

Abstract
Breaking down lignin into smaller units is the key to generate high value‐added products. Nevertheless, dissolving this complex plant polyphenol in an environment‐friendly way is often a challenge. Levulinic acid, which is formed during the hydrothermal processing of lignocellulosic biomass, has been shown to efficiently dissolve lignin. Herein, levulinic acid was evaluated as a medium for the reductive electrochemical depolymerization of the lignin macromolecule. Copper was chosen as the electrocatalyst due to the economic feasibility and low activity towards the hydrogen evolution reaction. After depolymerization, high‐resolution mass spectrometry and nuclear magnetic resonance spectroscopy revealed lignin‐derived monomers and dimers. A predominance of aryl ether and phenolic groups was observed. Depolymerized lignin was further evaluated as an anti‐corrosion coating, revealing enhancements on the electrochemical stability of the metal. Via a simple depolymerization process of biomass waste in a biomass‐based solvent, a straightforward approach to produce high value‐added compounds or tailored biobased materials was demonstrated.
Keywords: coating, depolymerization, electrocatalysis, levulinic acid, lignin
Lignin breakdown: The valorization of lignin is key to generate value‐added compounds. For this, high‐yield and green strategies to convert lignin to fine chemicals are intensively investigated. A reductive electrochemical route provides a feasible and sustainable way to obtain lignin‐derived aromatic monomers and dimers from the solubilization and further depolymerization of lignin in levulinic acid, a biomass‐based solvent.

Introduction
While facing a global population increase, society's dependence on non‐renewable sources represents a tremendous concern. Therefore, the sustainable production of fuels and valuable chemicals based on the efficient use of renewable resources has become an urgent matter.[ 1 , 2 , 3 ] In this context, lignocellulose biorefinery has been recognized as a potential candidate to replace petroleum refinery, while being a carbon‐neutral and non‐food feedstock.[ 4 , 5 , 6 , 7 ] Lignocellulosic biomass consists mainly of cellulose, lignin, hemicelluloses, and extractives. Comprising three different phenylpropane monomer units (monolignols), lignin is the second most abundant macromolecule on earth, while its annual production is estimated in the range of 100 million tons. [8] In this framework, technical lignins find a prominent space as potential resources to produce aromatic chemicals and phenolic‐containing macromolecules.[ 9 , 10 , 11 , 12 , 13 , 14 ]
The lignin macromolecule, often referred to as a polymer (or biopolymer), can be successively broken (or “depolymerized”) to generate high value‐added products.[ 9 , 10 ] This depolymerization process has represented a challenge since the 1930s, when the first findings in the field were published.[ 9 , 12 ] Given its heterogeneous nature, a wide range of phenolic‐containing polymers and aromatic compounds can be obtained via lignin depolymerization.[ 12 , 13 , 14 ]
For further valorization, efficient depolymerization strategies have been investigated to generate functionalized aromatic compounds at room temperature and with good yields. [16] Among different catalytic approaches, electrocatalysis (EC) has found a prominent space due to a better integration with the principles of green chemistry.[ 10 , 17 , 18 ] Furthermore, while being possible to control cell potential, EC enhances the catalyst selectivity and activity and allows for the use of mild reaction conditions, such as room temperature and green solvents.[ 9 , 19 , 20 ] Due to the possibility of carbonylation of α‐hydroxy groups present in the lignin chain (Figure 1), electrocatalytic oxidation (ECO) is currently the most reported approach for lignin depolymerization.[ 21 , 22 ] Besides inhibiting condensation, the presence of α‐carbonyl allows bond cleavage.[ 19 , 23 ] Nevertheless, the major downside of this process is the occurrence of overoxidation towards organic acids and CO2. [24] Conversely, electrocatalytic hydrogenation (ECH) appears as an alternative to the thermal catalytic hydrogenation, while generating final products with reduced oxygen content.[ 10 , 25 ] Ideally, the complete reduction of the oxygenated carbonylic groups would be advantageous towards the production of bio‐fuels. However, when reacted in open cell, the ECH is exposed to oxygenated products generated on the anodic surface of the counter electrode. [10]
Figure 1.
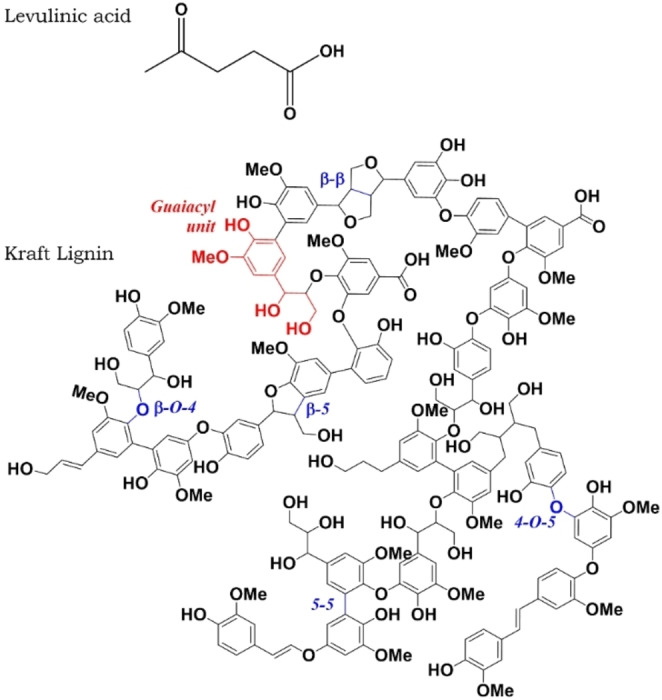
A representation of the structure of levulinic acid and the insoluble fraction of Kraft lignin, proposed by Crestini et al. [15] The typical interunit bonds are highlighted in blue.
One of the main advantages of using electrocatalysis lies in the green aspects of the process, which can be performed under atmospheric pressure and room temperature. These conditions reduce the energetic requirements, following the principles of green chemistry proposed by Anastas et al.[ 26 , 27 ] It can be also noted that the energetic source utilized can be generated from renewable technologies, such as wind or solar power. However, the need for metallic catalysts may simultaneously lead to the hydrogen evolution reaction (HER), which is a competing reaction often present in ECH processes. Therefore, the choice of the working electrode plays a paramount role in the system design. Herein, as in our previous study on the electrochemical reductive cleavage of a lignin model compound, copper was chosen as the working electrode. [10] While presenting lower activity towards HER, comparing to noble metals [28] such as Ir, Pt, and Ru, Cu is considerably more available and cheaper.
Ideally, a sustainable solvent system for lignin dissolution should not be only highly efficient, but also nontoxic, cost‐effective, and with low viscosity and volatility. To date, many investigations have been dealing with ionic liquids (ILs)[ 29 , 30 , 31 ] and deep eutectic solvents (DES) as promising[ 32 , 33 ] “green solvents” for the lignin pre‐treatment and/or dissolution. Besides the high cost, the recycling of pure ILs, in general, is energy intensive. In addition, extensive water input in the downstream process and high viscosity during dissolution can represent a serious drawback for electrocatalytic processes. Finally, some types of ILs, such as the imidazole‐based ones, present serious toxicity concerns; while being poorly biodegradable, they are usually toxic to microorganisms and can form hazardous products during hydrolysis.[ 30 , 34 ] While the use of DES overcomes most of the concerns related to ILs, such as toxicity, recyclability, and cost, undesired lignin condensation is a downside for most acid‐based DES fractionation processes. [35]
Due to the its documented ability to dissolve kraft lignin, levulinic acid represents a promising choice as a sustainable biobased solvent. [36] While being obtained from the hydrothermal processing of lignocellulosic biomass, this organic acid was cited as one of the top 12 value‐added chemicals from biomass by the United States Department of Energy. [37] The levulinic acid structure (Figure 1) contains aketone group, to which the good efficiency to dissolve kraft lignin is attributed. This structure has the ability of solvating both the hydrophobic and hydrophilic groups. The breaking and weakening of some of intermolecular interactions would also facilitate the disaggregation of lignin macromolecules in solution. In addition, the high content of carbonyl groups is beneficial for enhanced protonic and electrical conductivities in electrochemical applications. Furthermore, the recovered lignin presents an increased content of carbonyl groups, which can also play an important role enhancing its electrical conductivity. [36] Both outcomes bring advantages to the use of levulinic acid as a sustainable solvent for the electrocatalyzed fractionation of lignin.
The present investigation aims to assess the reductive electrochemical depolymerization of kraft lignin in concentrated aqueous solutions of levulinic acid. By relying on sustainable and biobased materials, the method described herein provides a straightforward approach to obtain a wide variety of phenolic‐containing polymers and aromatic compounds. Furthermore, in order to overcome one of the main limitations in the scaling‐up of electrochemical processes, the high cost of noble metal electrodes, copper was chosen as the electrocatalyst due to the economic feasibility and low activity towards the HER. Lastly, we present a preliminary assessment of the performance of the final chemicals as coatings for metallic surfaces, in order to enhance their corrosion resistance.
Results and Discussion
Electrochemical depolymerization of lignin
Here, we explored a one‐step approach for the reductive catalytic fractionation (RCF) of lignin, which consisted in its solubilization/depolymerization in levulinic acid, selectively forming aromatic monomers. As levulinic acid can be also reduced via different methods,[ 38 , 39 , 40 ] including electrochemical routes,[ 41 , 42 , 43 , 44 , 45 ] the 5.17 m aqueous solution of levulinic acid, with and without kraft lignin, was tested in a linear sweep voltammetry (LSV) experiment (Figure 2). Potentials over −1.5 V exhibited different current densities, with the highest value for the solution containing lignin. A similar behavior was observed by da Cruz et al. using 2‐phenoxyacetophenone as a lignin model compound in a DES. [10] From this, one can confirm that the electrochemical activity is occurring towards lignin linkages and not only in the levulinic acid structure.
Figure 2.

(a) LSV (scan rate 10 mV s−1) and (b) constant potential ECH at −1.7 V vs. Ag/AgCl (saturated KCl) of Kraft lignin in 5.17 m of levulinic acid.
The reductive fractionation was then performed through chronoamperometry at E=−1.7 V, which resulted in an average current density of around 4.5 and 9 mA cm−2 for the concentrated levulinic acid solution and the same solution with kraft lignin, respectively. This reveals the electrolytical activity of levulinic acid in the presence of NaCO3 as additive, and furthermore, the increase of current density for the kraft lignin reaction confirms the aforementioned activity towards lignin linkages.
The depolymerization products were extracted via liquid–liquid extraction using methyl isobutyl ketone (MIBK) as a selective extractant. Since this solvent is also selective towards levulinic acid,[ 46 , 47 ] it was possible to decrease the acid concentration in the final product to better analyze the outcomes. A major advantage of using MIBK is the possibility of reuse for further extractions, in addition to the fact that it is considered a safe and recommended solvent by CHEM21. [48]
It is well established that lignin depolymerization is a process with low selectivity. Therefore, the characterization of the final products by direct injection high‐resolution mass spectrometry (MS) often reveals a wide spectrum of compounds, while their quantification remains a challenge. The complex range of obtained products hinders the determination of important reaction parameters, such as yield and selectivity towards specific compounds. As a result, determining the sustainability of the process using green chemistry metrics, such as the atom economy, is not a simple task. Previous studies considering the conversion of lignin to lignin‐derived monomers present the E‐factor as a reliable start point in the design process. [49] Herein, the calculated E‐factor, determined according to Equation (1), was estimated as 3.82. This result is consistent with those summarized in the literature by Fadlallah et al. for lignin‐derived products and is applicable to the production of vanillin‐derived monomers and bisphenols. [49]
Lignin‐derived monomers and dimers were identified by direct injection high‐resolution mass spectrometry characterization, among dimers and trimers formed by levulinic acid polymerization, as the identified compounds presented in Figure 3. One of the main goals of choosing the reductive fractionation is avoiding overoxidation and condensation of the products.[ 10 , 20 , 43 , 50 ] In our process, we identified a predominance of aryl ether and phenolic groups. Studies show that late‐stage lignin catalytic conversion results in rich monomer yield of products.[ 50 , 51 , 52 ] Herein, our approach led to some of these monomers and also lignin dimers, which match those obtained from RCF,[ 51 , 53 , 54 ] comprising a low‐molecular‐weight and highly functional lignin oil.
Figure 3.

Main products of kraft lignin depolymerization in levulinic acid identified by direct injection high‐resolution MS.
Nuclear magnetic resonance (NMR) structural characterization is also often employed to determine the functional groups present in lignin and lignin‐based compounds. [55] The 1H and 13C NMR spectra (Figure 4a) confirmed the functional groups observed in the main compounds identified via direct injection high‐resolution MS. The peak at 6.2 ppm, from the phenolic groups, remarkably increased after 20 h of reaction, proving the high content of this functional group in the products due to ECH. The decrease of the peak at 209 ppm (Figure 4b) after the depolymerization evidences the lower concentrations of acetylated groups due to its hydrogenation.
Figure 4.

(a) 1H and (b) 13C NMR spectra of reacted 5.17 m solution of levulinic acid and kraft lignin solution in levulinic acid before and after depolymerization.
The similarity between the spectra for both 1H and 13C of the concentrated solution reacted and the dissolved lignin before the reaction shows that the solvent is stable during the electrocatalytic process. Besides that, it is noted that the simple dissolution of the macromolecule does not lead to any type of fractionation, evidencing that the identified products were obtained only after the electrocatalysis.
Wijaya et al. [50] described two different electron transfer mechanisms occurring on the ECH, namely electron transfer (ET), which is directly related to ECH, and proton transfer (PT), which occurs through electronation–protonation (EP) reaction (Figure 5). The former involves the adsorption of atomic hydrogen (Hads) as the proton source, which leads to the generation of alcoholic groups linked to the hydrocarbon chain. The latter occurs mainly in the bulk solution and is characterized by a high H2 overpotential, which is related to the HER leading to unsaturated hydrocarbon chains. By choosing an electrode with low activity towards HER, this effect can be therefore diminished. In this context, Cu, Ag, and Au represent good alternatives for this purpose.[ 10 , 28 , 56 , 57 ] The scanning electron microscopy (SEM) energy‐dispersive X‐ray spectroscopy (EDS) analysis of the Cu electrode before and after the reaction (Figure S1, Table S1) revealed the stability of the metal during the reaction. A small area suffered carbon deposition, but the polished surface kept its morphology and the metal concentration.
Figure 5.
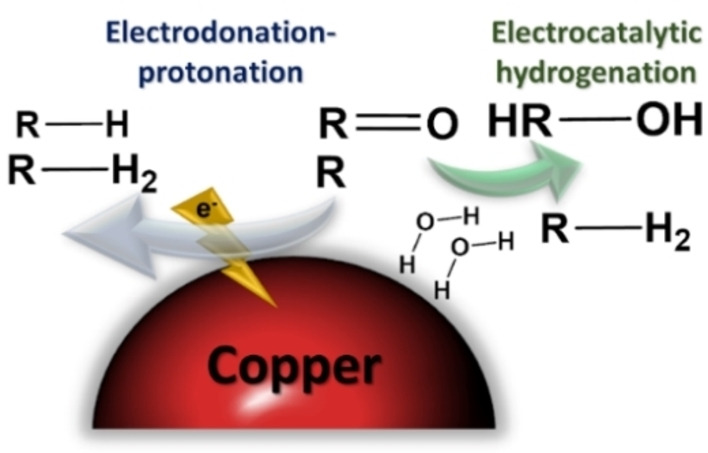
Possible mechanisms of electron transfer in ECH reaction on Cu surface: electronation–protonation and electrocatalytic hydrogenation.
HER is inherently a side reaction of the lignin ECH, which impairs the process efficiency. Hence, the EP mechanism is favored over PT, generating alcoholic‐rich products. This behavior was indeed observed in the NMR spectra (Figure 4). The peak at 6.2 ppm increased considerably compared to those related to aryl ether and aromatic rings. Thus, the C−C bonds were preserved, while the reduction occurred mainly on the surface of the electrode through Hads.
Commonly obtained from lignin through hydrothermal process at mild‐to‐high pressure and temperature, phenols are among the top value‐added chemicals from biomass, according to the United States Department of Energy. [37] Electrocatalyzed fractionation of lignin is a way to maintain the selectivity to these compounds via simple and cheap setups.
Corrosion resistance tests
Protection against corrosion is usually provided by coatings based on inorganic materials, such as chromates, [58] cerium, [59] or other metals,[ 60 , 61 ] and organic materials.[ 62 , 63 , 64 ] Aluminum alloys are widely used in industry and transportation due to their combination of low cost, high strength/weight ratio, and good mechanical properties. In particular, Al6061 is widely used in marine applications due to its intrinsic high corrosion resistance.[ 65 , 66 ]
Recently, the focus has been shifted to sustainable materials, such as biobased coatings, for example, linseed oil, chitosan/alginate, and lignin derivatives.[ 67 , 68 , 69 , 70 ] In our case, depolymerized lignin (DL) presents a wide range of organic molecules with easily accessible primary and secondary hydroxy groups, which can facilitate the DL adsorption on the surface of metals without any previous chemical modification.
The potential use of DL as an anticorrosive coating for Al6061 was assessed by electrochemical techniques. Potentiodynamic polarization (PDP) measurements were carried out in 5 % NaCl, with 1 cm of the Al plate immersed and a scanning rate of 1 mV s−1 in the anodic direction. Prior to each measurement, open‐circuit voltage (OCV) was recorded until a stable potential was reached. PDP curves are shown in Figure 6, and values of E corr and i corr obtained by the tangent extrapolation method are presented in Table S2. Despite a positive trend in the E corr values with increased DL concentrations of the coating, the differences remained in the 20 mV range, which does not allow for clear conclusions to be drawn. However, values of i corr showed a clear decrease, with the lowest values of 0.8 and 0.74 μA cm−2 being reached for Al20 and Al50, translating to respective inhibition efficiency (IE %) values of 70 and 74 %.
Figure 6.

PDP curves of uncoated and coated Al samples recorded in 5 % NaCl at a 1 mV s−1 scan rate. Tafel analysis of these curves showed a clear decrease in i corr values for coated samples, demonstrating an inhibition of the corrosion process.
The performance of the DL based coating for Al corrosion was also investigated using electrochemical impedance spectroscopy (EIS). The Nyquist plots and the circuit equivalent used for the data fitting of pure Al (i) and coated Al (ii) are displayed in Figure 7a. The circuit equivalent consists of R s (solution resistance) and R ct (charge transfer resistance) components. The double‐layer capacitance (C dl) component used for the Al sample is changed for a constant phase element in the case of coated Al, which describes best the properties of the organic layer.
Figure 7.
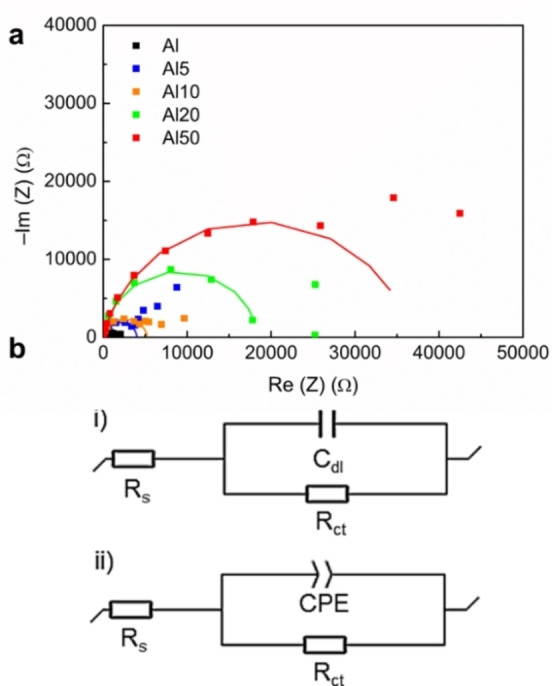
(a) Nyquist plots for uncoated and coated Al samples. (b) Circuit equivalent used for the data fitting of the uncoated (i) and coated samples (ii). The Nyquist plots show an increase of the diameter of the semicircle for coated samples, which is correlated to an increase of the charge transfer resistance (R ct), and the corrosion inhibition.
Nyquist plots for the Al and DL coated Al samples showed a typical depressed semicircle shape for the high‐frequency zone. Fitting of these plots using the circuit equivalent presented in Figure 7b gave the values of R ct reflecting the resistance of the sample to corrosion.
The diameter of the semicircles, corresponding to R ct, increased drastically in line with the percentage of DL in the coating, as shown in Table S2. This suggests a good resistance to charge transfer. The values of IE % obtained from impedance data showed a similar trend to those obtained from PDP measurement, pointing out to an increased corrosion resistance with higher percentage of DL, and plateauing at 50 wt %. However, the values are significantly higher, with up to 96.3 % (impedance) against 74 % (polarization) for Al50, which is closer to the recent values reported for lignin‐based anticorrosive coatings for Al.[ 68 , 69 ]
To directly observe the efficiency of the coating for corrosion protection, SEM micrographs were recorded on pure and coated Al sample before and after PDP measurements (Figure 8). The surface of the Al sample before corrosion tests showed parallel striations and some irregularities at higher magnification due to hand polishing. After PDP measurements, the surface of the Al20 coated sample showed a smooth surface and a good coverage with occasional defects (circled in yellow). In the case of the uncoated sample, a clear degradation of the surface is observed including surface pitting, indicative of corrosion.
Figure 8.

SEM micrographs of aluminum plate (a, d) before and (b, c, e, f) after PDP scan. After scanning to potential higher than E corr, uncoated sample (c, e) shows distinct pitting features due to corrosion; the coated sample Al20 (c, f) shows no such features and exhibits a smooth surface, demonstrating the ability of the coating to shield the surface from corrosion occurring during the PDP measurement. Circled in yellow (c) shows a defect area in the coating, created during sample preparation showing an underlying Al surface without corrosion features, further demonstrating the protecting ability of the coating.
These differences provide a confirmation that the coating acted as good protection against corrosion of the surface. Closer observation of the defect area shows an underlying surface without pitting, which could indicate that the defect was created after the corrosion measurement, possibly by a mechanical action (Figure 9).
Figure 9.

SEM micrograph of Al20 on a defect area after corrosion measurement; the higher magnification allows to distinct the smooth Al surface. EDS analysis confirmed the organic nature of the coating and only showed Al signal for the underlying surface.
To gain further insight on the composition of the surface, EDS analysis was carried out in the vicinity of one of these boundaries (data not shown). The spectrum of the coated area showed only peaks corresponding to Al, C, and O atoms, which is expected for an aluminum substrate coated with oxygen‐rich organic molecules such as DL.
Conclusion
Herein, we discovered an approach for the electrochemical depolymerization of lignin using levulinic acid as a solvent. Within the framework of green chemistry, the solvent choice occupies a prominent place. In this context, there is an urgent need for innovative alternatives for the replacement of volatile organic solvents in catalytic processes. Generated during the hydrothermal process of the lignocellulosic biomass, levulinic acid has been highlighted as one of the top 12 value‐added chemicals from biomass by the US Department of Energy. The critical advancement of this work is the demonstration that levulinic acid can act both as an efficient solvent for lignin and reaction media for the reductive electrochemical depolymerization of this complex polyphenol.
Our results showed that after 20 h of lignin depolymerization in levulinic acid, lignin‐derived monomers and dimers were obtained. This process resulted in compounds in which aryl ether and phenol groups predominated. The extraction of such compounds from lignin represents a key step towards the production of renewable chemicals for the coating, fuel, and lubricant industries. Using a biomass‐based solvent and copper, a cheap transition metal, as an electrocatalyst, we have demonstrated a straightforward approach to produce phenolic‐containing and aromatic compounds based on an abundant industrial side‐stream. We believe that this approach can be promising for the creation of functional biogenic materials that incorporates Nature's biodegradation design.
Experimental Section
Materials
Kraft lignin obtained from industrial pulping spent liquor was used as starting material. Levulinic acid and 4‐methyl‐2‐pentanone (MIBK) (technical grade) were purchased from Sigma‐Aldrich Co. (St Louis, MO, USA). All chemicals were used as received. The copper foil was acquired from WAT Venture, Poland. Before reaction, the foil was ultrasonically washed with a H2SO4 solution, followed by deionized (DI) water rinsing. Al 6061 was purchased from Alfa Aesar; the typical composition of the alloy given by the manufacturer is : Al 95.8–98.6 %, Mg 0.8–1.2 %, Si 0.4–0.8 %, Cr 0.04–0.35 %, Cu 0.15–0.4 %, Fe 0.7 % max, Zn 0.25 % max, Mn 0.15 % max, Ti 0.15 % max.
Lignin depolymerization via ECH system
The Kraft lignin was dissolved in a 5.17 m aqueous solution of levulinic acid at 3 g L−1 by mechanical stirring at 500 rpm. The electrochemical measurements were conducted using an Interface 5000E potentiostat (Gamry Instruments) in a three‐electrode electrochemical cell. The Cu foil working electrode (1 cm2), the platinum wire counter electrode, and the Ag/AgCl (saturated KCl) reference electrode were immersed in the solution, and LSV was carried out with scan rate of 10 mV s−1 over a range from 1.0 to −2.5 V (vs. Ag/AgCl). The chronoamperometry was performed using a potential of −1.7 V vs. Ag/AgCl (saturated KCl) for 20 h. All the experiments were performed at room temperature.
Product recovery and extraction
After depolymerization, products were extracted by precipitation of insoluble fractions followed by liquid–liquid extraction (LLE) of the soluble ones. MIBK was used as organic extractant phase for the LLE process. The mixture was stirred at 250 rpm overnight in an orbital shaker. Two phases were obtained, which were further separated using a separating funnel. In the last step, the liquid fraction was extracted from the organic phase using a rotary evaporator from where the solvent was recovered. A reference sample of the solution before the reaction was also extracted following the same procedure. The E‐factor was determined according to Equation (1), without considering the water in the levulinic acid solution. The MIBK used for LLE was not added to the calculation as it is reused for further processes.
| (1) |
Characterization
Direct injection high‐resolution MS samples were dissolved in a chloroform/methanol mixture (3 : 7, dilution factor of 1 : 1000). Samples were introduced into a LTQ Orbitrap XL hybrid tandem high‐resolution MS from Thermo Fisher Scientific (Bremen, Germany) by direct infusion applying a flow rate of 5 μL min−1. The instrument was fitted with electrospray ionization (ESI) ion source and operated in positive or negative ion mode. Nitrogen was used as a sheath gas. Helium was used both as a buffer and a collision gas in the linear ion trap section, where lower energy collision‐induced dissociation (CID) was performed. For the identification of chemical structures by tandem MS, product ions were generated in the linear ion trap via CID and detected by the high‐resolution orbitrap section of the instrument at a resolution of 60000 (full width at half maximum, FWHM). The mass measurements were acquired with a mass accuracy of 5 ppm or better. Data processing and interpretation were performed using the software tools Xcalibur version 2.0.7 and Mass Frontier version 6.0 from Thermo Fisher Scientific (Bremen, Germany). m/z describes the mass‐to‐charge ratio of the detected ions. As all negatively charged ions analyzed were single charged species, m/z also referred to the monoisotopic molecular masses of the detected ions. It is noted that ionization capability of chemical compounds is dependent on the chemical structure. 1H NMR spectra were recorded on a Bruker DRX 4OO NMR instrument at 25 °C in DMSO‐d6 containing tetramethylsilane as an internal standard. An inversed gated proton decoupling with 90° pulse angle, and delay time of 10 s was applied. The spectra were recorded in 32 scans with a 10 s delay.
Corrosion resistance of Aluminum 6061 with DL‐based coating
The aluminum plate (300×300×2.3 mm) was cut in small pieces (30×10×2.3 mm) and hand polished with SiC grinding paper (purchased from Buehler) with grain from P120 to P4000. Resulting pieces were washed by ultrasonication in acetone and absolute ethanol. A solution of DL in levulinic acid was diluted in absolute ethanol to the desired concentrations, 5, 10, 20, and 50 wt %. Electrodes were then coated by drop casting and the resulting samples were dried at 60 °C up to 2 h, followed by a curing step of 12 h at 80 °C. Samples were then labelled as Al5, Al10, Al20, and Al50. The electrochemical experiments were performed using a Biologic SP150 potentiostat in a typical three‐electrode setup. The working electrode was the coated/non‐coated aluminum specimen (immersed 1 cm), the reference was Ag/AgCl (3 m KCl), and the counter was a platinum wire (7 cm2). PDP and EIS plots were obtained and analyzed using the EC‐Lab software. EIS measurements were carried out with an AC amplitude of 15 mV at open circuit potential in the frequency range 500 kHz to 50 mHz. The efficiency was calculated accordingly to Equations (2) and 3:
| (2) |
| (3) |
where IE % is the inhibition efficiency (dimensionless), i corr,ref and i corr,inh represent the current density of unprotected and protected substrate extracted from PDP curves, respectively, and R ct,inh and R ct,ref represent the charge transfer resistance of coated and uncoated samples, respectively. SEM micrographs were recorded on a JEOL JSM‐7000F, with an accelerating voltage of 15 kV for Al samples and 10 kV for coated Al samples.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors thank the Swedish Foundation for Strategic Environmental Research (Mistra: project Mistra SafeChem, project number 2018/11) for financial support. M. G. D. C. thanks the Carl Tryggers Stiftelse för Vetenskaplig Forskning for a postdoctoral fellowship (grant CTS 39: 194). We acknowledge additional financial support by the COMET InTribology project (FFG No. 872176, project coordinator: AC2T research GmbH). The work was carried out in collaboration with “Excellence Centre of Tribology” AC2T research GmbH. We thank Joseph Samec for fruitful discussions. Open Access funding enabled and organized by Projekt DEAL.
M. G. A. da Cruz, R. Gueret, J. Chen, J. Piątek, B. Beele, M. H. Sipponen, M. Frauscher, S. Budnyk, B. V. M. Rodrigues, A. Slabon, ChemSusChem 2022, 15, e202200718.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐9164j).
Contributor Information
Prof. Bruno V. M. Rodrigues, Email: bruno.manzolli@mmk.su.se.
Prof. Adam Slabon, Email: slabon@uni-wuppertal.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Kang S., Miao R., Guo J., Fu J., Catal. Today 2021, 374, 61–76. [Google Scholar]
- 2. Yadav V. G., Yadav G. D., Patankar S. C., Clean Technol. Environ. Policy 2020, 22, 1757–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim H., Lee S., Ahn Y., Lee J., Won W., ACS Sustainable Chem. Eng. 2020, 8, 12419–12429. [Google Scholar]
- 4. Zhou S.-J., Wang H.-M., Xiong S.-J., Sun J.-M., Wang Y.-Y., Yu S., Sun Z., Wen J.-L., Yuan T.-Q., ACS Sustainable Chem. Eng. 2021, 9, 12017–12042. [Google Scholar]
- 5. Ashokkumar V., Venkatkarthick R., Jayashree S., Chuetor S., Dharmaraj S., Kumar G., Chen W.-H., Ngamcharussrivichai C., Bioresour. Technol. 2022, 344, 126195. [DOI] [PubMed] [Google Scholar]
- 6. Xu G. R., An Z. H., Xu K., Liu Q., Das R., Zhao H. L., Coord. Chem. Rev. 2021, 427, 213554. [Google Scholar]
- 7. Gopakumar A., Ren P., Chen J., Manzolli Rodrigues B. V., Vincent Ching H. Y., Jaworski A., Van Doorslaer S., Rokicińska A., Kuśtrowski P., Barcaro G., Monti S., Slabon A., Das S., J. Am. Chem. Soc. 2022, 144, 2603–2613. [DOI] [PubMed] [Google Scholar]
- 8. Bajwa D. S., Pourhashem G., Ullah A. H., Bajwa S. G., Ind. Crops Prod. 2019, 139, 111526. [Google Scholar]
- 9. Sun Z., Fridrich B., de Santi A., Elangovan S., Barta K., Chem. Rev. 2018, 118, 614–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. da Cruz M. G. A., Rodrigues B. V. M., Ristic A., Budnykb S., Das S., Slabon A., Green Chem. Lett. Rev. 2022, 15, 153–161. [Google Scholar]
- 11. Weng C., Peng X., Han Y., Biotechnol. Biofuels 2021, 14, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang C., Wang F., Acc. Chem. Res. 2020, 53, 470–484. [DOI] [PubMed] [Google Scholar]
- 13. Shuai L., Amiri M. T., Questell-Santiago Y. M., Héroguel F., Li Y., Kim H., Meilan R., Chapple C., Ralph J., Luterbacher J. S., Science 2016, 354, 329–333. [DOI] [PubMed] [Google Scholar]
- 14. Subbotina E., Rukkijakan T., Marquez-Medina M. D., Yu X., Johnsson M., Samec J. S. M., Nat. Chem. 2021, 13, 1118–1125. [DOI] [PubMed] [Google Scholar]
- 15. Crestini C., Lange H., Sette M., Argyropoulos D. S., Green Chem. 2017, 19, 4104–4121. [Google Scholar]
- 16. Du X., Zhang H., Sullivan K. P., Gogoi P., Deng Y., ChemSusChem 2020, 13, 4318–4343. [DOI] [PubMed] [Google Scholar]
- 17. Garedew M., Lin F., Song B., DeWinter T. M., Jackson J. E., Saffron C. M., Lam C. H., Anastas P. T., ChemSusChem 2020, 13, 4214–4237. [DOI] [PubMed] [Google Scholar]
- 18. Du X., Zhang H., Sullivan K. P., Gogoi P., Deng Y., ChemSusChem 2020, 13, 4318–4343. [DOI] [PubMed] [Google Scholar]
- 19. Garedew M., Lam C. H., Petitjean L., Huang S., Song B., Lin F., Jackson J. E., Saffron C. M., Anastas P. T., Green Chem. 2021, 23, 2868–2899. [Google Scholar]
- 20. Garedew M., Lin F., Song B., DeWinter T. M., Jackson J. E., Saffron C. M., Lam C. H., Anastas P. T., ChemSusChem 2020, 13, 4214–4237. [DOI] [PubMed] [Google Scholar]
- 21. Zirbes M., Waldvogel S. R., Curr. Opin. Green Sustain. Chem. 2018, 14, 19–25. [Google Scholar]
- 22. Breiner M., Zirbes M., Waldvogel S. R., Green Chem. 2021, 23, 6449–6455. [Google Scholar]
- 23. Rauen A. L., Weinelt F., Waldvogel S. R., Green Chem. 2020, 22, 5956–5960. [Google Scholar]
- 24. Di Marino D., Jestel T., Marks C., Viell J., Blindert M., Kriescher S. M. A., Spiess A. C., Wessling M., ChemElectroChem 2019, 6, 1434–1442. [Google Scholar]
- 25. Garedew M., Young-Farhat D., Bhatia S., Hao P., Jackson J. E., Saffron C. M., Sustain. Energy Fuels 2020, 4, 1340–1350. [Google Scholar]
- 26. Anastas P. T., Eghbali N., Chem. Soc. Rev. 2010, 39, 301–312. [DOI] [PubMed] [Google Scholar]
- 27. Anastas P. T., Warner J. C., in Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998. [Google Scholar]
- 28. Nørskov J. K., Bligaard T., Logadottir A., Kitchin J. R., Chen J. G., Pandelov S., Stimming U., J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar]
- 29. Geniselli da Silva V., Bioresour. Technol. Reports 2021, 16, 100824. [Google Scholar]
- 30. Pu Y., Jiang N., Ragauskas A. J., J. Wood Chem. Technol. 2007, 27, 23–33. [Google Scholar]
- 31. Espinoza-Acosta J. L., Torres-Chávez P. I., Carvajal-Millán E., Ramírez-Wong B., Bello-Pérez L. A., Montaño-Leyva B., BioResources 2014, 9, 3660–3687. [Google Scholar]
- 32. Lynam J. G., Kumar N., Wong M. J., Bioresour. Technol. 2017, 238, 684–689. [DOI] [PubMed] [Google Scholar]
- 33. Liu Y., Chen W., Xia Q., Guo B., Wang Q., Liu S., Liu Y., Li J., Yu H., ChemSusChem 2017, 10, 1692–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoo C. G., Pu Y., Ragauskas A. J., Curr. Opin. Green Sustain. Chem. 2017, 5, 5–11. [Google Scholar]
- 35. Shimada K., Hosoya S., Ikeda T., J. Wood Chem. Technol. 1997, 17, 57–72. [Google Scholar]
- 36. Melro E., Filipe A., Valente A. J. M., Antunes F. E., Romano A., Norgren M., Medronho B., Int. J. Biol. Macromol. 2020, 164, 3454–3461. [DOI] [PubMed] [Google Scholar]
- 37. Werpy T., Petersen G., Top Value Added Chemicals from Biomass Volume I, 2004. United States Department of Energy, Energy Efficiency and Renewable Energy. [Google Scholar]
- 38. Roa D. A., Garcia J. J., Inorg. Chim. Acta 2021, 516, 120167. [Google Scholar]
- 39. Sosa L. F., da Silva V. T., de Souza P. M., Catal. Today 2021, 381, 86–95. [Google Scholar]
- 40. Habe H., Sato Y., Kirimura K., Appl. Microbiol. Biotechnol. 2020, 104, 7767–7775. [DOI] [PubMed] [Google Scholar]
- 41. Latsuzbaia R., Bisselink R. J. M., Crockatt M., van der Waal J. C., Goetheer E. L. V., Biomass Valorization 2021, 225–264. [Google Scholar]
- 42. Mürtz S. D., Kurig N., Holzhäuser F. J., Palkovits R., Green Chem. 2021, 23, 8428–8433. [Google Scholar]
- 43. Lucas F. W. S., Grim R. G., Tacey S. A., Downes C. A., Hasse J., Roman A. M., Farberow C. A., Schaidle J. A., Holewinski A., ACS Energy Lett. 2021, 6, 1205–1270. [Google Scholar]
- 44. Rehbein M. C., Guschakowski M., Sauter W., Kunz J., Schröder U., Scholl S., Front. Energy Res. 2020, 8, 242. [Google Scholar]
- 45. Du Y., Chen X., Qi J., Wang P., Liang C., Catalysts 2020, 10, 692. [Google Scholar]
- 46. Kumar A., Shende D. Z., Wasewar K. L., Chem. Data Collect. 2020, 28, 100417. [Google Scholar]
- 47. Gajula S., Inthumathi K., Arumugam S. R., Srinivasan K., ACS Sustainable Chem. Eng. 2017, 5, 5373–5381. [Google Scholar]
- 48. Prat D., Wells A., Hayler J., Sneddon H., McElroy C. R., Abou-Shehada S., Dunn P. J., Green Chem. 2015, 18, 288–296. [Google Scholar]
- 49. Fadlallah S., Sinha Roy P., Garnier G., Saito K., Allais F., Green Chem. 2021, 23, 1495–1535. [Google Scholar]
- 50. Wijaya Y. P., Smith K. J., Kim C. S., Gyenge E. L., Green Chem. 2020, 22, 7233–7264. [Google Scholar]
- 51. Song Y., in Chemical Catalysts for Biomass Upgrading, 2019, pp. 395–437. [Google Scholar]
- 52. Fang Z., Flynn M. G., Jackson J. E., Hegg E. L., Green Chem. 2021, 23, 412–421. [Google Scholar]
- 53. Liao Y., Koelewijn S. F., van den Bossche G., van Aelst J., van den Bosch S., Renders T., Navare K., Nicolaï T., van Aelst K., Maesen M., Matsushima H., Thevelein J. M., van Acker K., Lagrain B., Verboekend D., Sels B. F., Science 2020, 367, 1385–1390. [DOI] [PubMed] [Google Scholar]
- 54. Supriyanto, Usino D. O., Ylitervo P., Dou J., Sipponen M. H., Richards T., J. Anal. Appl. Pyrolysis 2020, 151, 104917. [Google Scholar]
- 55. Shi Z., Xu G., Deng J., Dong M., Murugadoss V., Liu C., Shao Q., Wu S., Guo Z., Green Chem. Lett. Rev. 2019, 12, 235–243. [Google Scholar]
- 56. Farinazzo Bergamo Dias Martins P., Papa Lopes P., Ticianelli E. A., Stamenkovic V. R., Markovic N. M., Strmcnik D., Electrochem. Commun. 2019, 100, 30–33. [Google Scholar]
- 57. Ma Z., Klimpel M., Budnyk S., Rokicińska A. R., Kuśtrowski P. K., Dronskowski R., Mathew A. P., Budnyak T., Slabon A., ACS Sustainable Chem. Eng. 2021, 9, 3658–3667. [Google Scholar]
- 58. Kendig M., Jeanjaquet S., Addison R., Waldrop J., Surf. Coat. Technol. 2001, 140, 58–66. [Google Scholar]
- 59. Valdez B., Kiyota S., Stoytcheva M., Zlatev R., Bastidas J. M., Corros. Sci. 2014, 87, 141–149. [Google Scholar]
- 60. Santa Coloma P., Izagirre U., Belaustegi Y., Jorcin J. B., Cano F. J., Lapeña N., Appl. Surf. Sci. 2015, 345, 24–35. [Google Scholar]
- 61. Mishra A. K., Balasubramaniam R., Mater. Chem. Phys. 2007, 103, 385–393. [Google Scholar]
- 62. Doğru Mert B., Corros. Sci. 2016, 103, 88–94. [Google Scholar]
- 63. Nnaji N., Nwaji N., Mack J., Nyokong T., J. Mol. Struct. 2021, 1236, 130279. [Google Scholar]
- 64. Zhao R., Rupper P., Gaan S., Coating 2017, 7, 133. [Google Scholar]
- 65. Wahid M. A., Siddiquee A. N., Khan Z. A., Mar. Syst. Ocean Technol. 2020, 15, 70–80. [Google Scholar]
- 66. Cohen S. M., Corrosion 1995, 51, 71–78. [Google Scholar]
- 67. Boucher D., Ladmiral V., Negrell C., Caussé N., Pébère N., Prog. Org. Coat. 2021, 158, 106344. [Google Scholar]
- 68. Moreno A., Liu J., Gueret R., Hadi E., Bergstrçm L., Slabon A., Sipponen M. H., Angew. Chem. Int. Ed. 2021, 60, 20897–20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Carlos De Haro J., Magagnin L., Turri S., Griffini G., ACS Sustainable Chem. Eng. 2019, 7, 6213–6222. [Google Scholar]
- 70. Coquery C., Carosio F., Negrell C., Caussé N., Pébère N., David G., Surf. Interfaces 2019, 16, 59–66. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.