members with or without the DSP c.2493delA p.(Glu831Aspfs*33) mutation
| Left ventricle | Right ventricle | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | DSP c.2493delA p.(Glu831Aspfs*33) | Age (M/F) | EF (%) | EDV (ml) | EDV / BSA (ml/m2) | SV (ml) | EF (%) | EDV (ml) | EDV / BSA (ml/m2) | SV (ml) | Trabeculation | LGE |
| Index IV:12 | + | 25F | 62 | 210 | 92 | 131 | 73 | 177 | 78 | 129 | − | subepicardial |
| IV:10 | + | 38F | 38 | 208 | 114 | 79 | 68 | 112 | 62 | 76 | + | subepicardial |
| IV:11 | + | 32 M | 33 | 420 | 203 | 137 | 61 | 223 | 108 | 135 | + | subepicardial |
| III:7 | + | 66F | 34 | 223 | 129 | 76 | 62 | 122 | 71 | 75 | + | transmural |
| III:6 | + | 50F | 60 | 160 | 94 | 96 | 65 | 150 | 88 | 98 | − | no |
| IV:8 | + | 23F | 66 | 156 | 87 | 103 | 63 | 159 | 89 | 100 | − | subepicardial |
| III:5 | + | 54F | 69 | 146 | 75 | 100 | 67 | 151 | 78 | 101 | − | no |
| III:10 | − | 59F | 60 | 150 | 80 | 90 | 69 | 129 | 69 | 89 | − | no |
| IV:3 | − | 32F | 57 | 128 | 61 | 72 | 51 | 140 | 67 | 72 | − | no |
| IV:4 | − | 34F | 58 | 161 | 84 | 94 | 61 | 152 | 78 | 92 | − | no |
| IV:2 | − | 19 M | 62 | 197 | 82 | 121 | 61 | 197 | 82 | 121 | − | no |
Symbols and abbreviations: MRI, magnetic resonance image; genotype +, heterozygous for DSP c.2493delA p.(Glu831Aspfs*33); − wild type allele.
NA, not available; age (M/F), age at evaluation; M, male; F, female; EF, ejection fraction (%); EDV, end diastolic volume (ml); EDV / BSA, end diastolic volume normalized to body surface area (ml/m2); SV, systolic volume (ml); Trabeculation +, pathogenic trabeculation fulfilling LVNC criteria; Trabeculation ‐, no pathologic trabeculation; LGE, late gadolinium enhancement.
Association of dermatologic and cardiac abnormalities in a multigenerational family
To further investigate the association of dermatologic and cardiac abnormalities with the DSP c.2493delA p.(Glu831Aspfs*33) mutation, a detailed family history revealed several relatives in multiple generations with similar PPK and cardiac symptoms, mainly mild arrhythmias (Figs 1 and 2). Among altogether 21 family members tested, including the index, the DSP c.2493delA p.(Glu831Aspfs*33) mutation was found in 9/21, 8/9 had plantar PPK of varying severity (Table 1 and Fig. 1). Focal PPK was seen in 7/9 while one had a more diffuse PPK. IV:8 did not have PPK at the age of 23. The earliest onset of PPK was at one year's age and the latest at age 30. Only patient III:7 had mild hyperkeratosis also on the palms but the others presented only pronounced palmar creasing (Figs 1 and 2). Epidermal hyperkeratosis was not seen elsewhere. 5/9 reported aquagenic whitening of the hyperkeratoses and only 2/9 palmoplantar hyperhidrosis.
Altogether 13 members of the family had wavy/curly hair, including all the patients with DSP mutation and four unaffected individuals (III:4, IV:2, IV:3 and IV:4). In affected individuals hair microscopy revealed trichodystrophy in one, but other six evaluated had no abnormalities (Table 1). No one had nail dystrophy and only patient III:7 reported dental problems with severe dental caries of the permanent teeth. Curly/wavy hair was not evident in all from early childhood, as one reported it from puberty and another one from 30 years of age.
No PPK was seen in eleven unaffected individuals but one of them (III:4) had slight hyperkeratosis on the soles, fitting due to normal friction (not shown).
Eight patients (III:1, III:5, III:6, III:7, IV:8, IV:10, IV:11, IV:12) with the DSP c.2493delA p.(Glu831Aspfs*33) mutation, including the index, underwent thorough cardiac evaluation (Tables 2 and 3 and Fig. S1), but for III:1 only previous medical records were available. DCM diagnostic criteria were fulfilled by 3/8 (III:7, IV:10, IV:11) of the patients. In addition, III:6 had a dilated left ventricle in echocardiography (Table 2) without systolic dysfunction. III:1 had chronic heart failure according to medical records but could not be assessed, and the cause of heart failure remained unknown to us. Two patients (III:5, IV:8) had neither dilated left ventricle nor systolic dysfunction. In ECG, 3/8 had lateral T‐inversions and one had lateral and inferior T‐inversions. 4/8 underwent Holter monitoring and all had >100 VES/24 h), three had also episodes of non‐sustained ventricular tachycardia (NSVT), and in addition two of them had >100 supraventricular extrasystoles (SVES)/24 h. None had severe ventricular arrhythmias (sustained ventricular tachycardia or ventricular fibrillation). 5/8 reported palpitations, but none reported chest pain or dyspnea. An implantable cardioverter‐defibrillator (ICD) was implanted in 2/8.
CMR was conducted on seven patients and it revealed end diastolic volume (EDV) over the normal range in four (Table 3), III:6 had borderline EDV, IV:8 had slightly abnormal findings, III:5 had normal findings. Three (III:7, IV:10, IV:11) had pathological trabeculation fulfilling LVNC criteria. 7 , 9 Five patients had LGE, and in four (III:7, IV:8, IV:10, IV:12) it was observed in the inferolateral wall in basal and mid‐ventricular regions. In the fifth patient (IV:11) extensive LGE was observed in left ventricular myocardium. Four of the five had subepicardial LGE and one (III:7) had transmural LGE. In those patients who had LGE, T1 native relaxation times were increased in the same regions.
Troponin I (TnI) levels were measured from eight patients and were above the normal range (<45 ng/L) in five (Table 2). Their TnI levels ranged from slightly elevated (67 ng/L) to high (437 ng/L). None had any signs of myocardial infarction nor myocarditis at that time.
To rule out other genetic factors predisposing to cardiac abnormalities in the family, five family members without the DSP mutation also underwent cardiac evaluation (Tables 2 and 3). Two reported palpitations, but none had chest pain or dyspnea. All five had normal findings in ECG. One underwent Holter monitoring due to clinical indications not related to our study, with normal findings. Four had no history of arrhythmias, one had history of Wolf‐Parkinson‐White (WPW) syndrome. IV:2 had a dilated left ventricle (112% of the approximated LVEDD. The approximated LVEDD was calculated using the Henry's formula) on echocardiography but in CMR the ventricle size was considered normal (Tables 2 and 3). 15 CMR was normal also in three other family members studied. None had pathologic trabeculation. TnI levels were measured from three, all with normal findings. None of the family members without the DSP mutation were considered affected.
Deceased family members
The age of death of the eight deceased family members varied from 52 to 81 years. Only one (III:3) died due to cardiac reasons. II:8 had atopic eczema and was the only one with dermatologic findings. 2/8 (II:8, III:8) had an enlarged heart, though ventricular dilatation was not specified. Neither of them had coronary artery disease or valvular defects in autopsy. II:3 had history of SVT and VES in medical records. III:3 died at the age of 59, he had enlarged LV and right atrium, LV hypertrophy and mild coronary artery disease in autopsy. II:1 had history of heart failure and VES, in autopsy enlarged heart, patchy fibrosis, LV hypertrophy and coronary artery disease were observed, cause of death was pulmonary embolism. II:2, II:4 and II:7 had no cardiac abnormalities in autopsy.
Discussion
We identified a large multigenerational family with a novel autosomal dominant (AD) DSP c.2493delA p.(Glu831Aspfs*33) mutation in exon 18 associated with focal PPK as an early cutaneous sign, and dilatation of the left ventricle or DCM with arrhythmia susceptibility as major cardiac manifestations. Focal PPK was evident by one year of age in two patients, by puberty in one patient and at the latest appeared by age 30. PPK preceded cardiac symptoms and findings in all. The manifestation of cardiac symptoms was variable and age‐dependent, varying from 23 to 66 years of age. Wavy/curly hair was detected in family members with and without the DSP mutation, thus this feature could not be definitely linked to the mutation. Three of the nine patients with the DSP mutation fulfilled DCM diagnostic criteria and all three had pathologic trabeculations. 7 , 8 The age at DCM diagnosis varied from 32 to 66 years. None met ARVC diagnostic criteria, and none had right ventricular dilatation or dysfunction in CMR or echocardiography. In CMR five of the patients had LGE and increased segmental T1 native mapping times referring to myocardial fibrosis. 16 Mid‐wall septal pattern of LGE is most often seen in DCM patients, however subepicardial, diffuse and focal patterns have also been reported. 17 In our patient group the pattern of LGE was mostly subepicardial. Subepicardial LGE has been reported with patients suffering from DSP cardiomyopathy and patients with left‐dominant arrhythmogenic cardiomyopathy. 18 , 19 , 20
The major cardiac findings in our subjects are dilatation of the left ventricle, elevated TnI concentration and tendency for VES as well as SVES. None of the patients had severe ventricular arrhythmias and none of the deceased family members experienced sudden cardiac death. The participants were relatively asymptomatic when diagnosed with DCM.
We found TnI levels elevated in five patients with the mutation, and one of them had elevated TnI levels for unknown reasons in multiple occasions. All patients with elevated TnI levels had normal renal function. Elevated TnI concentrations are used for diagnosing myocardial infarct or myocarditis, however, elevated TnI concentrations reflect myocardial damage but do not directly reveal the aetiology. In our patients the elevated TnI levels could be explained by the cardiomyopathy. Especially in young patients with clinical suspicion of myocarditis, fluctuating TnI concentrations could be a sign for clinicians that further cardiac evaluation for possible cardiomyopathy is needed. The possibility of inflammatory episodes related to DSP cardiomyopathy has been proposed. 21 and the inflammatory aspect has been noted by other research groups as well. 18 , 22 Reichl et al. reported a case of a young patient with acute myocarditis as the first presentation of cardiomyopathy. 22 Smith et al. reported cardiomyopathy patients with DSP mutation having episodes of myocardial injury and troponin elevations and some patient were misdiagnosed with myocarditis or sarcoidosis initially. 18
DSP mutations are associated with highly variable AD or autosomal recessive (AR) inherited cutaneous disorders with or without cardiac manifestations. However, isolated PPK, as reported here with the DSP c.2493delA p.(Glu831Aspfs*33) mutation, has not been associated with cardiac involvement and thus warranting further cardiac evaluation. 2 Carvajal syndrome, with striate PPK, congenital woolly hair, dilated cardiomyopathy and sometimes skin fragility, was the first AR syndrome related to DSP mutations. 2 , 23 , 24 In a similar AD inherited form, additional oligodontia and onychodystrophy were also reported. 25 Furthermore, erythrokeratodermia‐cardiomyopathy syndrome is characterized by general erythrokeratodermia, alopecia, onychodystrophy, enamel defects and left ventricular dilatation. 26 Woolly hair or other major structural hair defects were undetectable in our patients. Nails and teeth abnormalities and skin fragility typical for some DSP mutations were absent. 2 DSP mutations cause also lethal acantholytic epidermolysis bullosa, a fatal AR disorder characterized by extensive postnatal shedding of the epidermis, universal alopecia, anonychia, malformed ears and possible cardiomyopathy. 27 DSP mutations causing the combination of PPK and hair shaft anomalies associated with severe left‐dominant arrhythmogenic cardiomyopathy, that usually appears later in life than skin symptoms, were proposed as a new clinical entity, thus sharing similarities with our family. 2 However, not all DSP mutations have been associated with cardiac dysfunction. AD inherited SAM‐like syndrome with severe dermatitis, multiple allergies, metabolic wasting or AR inherited DSP mutations with focal or diffuse PPK with woolly hair and skin fragility do not appear to involve cardiac anomalies. 28 , 29 , 30
Missense, loss‐of function, insertion, intronic and truncating DSP mutations have been previously described associated with dermatological and/or cardiac features. 1 Most of such mutations are AR homozygous or compound heterozygous and have been described in exon 24 with variable phenotypes containing skin fragility, woolly hair, acantholytic epidermolysis bullosa, Carvajal syndrome, ARVC, PPK and mucocutaneus blistering. In addition, mutations have also been described in intron 7 and exons 7–9,14–18,20 and 23. 1 , 2 Cardiocutaneous syndromes, mainly Carvajal syndrome with truncating AR DSP mutations have been described with PPK, woolly hair or hypotrichosis and ARVC with or without skin fragility. 1 , 2 Haploinsufficiency has previously been described as well with DSP mutations. 1 A heterozygous DSP c.6310delA, p.(Thr2104Glnfs*12) mutation has previously been described with left ventricular dilatation, dysfunction and major ventricular arrhythmias, but dermatological examination was not performed for the affected patients. 21 An AD nonsense DSP c.991C > T mutation has been described in striate PPK without cardiac involvement and an AD splice site mutation c.939 + 1A > G has been linked to striate PPK, left‐dominant arrhythmogenic cardiomyopathy and ARVC. 1 , 2 Phenotypic variation of the dermatological symptoms and the presence of cardiac involvement does not seem to correlate with the location or the type of DSP mutation. 1 , 2 It is also possible that the cardiac involvement in patients with DSP mutations and PPK has been overlooked as cardiac findings and symptoms have been reported to occur later at an adult age, supported by our findings as well. 21
DSP mutations can cause cardiomyopathy without dermatologic manifestations, but it is possible that milder dermatologic manifestations may have been overlooked in patients. DSP mutations have been reported to cause ARVC, but they are also known to cause DCM. 20 , 31 , 32 , 33 , 34 , 35 , 36 , 37 Recent studies have shown that mutations in DSP seem to cause biventricular or left‐dominant forms of ARVC instead of the well‐known right‐dominant disease pattern. 32 , 38 , 39 , 40 In one genotype–phenotype study, LV dysfunction was more common in ARVC patients with a truncating DSP variant. 32 Consensus criteria for all ARVC phenotypes are still lacking. In a recent study, Smith et al. characterize the phenotype of DSP cardiomyopathy as cardiomyopathy including systolic dysfunction, episodes of myocardial inflammation and fibrosis. 18
In conclusion, the DSP c.2493delA p.(Glu831Aspfs*33) mutation causes a phenotype of focal PPK and arrhythmogenic cardiomyopathy that may be present with classical features of DCM or mild to severe arrhythmias without structural and functional changes seen in cardiomyopathy. PPK alone should alert genetic testing as DSP mutations might cause only focal PPK without wavy/curly hair. When a likely pathogenic or pathogenic DSP variant is found, thorough cardiac evaluation (ECG, Holter‐monitoring, TnI and pro‐BNP levels, echocardiography and CMR) is warranted even in asymptomatic patients. This study highlights the importance of genetic testing in PPK patients, by a novel truncating DSP mutation causing focal PPK with varying severity with DCM and arrhythmias.
Supporting information
Appendix S1 Supplementary material.
Figure S1 Cardiac MRI of patient IV:11 with DSP c.2493delA p.(Glu831Aspfs*33) mutation.
Acknowledgements
We thank the family for participating in this study. We would also like to acknowledge Ms. Alli Tallqvist and Ms. Auli Saarinen for their technical assistance. The patients in this manuscript have given written informed consent to publication of their case details.
Funding sources
This study was supported by Helsinki University Hospital (HUS) research fund projects TYH2018221, TYH2020210, Y2020SK004, Y1016SK004, TYH2021105, Y780021046, Y780020063 (AR, TH, KHJ, KK), the Finnish Dermatological Society (VK, LH), Emil Aaltonen foundation (LH), the Finnish Medical Foundation (LH), Finnish Cultural Foundation (LH), Sigrid Jusélius Foundation (JK), Swedish Psoriasis Foundation (JK), Jane and Aatos Erkko Foundation (JK), the Swedish Research Council (JK), the Finnish Foundation for Cardiovascular Research (KH, TH), and Aarne Koskelo Foundation (TH). None of the funders were involved in the study design, data collection, data analysis, or manuscript preparation.
Conflicts of interest
Dr Koskenvuo is a co‐founder and previous shareholder and current full‐time employee of Blueprint Genetics, which offers genetic diagnostic services. Dr Hannula‐Jouppi is a clinical consultant for Blueprint Genetics. The other authors report no conflicts of interest.
Data availability statement
Data available on request due to privacy/ethical restrictions.
References
- 1. Pigors M, Schwieger‐Briel A, Cosgarea R et al. Desmoplakin mutations with palmoplantar keratoderma, woolly hair and cardiomyopathy. Acta Derm Venereol 2015; 95: 337–340. [DOI] [PubMed] [Google Scholar]
- 2. Polivka L, Bodemer C, Hadj‐Rabia S. Combination of palmoplantar keratoderma and hair shaft anomalies, the warning signal of severe arrhythmogenic cardiomyopathy: a systematic review on genetic desmosomal diseases. J Med Genet 2016; 53: 289–295. [DOI] [PubMed] [Google Scholar]
- 3. Guerra L, Castori M, Didona B, Castiglia D, Zambruno G. Hereditary palmoplantar keratodermas. Part II: syndromic palmoplantar keratodermas ‐ diagnostic algorithm and principles of therapy. J Eur Acad Dermatol Venereol 2018; 32: 899–925. [DOI] [PubMed] [Google Scholar]
- 4. Has C, Technau‐Hafsi K. Palmoplantar keratodermas: clinical and genetic aspects. J Dtsch Dermatol Ges 2016; 14: 123–139. [DOI] [PubMed] [Google Scholar]
- 5. Samuelov L, Sarig O, Harmon RM et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet 2013; 45: 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Genomics England PanelApp. https://panelapp.genomicsengland.co.uk/panels/215/. Last accessed October 27 2021.
- 7. Petersen SE, Selvanayagam JB, Wiesmann F et al. Left ventricular non‐compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol 2005; 46: 101–105. [DOI] [PubMed] [Google Scholar]
- 8. Grothoff M, Pachowsky M, Hoffmann J et al. Value of cardiovascular MR in diagnosing left ventricular non‐compaction cardiomyopathy and in discriminating between other cardiomyopathies. Eur Radiol 2012; 22: 2699–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alfakih K, Plein S, Thiele H, Jones T, Ridgway JP, Sivananthan MU. Normal human left and right ventricular dimensions for MRI as assessed by turbo gradient echo and steady‐state free precession imaging sequences. J Magn Reson Imaging 2003; 17: 323–329. [DOI] [PubMed] [Google Scholar]
- 10. Reiter G, Reiter C, Kräuter C, Fuchsjäger M, Reiter U. Cardiac magnetic resonance T1 mapping. Part 1: aspects of acquisition and evaluation. Eur J Radiol 2018; 109: 223–234. [DOI] [PubMed] [Google Scholar]
- 11. Haaf P, Garg P, Messroghli DR, Broadbent DA, Greenwood JP, Plein S. Cardiac T1 mapping and extracellular volume (ECV) in clinical practice: a comprehensive review. J Cardiovasc Magn Reson 2016; 18: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. ClinVar. https://www.ncbi.nlm.nih.gov/clinvar. Last accessed March 28 2021.
- 13. Karczewski KJ, Francioli LC, Tiao G et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581: 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mestroni L, Maisch B, McKenna WJ et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative research Group of the European Human and Capital Mobility Project on familial dilated cardiomyopathy. Eur Heart J 1999; 20: 93–102. [DOI] [PubMed] [Google Scholar]
- 15. Henry WL, Gardin JM, Ware JH. Echocardiographic measurements in normal subjects from infancy to old age. Circulation 1980; 62: 1054–1061. [DOI] [PubMed] [Google Scholar]
- 16. Yanagisawa F, Amano Y, Tachi M, Inui K, Asai K, Kumita S. Non‐contrast‐enhanced T1 mapping of dilated cardiomyopathy: comparison between native T1 values and late gadolinium enhancement. Magn Reson Med Sci 2019; 18: 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parsai C, O'Hanlon R, Prasad SK, Mohiaddin RH. Diagnostic and prognostic value of cardiovascular magnetic resonance in non‐ischaemic cardiomyopathies. J Cardiovasc Magn Reson 2012; 14: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith ED, Lakdawala NK, Papoutsidakis N et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 2020; 141: 1872–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Augusto JB, Eiros R, Nakou E et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype‐imaging phenotype study. Eur Heart J Cardiovasc Imaging 2020; 21: 326–336. [DOI] [PubMed] [Google Scholar]
- 20. Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol 2011; 9: 223–233. [DOI] [PubMed] [Google Scholar]
- 21. Heliö K, Kangas‐Kontio T, Weckström S et al. DSP p.(Thr2104Glnfs*12) variant presents variably with early onset severe arrhythmias and left ventricular cardiomyopathy. BMC Med Genet 2020; 21: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reichl K, Kreykes SE, Martin CM, Shenoy C. Desmoplakin variant‐associated arrhythmogenic cardiomyopathy presenting as acute myocarditis. Circ Genom Precis Med 2018; 11: e002373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carvajal‐Huerta L. Epidermolytic palmoplantar keratoderma with woolly hair and dilated cardiomyopathy. J Am Acad Dermatol 1998; 39: 418–421. [DOI] [PubMed] [Google Scholar]
- 24. Norgett EE, Hatsell SJ, Carvajal‐Huerta L et al. Recessive mutation in desmoplakin disrupts desmoplakin‐intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet 2000; 9: 2761–2766. [DOI] [PubMed] [Google Scholar]
- 25. Norgett EE, Lucke TW, Bowers B, Munro CS, Leigh IM, Kelsell DP. Early death from cardiomyopathy in a family with autosomal dominant striate palmoplantar keratoderma and woolly hair associated with a novel insertion mutation in desmoplakin. J Invest Dermatol 2006; 126: 1651–1654. [DOI] [PubMed] [Google Scholar]
- 26. Boyden LM, Kam CY, Hernández‐Martín A et al. Dominant de novo DSP mutations cause erythrokeratodermia‐cardiomyopathy syndrome. Hum Mol Genet 2016; 25: 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bolling MC, Veenstra MJ, Jonkman MF et al. Lethal acantholytic epidermolysis bullosa due to a novel homozygous deletion in DSP: expanding the phenotype and implications for desmoplakin function in skin and heart. Br J Dermatol 2010; 162: 1388–1394. [DOI] [PubMed] [Google Scholar]
- 28. McAleer MA, Pohler E, Smith FJ et al. Severe dermatitis, multiple allergies, and metabolic wasting syndrome caused by a novel mutation in the N‐terminal plakin domain of desmoplakin. J Allergy Clin Immunol 2015; 136: 1268–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vakkilainen S, Puhakka L, Klemetti P et al. Novel DSP Spectrin 6 region variant causes neonatal erythroderma, failure to thrive, severe herpes simplex infections and brain lesions. Acta Derm Venereol 2019; 99: 789–796. [DOI] [PubMed] [Google Scholar]
- 30. Whittock NV, Wan H, Morley SM et al. Compound heterozygosity for non‐sense and mis‐sense mutations in desmoplakin underlies skin fragility/woolly hair syndrome. J Invest Dermatol 2002; 118: 232–238. [DOI] [PubMed] [Google Scholar]
- 31. Campuzano O, Alcalde M, Allegue C et al. Genetics of arrhythmogenic right ventricular cardiomyopathy. J Med Genet 2013; 50: 280–289. [DOI] [PubMed] [Google Scholar]
- 32. Castelletti S, Vischer AS, Syrris P et al. Desmoplakin missense and non‐missense mutations in arrhythmogenic right ventricular cardiomyopathy: genotype‐phenotype correlation. Int J Cardiol 2017; 249: 268–273. [DOI] [PubMed] [Google Scholar]
- 33. Rampazzo A, Nava A, Malacrida S et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet 2002; 71: 1200–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mazzarotto F, Tayal U, Buchan RJ et al. Reevaluating the genetic contribution of monogenic dilated cardiomyopathy. Circulation 2020; 141: 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Akinrinade O, Ollila L, Vattulainen S et al. Genetics and genotype‐phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J 2015; 36: 2327–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McNally EM, Mestroni L. Dilated cardiomyopathy: genetic determinants and mechanisms. Circ Res 2017; 121: 731–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Elliott P, O'Mahony C, Syrris P et al. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ Cardiovasc Genet 2010; 3: 314–322. [DOI] [PubMed] [Google Scholar]
- 38. López‐Ayala JM, Gómez‐Milanés I, Sánchez Muñoz JJ et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: characterizing a phenotype. Europace 2014; 16: 1838–1846. [DOI] [PubMed] [Google Scholar]
- 39. Bhonsale A, Groeneweg JA, James CA et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy‐associated mutation carriers. Eur Heart J 2015; 36: 847–855. [DOI] [PubMed] [Google Scholar]
- 40. Towbin JA, McKenna WJ, Abrams DJ et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019; 16: e301–e372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplementary material.
Figure S1 Cardiac MRI of patient IV:11 with DSP c.2493delA p.(Glu831Aspfs*33) mutation.
Data Availability Statement
Data available on request due to privacy/ethical restrictions.