

Abstract
Precision medicine is revolutionising patient care in cancer. As more knowledge is gained about the impact of specific genetic lesions on diagnosis, prognosis and treatment response, diagnostic precision and the possibility for optimal individual treatment choice have improved. Identification of hallmark genetic aberrations such as the BCR::ABL1 gene fusion in chronic myeloid leukaemia (CML) led to the rapid development of efficient targeted therapy and molecular follow‐up, vastly improving survival for patients with CML during recent decades. The assessment of translocations, copy number changes and point mutations are crucial for the diagnosis and risk stratification of acute myeloid leukaemia and myelodysplastic syndromes. Still, the often heterogeneous and complex genetic landscape of haematological malignancies presents several challenges for the implementation of precision medicine to guide diagnosis, prognosis and treatment choice. This review provides an introduction and overview of the important molecular characteristics and methods currently applied in clinical practice to guide clinical decision making in haematological malignancies of myeloid and lymphoid origin. Further, experimental ways to guide the choice of targeted therapy for refractory patients are reviewed, such as functional precision medicine using drug profiling. An example of the use of pipeline studies where the treatment is chosen according to the molecular characteristics in rare solid malignancies is also provided. Finally, the future opportunities and remaining challenges of precision medicine in the real world are discussed.
Keywords: drug screening, haematology, MRD, precision medicine

Introduction
The term precision medicine refers to the strategy of predicting which patients are most likely to respond to specific cancer therapies based on high‐resolution molecular diagnostics and, increasingly, functional and mechanistic understanding of individual tumours [1]. The field of haematology has been pivotal in the development and implementation of precision medicine. This is partly because tumour material is readily available in the blood and bone marrow. Further, haematologic malignancies have been suitable candidates for pioneering precision medicine due to the presence of some disorders with a homogeneous genetic landscape and characteristic driver mutations. For example, one of the first largely successful targeted cancer therapies was tyrosine‐kinase inhibitors (TKIs) for chronic myeloid leukaemia (CML) [2]. While genetic analyses such as karyotyping have been essential diagnostic procedures for a long time in haematology [2, 3], gene panel sequencing has been introduced more recently in clinical routine, to identify genetic alterations with diagnostic, prognostic and predictive impact [4, 5, 6, 7]. In addition, more sensitive techniques to assess minimal residual disease (MRD) are being developed. However, since the first whole‐genome sequencing (WGS) of leukaemia in 2008 [8], the mutational landscape of haematological malignancies has proven to be both much more complex and heterogeneous with a shortage of targets as uniformly present and druggable as in CML. Also, intratumour heterogeneity and clonal evolution are increasingly recognised and likely to play a role in tumour progression as well as therapy resistance [9, 10, 11, 12].
Despite rapid advances, a multitude of challenges remains in implementing precision medicine in clinical routine. How do we best identify genetic alterations of importance for an individual patient? How do we ascertain who will benefit from novel and often costly targeted therapies? How do we best evaluate the treatment effect and progression to stay ‘on top’ of the tumour and modify treatment schemes if needed?
This review provides an overview and examples of the key methods of molecular characterisation currently applied to guide diagnosis, prognosis and individual treatment choice in myeloid and lymphoid malignancies in real‐world clinical practice. Whereas genetic characterisation is a cornerstone of the clinical work‐up of myeloid malignancies, we have yet to understand the full benefit in several lymphoid tumour subtypes. Further, we will also provide examples of more experimental ways to choose optimal treatment using functional precision medicine (FPM; drug profiling) and examples of how precision medicine has been used to improve survival in rare solid malignancies. Finally, we will discuss the remaining challenges and future opportunities with precision medicine in haematological malignancies.
Current precision diagnostics in myeloid haematological malignancies
For the diagnosis, classification, prognostication and therapy selection of myeloid malignancies, a comprehensive genetic workup is required in current clinical practice. Currently, a combination of different techniques such as chromosome banding analysis, fluorescence in situ hybridisation (FISH) and panel sequencing is performed to comprehensively assess translocations, copy number changes and point mutations. The extent of the genetic analyses, especially of the set of genes to be analysed, is dependent on the disease entity as the diagnostic relevance of translocations, copy number changes and point mutations vary between entities. The WHO classification and several entity‐specific guidelines provide recommendations [13].
CML is an example of a disease in which both diagnostics and therapeutics have been revolutionised by advances in genetic characterisation [2]. This is largely thanks to the fact that CML is quite a simple disease from a genetic point of view, driven by the BCR::ABL1 fusion. Following the availability of TKIs, young patients with CML responding optimally to TKI therapy have a near‐normal life expectancy [14, 15, 16]. Thus, at diagnosis, a combination of chromosome banding analysis and molecular techniques to confirm the presence of the BCR::ABL1 fusion is sufficient for diagnosis and treatment initiation [17]. For follow‐up, measurement of the BCR::ABL1 real‐time quantitative reverse transcriptase polymerase chain reaction (PCR) is the current gold standard [17]. In the case of TKI resistance, a more comprehensive genetic workup including sequencing of BCR::ABL1 and the detection of additional structural variants, copy number alterations (CNAs) and single nucleotide variants (SNVs) are helpful for further treatment decisions. However, the main focus in personalizing treatment in CML is currently to select TKIs based on patients’ comorbidities and to monitor treatment response [18].
The group of BCR::ABL1 negative myeloproliferative neoplasms (e.g., polycythaemia vera, essential thrombocythaemia, primary myelofibrosis) is much more heterogeneous from a clinical point of view but also in terms of genetics. Much progress has been made in the genetic characterisation of these diseases and the presence of point mutations in genes such as JAK2, CALR and MPL is now crucial for diagnosis and prognosis, but translation into targeted treatment approaches based on genetic markers has yet to occur [19].
Myelodysplastic syndrome (MDS) and acute myeloid leukaemia (AML) are very complex disease groups including subtypes with rather few genetic abnormalities up to more complex changes including a variety of structural variants, CNAs and SNVs. The genetic landscape of MDS and AML has been evaluated in detail by several groups [4, 20, 21]. Internationally accepted prognostication scoring systems have been established for both diseases that guide treatment decisions—in MDS including karyotype and AML based on karyotype and molecular mutations [22, 23, 24]. More recent prognostication models in MDS also include gene mutations [25].
Examples of specific genetic abnormalities that currently guide treatment choice in MDS include deletion 5q as the sole abnormality and SF3B1 mutations in MDS with less than 5% blasts, influencing selection to treatment with lenalidomide and luspatercept, respectively [26, 27]. In AML, treatment assignment based on genetic markers such as FLT3‐ITD and mutations in IDH1 and IDH2 is both implemented in clinical routine and studied in ongoing trials [28, 29].
Due to the genetic complexity of MDS and AML, a combination of chromosome banding analysis and panel sequencing is required to capture the relevant genetic alterations. WGS could be a great step forward in the genetic characterisation of myeloid malignancies as it captures all different types of genetic aberrations in one assay. Recently, a cohort of AML and MDS patients was prospectively analysed by WGS and results were compared to the gold standard genetic work‐up demonstrating the feasibility and clinical utility of WGS [30].
While current diagnostics to aid precision medicine are focused mainly on identifying variants at the DNA level, the ability to identify gene fusions and to measure gene expression by RNA sequencing, also referred to as whole‐transcriptome sequencing (WTS), holds great promises, as recently reviewed [31]. For example, many gene fusions in AML constitute class‐defining and treatment predictive entities, for example, PML::RARA, BCR::ABL1, CBFB::MYH11 and RUNX1::RUNX1T1, that are readily detectable by WTS [32]. In addition, data are forthcoming that classification based on transcriptional profiles can be used to subdivide class‐defining subtypes (e.g., NPM1‐mutated AML) into separate entities of prognostic and possibly therapeutic relevance and as a proxy to predict response to a large variety of drugs [33, 34].
Collectively, depending on the entity of myeloid malignancies, the complexity and spectrum of genetic aberration vary as well as the progress in adapting treatment decisions based on genomics. Depending on these parameters, the value of comprehensive diagnostics up to WGS varies and needs to be re‐evaluated and adjusted periodically based on the evolution of knowledge and availability of new drugs.
Current precision diagnostics in lymphoid haematological malignancies
For lymphoid haematological malignancies, genetic categorisation and treatment choice have come the furthest in chronic lymphocytic leukaemia (CLL). In CLL, the prognostic role of genetic markers has been established for a long time and the presence of TP53 alterations as well as the IGHV mutation status have been included in the international prognostic index for patients with CLL [35]. Current clinical practice guidelines state that TP53 mutations (analysed by Sanger sequencing or next generation sequencing (NGS)) and del(17p) (evaluated by FISH) are of the strongest prognostic and predictive relevance. Strong prognostic evidence and predictive evidence for chemoimmunotherapy (CIT) have been confirmed for the IGHV mutation status. A complex karyotype determined by chromosome banding analysis was categorised as possible prognostic and predictive relevance still requiring prospective clinical trials for confirmation [36].
Additional genetic markers have been analysed in CLL for their prognostic impact by different techniques such as FISH [37, 38], genomic arrays [39, 40] or whole exome sequencing (WES)/WGS [41, 42, 43]. The next step is to evaluate these genetic markers in relation to new therapeutic concepts. For gene mutations such as NOTCH1, SF3B1, BIRC3 or RPS15, as well as complex karyotype, it was demonstrated that they predict an unfavourable prognosis also in the absence of TP53 deletion/mutation, but it still has to be determined in clinical trials which drug/drug combinations can improve outcome in a certain genomic setting [36].
Another lymphoid disease in which genetic aberrations guide diagnosis and prognosis is acute lymphoblastic leukaemia (ALL) [1, 44]. According to the current WHO classification, nine genetic subgroups of B‐cell precursor ALL are defined as requiring a comprehensive genetic work‐up currently based mainly on the karyotype and the detection of gene fusions by FISH or molecular techniques [45]. However, alterations below the resolution of chromosome banding analysis such as submicroscopic gene deletions or even intragenic deletions such as deletion within IKZF1 were also shown to be relevant for the pathogenesis and prognosis [1, 46, 47]. Also, SNVs were shown to be relevant [1]. However, in contrast to AML, screening for mutations in a panel of genes is not yet considered standard in ALL. While risk assessment and risk‐adapted treatment based on genomics have been implemented for many years in paediatric ALL, this approach is lagging in adult ALL [44, 48, 49]. The availability of new drugs and the need to identify which patients will benefit the most from these will likely require increasingly detailed genomic diagnostics. As ALL harbours a broad spectrum of structural variants, CNAs and SNVs, technologies that can assess all of these, such as WGS, might turn out to be the method of choice, especially as chromosome banding analysis is hampered by low in vitro proliferation of certain ALL subtypes as well as the cytogenetically cryptic nature of specific abnormalities such as ETV6::RUNX1, IGH::CRLF2, EP300::ZNF384, DUX4‐rearranged and others [31, 50]. In this context, more widespread WTS in a clinical setting is likely to improve precision diagnostics further given that ALL is a disease entity characterised by very distinct gene expression signatures and a large number (greater than 150) of recurrent gene fusions [31, 51–53]. Of particular importance for improved precision medicine in the management of ALL is the detection of gene expression signatures (e.g., Philadelphia‐like or BCR::ABL1‐like ALL) or gene fusions involving CRLF2(IGH::CRLF2, P2RY8::CRLF2), ABL‐class genes (ABL1, ABL2, CSF1R, LYN, PDGFRA, PDGFRB) or alterations resulting in the activation of JAK‐STAT and RAS signalling [54, 55, 56]. Clinical trials are currently ongoing in which kinase inhibitors against ABL‐class and JAK1/2 family members are tested as treatment additions depending on the underlying genetic alteration (ClinicalTrials.gov: NCT03117751, NCT02883049 and NCT02723994).
In lymphomas, detection of chromosomal translocations through FISH has been an important part of the diagnostic workup for several decades, including, t(14;18) in follicular lymphoma, t(11;14) in mantle cell lymphoma and t(8;14) in Burkitt lymphoma. Since then, the broader WES and WGS techniques and their detection of both translocations, SNV and CNAs, have paved the way for further understanding of the molecular biology of the diseases and refinements of the lymphoma subtype classification. About 10 years ago, the BRAFV600E point mutation was identified as a molecular hallmark of hairy cell leukaemia and its detection is now used in routine diagnostics by real‐time PCR or targeted NGS [57]. Additionally, this finding has created possibilities for potential treatment of relapsed/refractory (R/R) cases with BRAF inhibitors [58]. In Waldenström's macroglobulinemia, the MYD88L265P point mutation has been found to be highly recurrent, although it is not exclusive to this lymphoma subtype, and may have implications for response to ibrutinib [59, 60]. The CXCR4 mutation has also been identified specifically in Waldenström albeit in a lower proportion of the cases (30%) [61].
In aggressive lymphomas, the distinction of high‐grade B‐cell lymphomas with translocations involving MYC and BCL2 and/or BCL6 (often referred to as double‐ or triple‐hit lymphomas) is of high clinical relevance as these patients are now recommended to receive more intensified treatment up front [62]. Also, gene‐expression profiling and exome sequencing analysis has shed additional light on the aggressive grey‐zone lymphomas [63]. This uncommon entity can further be dichotomised into a more primary mediastinal B‐cell lymphoma‐like and a more diffuse large B‐cell lymphoma (DLBCL)‐like molecular subtype strongly correlated with the presence or absence of anterior mediastinal involvement, and with implications for the choice of treatment. The former displays a mutational profile resembling classical Hodgkin lymphoma with SOCS1 and B2M mutations, whereas the latter nonmediastinal subtype commonly displays mutations in TP53 and BCL2 [63]. In another subtype of DLBCL, the T‐cell/histiocyte‐rich large B‐cell lymphoma, PD‐L1/PD‐L2 alterations are common and provide a genetic basis for immune evasion, and treatment with immune checkpoint inhibitors [64]. These alterations resemble those of Hodgkin Reed–Sternberg cells where gains of chromosome band 9p24.1 encompassing PD‐L1/PD‐L2/JAK2 contribute to an inefficient antitumour microenvironment and an ‘exhausted’ T‐cell phenotype. It is possible that a broad gene panel or WGS incorporated in the early diagnostics of lymphoid malignancies will prove to be efficient in both recognising rare subtypes early on as well as guiding primary and/or secondary treatment lines [65].
Precision medicine in the clinical routine in AML—Progress and challenges
The intensive regimen of a combination of cytarabine and anthracycline, termed ‘7 + 3’ chemotherapy, has long been considered a standard of care for ‘fit’ younger patients, yet the long‐term cure rates remain at only 30%, with consistently poor outcomes in patients aged >65 years. The examples of data‐driven therapy for molecular subsets were, for a long time, limited to the use of all‐trans retinoic acid/arsenic trioxide in acute promyelocytic leukemia [66] and subsequent recognition of the benefits of anti‐CD33 antibody‐drug conjugate gemtuzumab ozogamicin (GO) in core‐binding factor leukaemias [67], followed by FDA re‐approval of GO in 2017. Recent advances in understanding the pathophysiology and the molecular basis of leukemogenesis led to accelerated discovery and introduction in clinical practice of novel agents for AML therapy.
The affordable and broad use of NGS has enabled the definition of refined molecular subsets of AML. This has not only improved risk assignment but also fine‐tuned and increased knowledge regarding prognostic markers to further understand the role of stem cell transplantation (SCT). Ultimately, it has also led to FDA approvals of several molecularly defined targeted agents clinically useful for patients of all ages. The era of new drug approvals in the setting of molecularly defined personalised medicine for AML was pioneered by the RATIFY trial, which showed a survival benefit of adding the first generation FLT3 inhibitor midostaurin to 7 + 3 chemotherapy for newly diagnosed patients with FLT3 mutations, followed by multiple trials with second and third generation FLT3 inhibitors [68]. From these, single‐agent gilteritinib was FDA approved as salvage therapy for FLT3‐mutated AML based on results of the phase 3 ADMIRAL trial, which demonstrated higher response rates and longer survival with gilteritinib therapy compared with salvage chemotherapy (complete remission [CR] rates of 54% vs. 22%, and median survival 9.3 vs. 5.6 months; hazard ratio 0.637; p = 0.0007) [69]. Several ongoing randomised studies are studying the efficacy of gilteritinib and other FLT3 inhibitors, such as quizartinib and crenolanib, when added to the 7 + 3 backbone.
Recurrent mutations in two IDH isoforms, IDH1 (6%–10%) and IDH2 (9%–13%), have been identified in AML and are more often seen in elderly patients. Specific inhibitors for AML with an IDH1 mutation (ivosidenib) and IDH2 mutation (enasidenib) are both FDA approved now for use in patients with R/R AML harbouring these respective mutations, based on acceptable safety and impressive activity with overall response rates of approximately 40% and median overall survival (OS) of 9 months [70, 71]. Furthermore, ivosidenib is also approved for frontline therapy based on efficacy in patients with newly diagnosed IDH1‐mutated AML [72]. These agents induce leukaemia cell differentiation, which may manifest as on‐target ‘differentiation syndrome’ toxicity.
Given that the median age at diagnosis of AML is high—approximately 68 years—a majority of AML patients are considered ‘older’ [73]. In ‘fit’ older patients, a phase III randomised trial has compared the intensive regimen CPX‐351 (liposomal daunorubicin and AraC) to the 7 + 3 regimen in patients aged 60–75 with secondary AML. With CPX‐351, both CR rates and OS were improved compared to 7 + 3 (2‐year OS 31.1% with CPX‐351 vs. 12.3% with 7 + 3), and 34% of patients treated with CPX‐351 underwent SCT [74]. This led to FDA approval of CPX‐351 for adult newly diagnosed therapy‐related AML and AML with MDS changes, with no age restriction. For older AML patients ineligible for intensive chemotherapy, the combination of hypomethylating agents (HMA) or low‐dose cytarabine (LDAC) and the BCL2 inhibitor venetoclax (VEN) has become a standard‐of‐care nonintensive option. AML cells are highly dependent on the anti‐apoptotic BCL2 family of proteins for survival, and venetoclax is a specific and potent inhibitor of the anti‐apoptotic protein BCL2 [75]. The addition of the BCL2 inhibitor venetoclax to HMA was highly effective in the pivotal randomised Phase III VIALE‐A study, extending OS from 9.6 months in the the AZA group to 14.7 months in the AZA/Ven group and increasing the CR/CRi (CR with incomplete recovery) rate from 28.3% to 66.4%, with an overall acceptable safety profile notable for higher rates of myelosuppression [76]. LDAC with venetoclax is also approved for AML based on VIALE‐C results [77]. Glasdegib is an oral inhibitor of the Hedgehog signalling pathway, which has been shown to play a role in maintaining the leukaemia stem cell compartment. Among AML patients, the addition of glasdegib to LDAC improved the median OS from 4.3 months to 8.3 months and improved the overall response rate (ORR) from 5.3% to 26.9% [78].
Apart from FDA‐approved novel agents, multiple exciting immunotherapy approaches are being exploited in AML clinical trials. T‐cell engaging molecules that co‐engage the CD3 receptor on T cells and a myeloid surface marker include the CD3‐CD123 DART flotetuzumab and the CD3‐CD33 BiTE AMG 330. In a phase I/II study of flotetuzumab in patients with R/R AML, the ORR in 30 patients with early relapse or primary induction failure was 30% [79]. Early phase clinical trials are currently assessing the role of CAR‐T cells against CD33 or CD123 in patients with R/R AML. The potential roles of anti‐CD47 antibody magrolimab in the treatment of TP53‐mutated AML, and of menin inhibitors in the treatment of KMT2A‐ and NPM1‐rearranged AML, are under evaluation [80]. The clear benefit of oral azacitidine (CC‐486) maintenance therapy in AML in the first CR in a QUAZAR AML‐001 study with a median survival of 24.7 months with CC‐486 versus 14.8 months with placebo (hazard ratio 0.69, p = 0.0009) heralded renewed interest of maintenance strategies as a valid therapeutic approach in AML armamentarium [81].
Despite these encouraging advances, several roadblocks await a resolution to improve the efficacy of novel and established therapies. Recognition of the complex molecular patterns of clonal architecture and mutation order in AML supports the notion of using rotating schedules of combination therapies, ideally focusing on driver molecular events [82, 83]. One example of such a strategy could be the rational use of ‘triplets’ with HMA/VEN backbone and targeted agents (FLT3, IDH inhibitors, immune therapies), with judicious attention to overlapping toxicities. Shared antigens between AML and normal haematopoietic stem cells yield risks of on‐target off‐tumour toxicity (cytokine‐release syndrome and profound myelosuppression) and antigen escape (leading to early relapse). For any type of therapy, utilisation of sensitive and specific assays of minimal residual disease (MRD) may improve the early detection of tumour escape and offer opportunities for MRD‐directed therapies. Identification of novel targets for both AML and immune microenvironment using single‐cell approaches will broaden our horizons of personalised therapy in AML.
Precision medicine in clinical routine in lymphoid malignancies—Progress and challenges
Although genetic aberrations have improved and been implemented in the diagnostic work‐up for several lymphoid disorders, few lymphoid malignancies are as yet managed with a precision medicine approach in the first‐line setting. One exception is CLL. As reviewed above, TP53 alterations are associated with the poorest prognosis if patients are treated with CIT. Prognosis has significantly improved under treatment with B‐cell receptor inhibitors, such as ibrutinib, and the BCL2 inhibitor venetoclax. Thus, the presence of TP53 deletion or mutation currently mandates treatment with BTK‐inhibitor ibrutinib in the first line [84, 85]. Moreover, VDJ mutation status impacts treatment choice with the combination of BCL2‐inhibitor venetoclax and the CD20 antibody obinutuzumab constituting a treatment option for IGVH unmutated patients [86].
Several studies now suggest that TP53 aberrations are also associated with worse prognosis in other lymphoma subtypes, most notably mantle cell lymphoma, but also, for example, Waldenström's macroglobulinemia [61]. Recently, four distinct clusters of mantle cell lymphoma were defined based on WES and RNA sequencing and the group with the worst outcome was characterised by TP53/del(17)p mutations, del(13q) and del(9p) and active MYC and hyperproliferation signatures [87]. However, we have yet to learn how these findings should be implemented in clinical practice. In follicular lymphoma, a prognostic model has been suggested, called the m7‐FLIPI, that integrates established clinical prognostic determinants such as advanced disease stage and number of involved lymph node regions with the mutation status of seven genes (EZH2, ARID1A, MEF2B, EP300, FOXO1, CREBBP and CARD11) [88]. However, the m7‐FLIPI was defined among patients treated with first‐line immunochemotherapy and was not validated in a cohort of patients treated with single rituximab [89].
The most commonly occurring lymphoma is DLBCL. That DLBCL constitutes an extremely genetic and phenotypic heterogeneous entity has made precision medicine a challenge in DLBCL [65]. Genetic studies have elucidated that DLBCL is much more heterogeneous than the previous stratification into germinal centre B‐cell (GCB) and non‐germinal centre (GC) subtypes [90]. The findings have led to the proposal of two major classifications according to genetic signature [91, 92]. These new classifications have led to the development of the ‘LymphGen’ classifier, in which cases of DLBCL are clustered according to probability algorithms belonging to a certain genetic subtype based on present genetic aberrations [65]. Approximately 5.7% of patients are classified as strongly belonging to more than one genetic subtype and classified as genetically composite, while approximately 35% remain unclassifiable due to a lack of mutations, atypical mutations or very few genetic aberrations identified [65]. Still, stratification according to these subgroups has already led to the identification of subgroups that may benefit from specific therapies. For example, in a subgroup analysis of the phase III PHOENIX trial, OS was 100% for patients classified in the genetic subgroup MCD and aged ≤60 years, when the BTK‐inhibitor ibrutinib was added to standard treatment with R‐CHOP [93]. As BTK activity was one of the essential survival mechanisms found for DLBCL in the MCD subtype, the augmented efficacy of ibrutinib in this setting appears plausible [65]. Thus, treatment stratifications according to genetic subgroups now represent a feasible future possibility. To enable this development, the LymphGen algorithm is publicly available for use to support treatment stratification according to genetic variants in future clinical trials [65]. Further supporting this claim are the interim results of a prospective study evaluating R‐CHOP + X, where X is chosen according to stratification with the LymphGen algorithm. With 128 patients included thus far, CR rates were 85% in the R‐CHOP + X group compared to 65% in the comparator arm [94].
Apart from the genetic heterogeneity, several challenges further remain in the application of precision medicine in lymphoid malignancies. To be clinically feasible, genetic analyses need to be succinct with a short turnaround, especially in aggressive lymphoma subtypes, to facilitate prompt treatment initiation. Further, some lymphomas are not easily accessible for biopsy. Here, analyses of circulating tumour DNA (ctDNA) in the blood may offer a potential solution [95, 96]. Lastly, the complex genetic landscape of lymphomas and the as‐yet‐unknown significance of specific aberrations highlight the need for extensive collaboration between clinicians, pathologists and geneticists through molecular tumour boards and easily accessible and updated support tools [97].
How to best assess treatment response and follow longitudinal tumour evolution?
Apart from guiding diagnosis and prognosis, genotypic and phenotypic characterisation of malignant clones at diagnosis provides prerequisites to monitor haematological disease during treatment. Capturing remaining disease at low levels offers an opportunity to follow treatment response and progression and to enable swift treatment initiation and adjustment, if needed. An increasingly utilised method to do this is by assessment of MRD. MRD refers to the presence of a detectable small population of malignant cells upon treatment and has revolutionised the possibility to assess disease progression and treatment response. MRD positivity indicates that there are still remaining malignant cells in the body while MRD negativity means that no cancer cells are detectable even using very sensitive methods. MRD negativity is associated with longer remissions and potentially longer rates of survival for certain haematological malignancies [23, 98–100]. In general terms, MRD testing can show how well patients respond to treatment and it can be used to monitor remission, predict relapse and identify patients in need of alternative therapies. Depending on the specific malignancy, some or all of these potential goals can be met when measuring MRD, but technological considerations and improvements, as well as careful clinical studies in specific disease entities, are warranted for successful clinical implementation [101, 102, 103, 104, 105].
By definition, MRD methods are highly sensitive and can detect very low amounts of malignant cells in a large background of normal cells, at levels under morphologic detection. The most widely used methods are based on cells using flow cytometry (FC), genome detection with NGS, quantitative real‐time PCR (qPCR) and droplet digital PCR (ddPCR), among others. The choice of MRD markers and choice of tissue to examine often varies by type of malignancy and specific clinical needs. FC is widely used in most haematological malignancies [106] and measures protein markers at the surface of individual malignant cells and often a fresh bone marrow sample is required. Immunophenotypic evaluation by multiparameter FC can detect one malignant cell in 103–105 normal cells [107], as it uses a combination of leukaemic specific markers. The advantages of FC methods include its fast turnaround time, within 1 day, but it requires skilled pathologists and standardised procedures.
PCR methods search for low amounts of specific aberrant genomic sequences characteristic of the leukaemic clone, which include SNVs to translocation breakpoints in DNA and fusion transcripts in RNA. Quantitative PCR methods can measure levels of the mutant genotype compared to the wildtype gene or a control gene and are widely applicable to many different types of genetic changes identified at diagnosis by NGS. qPCR methods are sensitive (103–105), well standardised and with established routines for quality assurance [99, 108]. Among the limitations are their low multiplexing potential and the need for standard curves for each specific assay. ddPCR can overcome the need for standard curves, as absolute quantification of targets can be achieved, but it also offers simpler and faster laboratory workflows [109, 110, 111, 112].
Considering the large number of genetic aberrations that the NGS method can identify at diagnosis, a sensitive follow‐up using NGS methods with high multiplexing capacity should be optimal. However, due to intrinsic errors in PCR and sequencing, variants with allele fractions under 1% are difficult to detect and measure using standard NGS [113]. Several tag‐based error correction approaches exist to distinguish errors from true mutations [114, 115] and can detect variant allele frequencies (VAF) as low as 0.1% VAF but these require ultra‐deep sequencing at a high cost or pooling of many samples in larger sequencing instruments, compromising fast turnaround times, often needed for follow‐up samples. Notwithstanding, novel NGS approaches that circumvent the need for high sequencing depths, able to detect variants in multiple genes at low VAF and affordable sequencing depths, are being developed [116]. The technological advances of NGS will certainly lead to easily standardised, sensitive and robust MRD methods. However, method development should be undertaken in the context of specific treatment protocols to fully evaluate their clinical utility and limitations. Furthermore, the value of MRD as a guide for therapy should be further evaluated in prospective studies.
How to choose the right treatment for the right patient?
Certain genetic aberrations have a direct impact on treatment choice, but so far the majority do not. Further, that some tumours lack mutations detectable with current methods is well known [65, 91, 92]. Despite treatment advances, there are still patients for whom all standard treatment options have been exhausted without obtaining disease control. Here, taking advantage of genetic characteristics to guide experimental treatments, ideally in a clinical trial setting, is appealing. In the following sections, novel methods to guide the choice of the right treatment for the right patient are discussed.
FPM as a complement to comprehensive molecular profiling
FPM means the direct measurements of drug effects on primary neoplastic cells to profile the potential efficacy of a specific treatment for a patient [117, 118, 119, 120]. This may be performed in situations where available treatment options have been exhausted to find other potential drugs with possible activity in an individual patient. Measuring drug effect on cells has been around since the 1950s; however, FPM brings forward important advances such as disease relevance by using primary cells ex vivo and by including controls to distinguish general toxicity [119, 120]. Often, the ex vivo drug tests are done in multiwell plates to test tens to hundreds of drugs in dose‐measuring cell viability or death as a readout [117, 119]. These bulk assays are useful to profile cell population‐level responses but are limited in providing information on the specific cell types that respond to the drug. To exclude general toxicity and to provide for disease selectivity, calculation of the difference to a control sample, such as healthy bone marrow, is often included in the hit scoring [117, 121]. Viability‐based FPM has been used to profile drug responses and to create large cohorts of leukaemia cases often including molecular omic profiling [34, 122, 123]. These systems biology data sets can readily be generated from both prospective fresh samples and viably frozen biobanked samples with good technological success rates. In one of the recent studies on AML, actionable drugs were found for 97% of the 252 included patient samples [123]. The same study also suggested individually tailored therapies for 37 relapsed or refractory AML patients and reported that 59% of the patients had objective responses, which are praising for the added value of the FPM approach.
In addition to measuring the cell viability by ATP–content, the apoptotic priming of a cell can be measured using dynamic BH3‐profiling (DBP) (Fig. 1) [124, 125]. The DBP assay is a pathway‐specific approach and interrogates the BCL2 family of proteins that regulates commitment to the mitochondrial pathway of apoptosis. This mode of program of cell death is commonly used by cancer cells in response to most chemotherapeutic agents [118]. The apoptotic priming is measured by the level of mitochondrial outer membrane permeabilisation induced by standardised concentrations of synthetic peptides derived from the BH3 domains of the pro‐apoptotic BCL2 family proteins. The DBP assays are generally shorter (4–24 h) in comparison to the ATP‐dependent viability assays (24–72 h) but have had limited throughput to tens of drugs tested. The throughput is important if one wishes to move beyond hypothesis‐driven drug testing and perform unbiased screening of compound libraries to discover new therapies or new indications for existing therapies. Recently, a novel high‐throughput DPB was developed using microscopy, thereby also offering single cellolution [126].
Fig. 1.
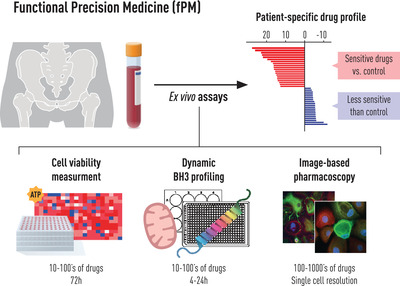
Graphic overview of the workflow in functional precision medicine.
Single‐cell resolution provides the ability to measure cell type–specific responses and use nonmalignant cells from the same specimens as general toxicity controls. This approach was pioneered by the pharmacoscopy imaging‐based assay on a retrospective cohort of 20 newly diagnosed and previously untreated patients with AML [127]. This microscopy‐based approach predicted the clinical response of AML patients to their initial therapy with 88% accuracy. Recently, the clinical feasibility of pharmacoscopy was shown in the first prospective single‐cell FPM (scFPM) trial called EXALT [128]. Here, 56 patients received scFPM guided treatment and 30 of them (54%) demonstrated clinical benefit of more than 1.3‐fold enhanced progression‐free survival (PFS) compared to their previous therapy. Twelve patients experienced exceptional responses lasting three times longer than expected for their respective diseases. This trial is a milestone for FPM by demonstrating the feasibility and clinical relevance in guiding treatment and thereby providing proof for a novel single‐cell resolution tool for precision haematology/oncology. In summary, today's FPM approaches have advanced away from the early days of drug testing when cancer cell lines were used to assess in vivo therapy [129]. All the described FPM assays have demonstrated an important contribution to the field of precision medicine and are particularly feasible for blood‐based cancer with easily attainable disease material. They are not costlier than genome‐based assays and have quick turnover times (hours to days). Implementation into a clinical routine can be done at diagnosis and/or more advanced disease stages whenever enough sample (blood, bone marrow) is available. FPM provides an important complement to genomic profiling by direct efficacy measurement of drugs on the malignant cells and could be used to design and assess treatment response throughout the disease.
Comprehensive molecular profiling as a tool for improving diagnostic precision and selection of treatment—Lessons learned from rare cancers
Although haematologic malignancies, for reasons outlined in the previous section, are disease entities that have pioneered precision diagnostics and medicine, several lessons can also be learned from other rare cancer types. The translational and clinical potential of comprehensive molecular profiling to improve the stratification of patients to treatments is well illustrated by the example of rare cancers. However, determining the clinical consequences of a tumour's molecular profile is not always straightforward. For example, the question often arises whether the ‘druggability’ of a genetic variant can be transferred from one tissue context to another. This problem is well illustrated by the example of monotherapy with the mutation‐specific BRAF inhibitor vemurafenib in more common malignancies with BRAFV600E mutations, such as melanoma, where these alterations were first described [130], which leads to an objective response in approximately half of the entities studied [131]. In contrast, in colorectal carcinoma, rational combination therapies that take into account the physiologic expression profile of the tissue of origin are required to suppress oncogenic RAF‐MEK‐ERK signalling [132]. Another barrier to implementing precision oncology approaches in rare cancers is that negative evidence of drug efficacy in unstratified clinical trials that followed a ‘one‐size‐fits‐all’ strategy likely underestimated the value of many therapies, leaving the efficacy of most approved targeted drugs in rare cancers unexplored [133].
The problem of context dependence complicates the study of targeted therapies in rare cancers, defined as entities with an incidence of less than six per 100,000 persons per year, and prevents their use despite known molecular alterations in these diseases that are, in principle, addressable. This caveat is also frequently present in haematological malignancies, as many are also very rare. To overcome these limitations and advance the discovery of effective biomarker–drug pairs, including their tissue dependence, new adaptive trial designs have been introduced [134]. To further address this unmet clinical need, the National Center for Tumor Diseases, the German Cancer Research Center and the German Cancer Consortium have established a multicentre, prospective observational study that uses a standardised workflow to inform clinical management of these patients and to identify starting points for molecularly stratified clinical trials (Fig. 2). This workflow ranges from patient selection, tissue processing and WGS/WES and RNA sequencing under accredited conditions to clinical decision making in semiweekly molecular tumour boards. The molecular profiles and clinical outcomes of more than 1300 patients first enrolled in this study, approximately three quarters of whom represented rare cancers and were enrolled by more than 100 partners from all cancer care settings in Germany, were recently published [135]. The results demonstrate that comprehensive molecular analysis has tangible diagnostic and therapeutic implications in this prognostically unfavourable patient population. First, the molecular profile was inconsistent with the clinical diagnosis in approximately 5% of the tumours studied, particularly in specific disease groups such as soft‐tissue sarcomas. This usually triggered a re‐evaluation of the particular case, sometimes leading to refinement or even revision of the diagnosis with immediate therapeutic consequences. Second, systematic analysis of autosomal dominant or recessive cancer predispositions in the germline revealed pathogenic variants in approximately 10% of patients. Most of these alterations were not previously known, had immediate consequences for affected families and were sometimes suitable therapeutic targets. Third, comprehensive molecular profiling had relevant therapeutic implications. Based on the categorisation of patients into distinct molecular intervention baskets using several hundred individual genes and six composite biomarkers, the molecular tumour board made recommendations for further clinical management in nearly 90% of the cases. These could be implemented in about one third of cases, indicating that access to medications and clinical trials is an unresolved issue for patients with rare cancers. Among patients who could be treated according to molecular tumour board recommendations, the disease control rate was 55% (response, 24%; stabilisation, 31%) and a PFS ratio >1.3 was achieved in 36% [135]. While these results are encouraging overall, given the prognostically unfavourable patient population with a median survival of less than 1 year and generally exhausted standard therapy, they also identify patient subsets, such as bone sarcomas, in which current precision oncology strategies have not yet been successful.
Fig. 2.
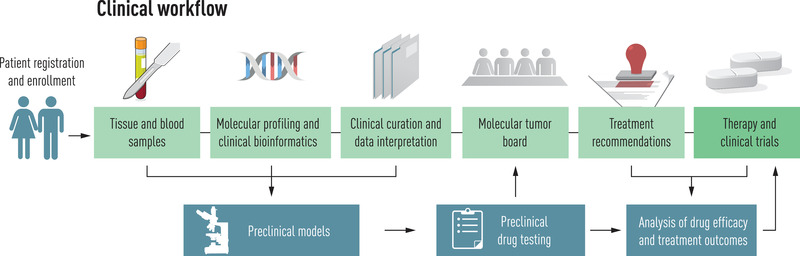
Workflow for guiding a therapeutic decision based on comprehensive genomic and transcriptomic profiling in patients with rare cancers, with courtesy of NCT Heidelberg [135].
Overall, these recent developments illustrate the feasibility and added value of team research in multicentre, sometimes even nationwide, precision oncology networks and demonstrate that WGS/WES and RNA sequencing can enable molecularly informed treatments that lead to clinical benefit in a substantial proportion of patients with rare cancers. In addition, they demonstrate how cross‐entity approaches can help understand which molecular alterations and pathways are valid pan‐cancer targets and which have strong tissue dependency. Hopefully, even stronger interactions between precision oncology in haematological and solid‐organ neoplasms will develop in this regard in the future.
Discussion and concluding remarks
It is clear that genetic characterisation is already pivotal for both diagnosis and prognosis and sometimes treatment choice for many haematological malignancies and implemented in clinical practice, as summarised in Table 1. A summary of techniques used for various haematological malignancies is summarised in Fig. 3. Still, a lot remains to be learned with regard to how best to choose treatment according to an individual patient's genetic tumour landscape. Drug screening and a structured categorisation pipeline, as described above, may offer two novel alternative options. Also, the application of novel therapies based on the presence of certain aberrations will require organised follow‐up and registration to ascertain which patients benefit from specific interventions and when, and to enable robust evaluations across centres.
Table 1.
Overview of advances in implementation of molecular characterisation and its impact on diagnosis, prognosis, follow‐up and treatment choice for major myeloid and lymphoid haematological malignancies
| For diagnosis | For prognosis | For longitudinal follow‐up | Targeted therapies in clinical practice | Impact on survival | |||||
|---|---|---|---|---|---|---|---|---|---|
| Disease entity | In use | Impact | In use | Impact | In use | Impact | In use | Examples | |
| Acute myeloid leukaemia | Yes | Required according to WHO classification | Yes | Improved risk stratification; guides treatment choice | Yes | MRD used to assess treatment response and identify treatment failure and relapse | Yes |
FLT3‐inhibitors IDH1/2‐inhibitors Anti‐CD33 antibody‐drug conjugate BCL2‐inhibitor |
Yes |
| Chronic myeloid leukaemia | Yes | Required according to WHO classification | Yes | Provides information regarding TKI resistance | Yes | MRD used to assess treatment response and identify treatment failure and relapse | Yes | TK‐inhibitors | Yes |
| Myelodysplastic syndrome | Yes | Required according to WHO classification | Yes | Improved risk stratification; guides treatment choice | Yes | MRD used to assess treatment response and identify treatment failure and relapse | Yes |
BCL2‐inhibitor Luspatercept Lenalidomide |
Yes |
| Myeloproliferative neoplasms | Yes | Required according to WHO classification | Yes | Improved risk stratification | No | Not yet in clinical practice | Yes | JAK2‐inhibitor | Yes |
| Chronic lymphocytic leukaemia | No | Not yet used in clinical practice for diagnosis but necessary for risk stratification | Yes | Improved risk stratification; guides treatment choice | Yes | Longitudinal addition of genetic alterations guides subsequent treatment choice | Yes |
BTK‐inhibitors, BCL2‐inhibitor, |
Yes |
| Acute lymphoblastic leukaemia | Yes | Improved diagnosis and of help to differentiate between ALL, AML and mixed phenotype leukaemia | Yes | Improved risk stratification | Yes | MRD used to assess treatment response and identify treatment failure and relapse | Yes |
CD19/CD3 BiTes Anti‐CD22 antibody‐drug conjugate TK‐inhibitors |
Yes |
| Aggressive lymphomas | Yes |
Identification of double‐hit lymphomas. Can help guide diagnosis in unclear cases |
Yes | Improved risk stratification; granular Diffuse large B‐cell lymphoma classifications underway using genetic alterations | No | Not yet used in clinical practice | Yes |
CD79‐antibody BTK‐inhibitor |
Yes |
| Mantle cell lymphoma | Yes | Presence of genetic aberrations function as diagnostic criteria (t[11;14], SOX11) | Yes | Improved risk stratification (TP53‐mutation) | No | MRD to guide follow‐up is being evaluated in clinical trials | Yes |
BTK‐inhibitors BCL2‐inhibitor |
Yes |
| Hairy cell leukaemia | Yes | Presence of genetic aberrations function as diagnostic criteria (BRAFV600E) | No | Not yet in clinical practice | No | Not yet in clinical practice | Yes | BRAF‐inhibitor | Yes |
| Waldenström macroglobulinemia | Yes | Presence of genetic aberrations function as diagnostic criteria (MYD88) | Yes | Can help guide response to BTK‐inhibitor (MYD88, CXCR4) | No | Not yet in clinical practice | Yes | BTK‐inhibitor | Yes |
| Follicular lymphoma | Yes | Presence of genetic aberrations function as diagnostic criteria (t[14;18]) | No | Not yet in clinical practice | No | Not yet in clinical practice | Yes | P13K‐inhibitor | Yes |
Abbreviations: ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; MRD, minimal residual disease; TKI, tyrosine‐kinase inhibitor; WHO, World Health Organisation.
Fig. 3.
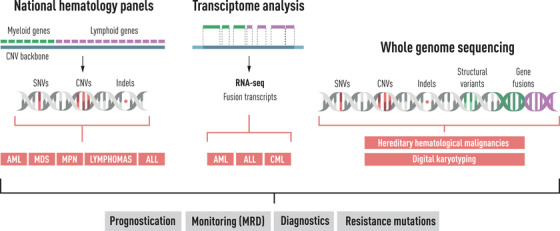
An overview of current workflows and available methods for genetic analyses in haematological diseases.
In addition to the tumour heterogeneity between patients, intratumour heterogeneity is increasingly recognised and a lot remains to be uncovered with regard to longitudinal changes in genetic aberrations. That subclones of a tumour harbour different genetic aberrations and that clonal evolution often drives disease progression and relapse have been demonstrated for several malignancies [10, 11, 12]. Thus, monitoring and precise characterisation of clonal evolution and diversity will be important to understand the development of relapse and resistance mechanisms and to choose subsequent second‐line treatment and enable a deeper understanding of the biology and evolution of haematological diseases. This can be achieved through close monitoring of MRD, for which there are several different techniques available, as covered in this review. Further refinement of methods to measure MRD may provide more accurate and sensitive treatment response information and increase its prognostic value [136]. Moreover, methods that allow measurement of genetic, epigenetic, transcriptomic and proteomic alterations as well as drug efficacies at the single‐cell level are already dramatically improving our understanding of the cellular ecosystem of haematologic malignancies, but several challenges remain before the implementation in a clinical setting [12, 83, 137, 138]. Increasing possibilities to measure disease burden from other sites than the tumour, through, for example, measurement of ctDNA, is also likely to become a valuable tool to capture genetic aberrations more comprehensively [96, 139]. This may especially be useful for diseases such as lymphomas, where recurrent analyses of tumour material may not readily be available [140, 141]. In addition, in studies using ctDNA, specific genetic lesions have been identified in samples from the blood of patients with aggressive lymphoma that were not found in the original tumour, indicating both intratumour heterogeneity and clonal evolution [96]. Still, most current approaches to personalised medicine do not account for intratumour heterogeneity. In studies using single‐cell sequencing to describe this phenomenon, a drastically different treatment response to ex vivo anticancer treatment was seen in malignant subpopulations from the same patient [10]. Further, the impact of the tumour environment and its diverse composition of cell types and the cell states of these (also possible to depict using single‐cell sequencing and transcriptome deconvolution methods) have shown prognostic impact and may also offer additional therapeutic opportunities [142].
A challenge in the implementation of precision medicine is its cost. TKI for the treatment of CML has proven to be cost effective [143]. For other disorders, it is less clear, although systematic reviews indicate that the application of precision medicine overall is cost neutral in comparison to standard care [144]. Evaluation of the cost versus both survival and the potential quality of life benefit of precision medicine in haematology, and continued efforts to assess which patients will benefit from which intervention, will be of paramount importance. This is ongoing in clinical studies, but real‐world evaluation will be needed to ultimately assess its role. Here, register‐based evaluation may be a feasible option and such efforts are ongoing, for example, in the Lymphoma Epidemiology of Outcomes (LEO) study in the United States [145] and the REAL‐LYSA study currently ongoing at lymphoma centres in France [146].
Overall, the easy access to blood to monitor leukaemic diseases provides a unique opportunity for longitudinal follow‐up of disease burden and genetic aberrations in many haematological malignancies and may offer a means to overcome the heterogeneity of tumours. Thus, we are likely to see several expansions to the field of precision medicine in the near future. As haematological malignancies are caused by an interplay of genetic, behavioural and environmental risk factors, models for prediction and diagnosis that incorporate genetic and nongenetic data will likely remain necessary for a long time.
Conflicts of interest
L. B. has held an advisory role and/or received honoraria from AbbVie, Amgen, Astellas, BristolMyers Squibb, Celgene, Daiichi Sankyo, Gilead, Hexal, Janssen, Jazz Pharmaceuticals, Menarini, Novartis, Pfizer, Roche, Sanofi and Seattle Genetics, and has received research support from Bayer and Jazz Pharmaceuticals.
M. K. has received grants from NIH, NCI, AbbVie, Genentech, Stemline Therapeutics, Forty‐Seven, Eli Lilly, Cellectis, Calithera, Ablynx and AstraZeneca; consulting/honorarium from AbbVie, Genentech, F. Hoffman La‐Roche, Stemline Therapeutics, Amgen, Forty‐Seven and Kisoji; clinical trial support from Ascentage and has stocks/royalties in Reata Pharmaceutical. T. F. is a cofounder and board member and owns stocks in Cantargia AB and Qlucore AB. The remaining authors have no conflicts of interest to declare.
Author contributions
K. E. S., T. F. and T. W. planned and prepared the manuscript structure. All authors contributed to the writing of the manuscript and critically reviewed and revised the manuscript. All authors approved the final manuscript.
Acknowledgements
T. W. was supported by Region Stockholm (clinical postdoctoral appointment).
Wästerlid T, Cavelier L, Haferlach C, Konopleva M, Fröhling S, Östling P, et al. Application of precision medicine in clinical routine in haematology—Challenges and opportunities. J Intern Med. 2022;292:243–261.
Content List – This is an article from the symposium: “Precision medicine in hematology”.
References
- 1. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan‐Smith E, Dalton JD, et al. Genome‐wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007;446:758–64. [DOI] [PubMed] [Google Scholar]
- 2. Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long‐term outcomes of imatinib treatment for chronic myeloid leukemia. N Engl J Med. 2017;376:917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sanz MA, Fenaux P, Tallman MS, Estey EH, Löwenberg B, Naoe T, et al. Management of acute promyelocytic leukemia: updated recommendations from an expert panel of the European LeukemiaNet. Blood. 2019;133:1630–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papaemmanuil E, Gerstung M, Bullinger L, Paschka P, Roberts ND, Potter NE, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Campo E, Cymbalista F, Ghia P, Jäger U, Pospisilova S, Rosenquist R, et al. Aberrations in chronic lymphocytic leukemia: an overview of the clinical implications of improved diagnostics. Haematologica. 2018;103:1956–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haferlach T. Advancing leukemia diagnostics: role of next generation sequencing (NGS) in acute myeloid leukemia. Hematol Rep. 2020;12:8957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Haferlach T, Schmidts I. The power and potential of integrated diagnostics in acute myeloid leukaemia. Br J Haematol. 2020;188:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ley TJ, Mardis ER, Ding L, Fulton B, Mclellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. García‐Sanz R, Jiménez C. Time to move to the single‐cell level: applications of single‐cell multi‐omics to hematological malignancies and Waldenström's macroglobulinemia—a particularly heterogeneous lymphoma. Cancers (Basel). 2021;13:1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roider T, Seufert J, Uvarovskii A, Frauhammer F, Bordas M, Abedpour N, et al. Dissecting intratumour heterogeneity of nodal B‐cell lymphomas at the transcriptional, genetic and drug‐response levels. Nat Cell Biol. 2020;22:896–906. [DOI] [PubMed] [Google Scholar]
- 11. Swanton C. Intratumor heterogeneity: evolution through space and time. Cancer Res. 2012;72:4875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Galen P, Hovestadt V, Wadsworth Ii MH, Hughes TK, Griffin GK, Battaglia S, et al. Single‐cell RNA‐Seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176:1265–81.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405. [DOI] [PubMed] [Google Scholar]
- 14. Malhotra H, Radich J, Garcia‐Gonzalez P. Meeting the needs of CML patients in resource‐poor countries. Hematology Am Soc Hematol Educ Program. 2019;2019:433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Radivoyevitch T, Weaver D, Hobbs B, Maciejewski JP, Hehlmann R, Jiang Q, et al. Do persons with chronic myeloid leukaemia have normal or near normal survival? Leukemia. 2020;34:333–5. [DOI] [PubMed] [Google Scholar]
- 16. Ector G, Visser O, Posthuma EFM, Westerweel PE, Janssen J, Blijlevens NMA, et al. Conditional relative survival among adult patients with chronic myeloid leukemia in the tyrosine kinase inhibitor era: a population‐based study in the Netherlands. Leukemia. 2021;35:3291–4. [DOI] [PubMed] [Google Scholar]
- 17. Hochhaus A, Saussele S, Rosti G, Mahon F‐X, Janssen J, Hjorth‐Hansen H, et al. Chronic myeloid leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2018;29:iv261. [DOI] [PubMed] [Google Scholar]
- 18. Cortes J. How to manage CML patients with comorbidities. Blood. 2020;136:2507–12. [DOI] [PubMed] [Google Scholar]
- 19. Grinfeld J, Nangalia J, Baxter EJ, Wedge DC, Angelopoulos N, Cantrill R, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. 2018;379:1416–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia‐Manero G, Solé F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nazha A, Komrokji R, Meggendorfer M, Jia X, Radakovich N, Shreve J, et al. Personalized prediction model to risk stratify patients with myelodysplastic syndromes. J Clin Oncol. 2021;39:3737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. List A, Dewald G, Bennett J, Giagounidis A, Raza A, Feldman E, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med. 2006;355:1456–65. [DOI] [PubMed] [Google Scholar]
- 27. Fenaux P, Platzbecker U, Mufti GJ, Garcia‐Manero G, Buckstein R, Santini V, et al. Luspatercept in patients with lower‐risk myelodysplastic syndromes. N Engl J Med. 2020;382:140–51. [DOI] [PubMed] [Google Scholar]
- 28. Burd A, Levine RL, Ruppert AS, Mims AS, Borate U, Stein EM, et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: feasibility and preliminary efficacy of the Beat AML Master Trial. Nat Med. 2020;26:1852–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Döhner H, Wei AH, Löwenberg B. Towards precision medicine for AML. Nat Rev Clin Oncol. 2021;18:577–90. [DOI] [PubMed] [Google Scholar]
- 30. Duncavage EJ, Schroeder MC, O'Laughlin M, Wilson R, Macmillan S, Bohannon A, et al. Genome sequencing as an alternative to cytogenetic analysis in myeloid cancers. N Engl J Med. 2021;384:924–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lilljebjörn H, Orsmark‐Pietras C, Mitelman F, Hagström‐Andersson A, Fioretos T. Transcriptomics paving the way for improved diagnostics and precision medicine of acute leukemia. Semin Cancer Biol. 2021. 10.1016/j.semcancer.2021.09.013 [DOI] [PubMed] [Google Scholar]
- 32. Arindrarto W, Borràs DM, de Groen RAL, Van Den Berg RR, Locher IJ, Van Diessen S, et al. Comprehensive diagnostics of acute myeloid leukemia by whole transcriptome RNA sequencing. Leukemia. 2021;35:47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mer AS, Heath EM, Madani Tonekaboni SA, Dogan‐Artun N, Nair SK, Murison A, et al. Biological and therapeutic implications of a unique subtype of NPM1 mutated AML. Nat Commun. 2021;12:1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature. 2018;562:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. International CLL‐IPI working group . An international prognostic index for patients with chronic lymphocytic leukaemia (CLL‐IPI): a meta‐analysis of individual patient data. Lancet Oncol. 2016;17:779–90. [DOI] [PubMed] [Google Scholar]
- 36. Eichhorst B, Robak T, Montserrat E, Ghia P, Niemann CU, Kater AP, et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2021;32:23–33. [DOI] [PubMed] [Google Scholar]
- 37. Döhner H, Stilgenbauer S, Benner A, Leupolt E, Krober A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343:1910–6. [DOI] [PubMed] [Google Scholar]
- 38. Rossi D, Rasi S, Spina V, Bruscaggin B, Monti S, Ciardullo C, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013;121:1403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Leeksma AC, Baliakas P, Moysiadis T, Puiggros A, Plevova K, Van der Kevie‐Kersemaekers A‐M, et al. Genomic arrays identify high‐risk chronic lymphocytic leukemia with genomic complexity: a multi‐center study. Haematologica. 2021;106:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cuneo A, Rigolin GM, Mecucci C. Genomic arrays for the identification of high‐risk chronic lymphocytic leukemia: ready for prime time? Haematologica. 2021;106:7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Landau DA, Carter SL, Stojanov PA, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152:714–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Landau DA, Tausch E, Taylor‐Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burns A, Alsolami R, Becq J, Stamatopoulos B, Timbs A, Bruce D, et al. Whole‐genome sequencing of chronic lymphocytic leukaemia reveals distinct differences in the mutational landscape between IgHV. Leukemia. 2018;32:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roberts KG. Genetics and prognosis of ALL in children vs adults. Hematology Am Soc Hematol Educ Program. 2018;2018:137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hoelzer D, Bassan R, Dombret H, Fielding A, Ribera JM, Buske C; ESMO Guidelines Committee . Acute lymphoblastic leukaemia in adult patients: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol. 2016;27:v69–82. [DOI] [PubMed] [Google Scholar]
- 46. Hamadeh L, Enshaei A, Schwab C, Alonso CN, Attarbaschi A, Barbany G, et al. Validation of the United Kingdom copy‐number alteration classifier in 3239 children with B‐cell precursor ALL. Blood Adv. 2019;3:148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LAA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rau RE, Loh ML. Using genomics to define pediatric blood cancers and inform practice. Hematology Am Soc Hematol Educ Program. 2018;2018:286–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Moorman AV. Time for ALL adults to catch up with the children. Blood. 2017;130:1781–3. [DOI] [PubMed] [Google Scholar]
- 50. Lilljebjörn H, Fioretos T. New oncogenic subtypes in pediatric B‐cell precursor acute lymphoblastic leukemia. Blood. 2017;130:1395–401. [DOI] [PubMed] [Google Scholar]
- 51. Gu Z, Churchman ML, Roberts KG. PAX5‐driven subtypes of B‐progenitor acute lymphoblastic leukemia. Nat Genet. 2019;51:296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Li JF, Dai YT, Lilljebjörn H. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci USA. 2018;115:E11711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mitelman F, Johansson B, Mertens F. Mitelman database of chromosome aberrations and gene fusions in cancer. https://mitelmandatabase.isb‐cgc.org (2022). Accessed 13 Dec 2021.
- 54. Roberts KG, Li Y, Payne‐Turner D, Harvey RC, Yang Y‐L, Pei D, et al. Targetable kinase‐activating lesions in Ph‐like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reshmi SC, Harvey RC, Roberts KG, Stonerock E, Smith A, Jenkins H, et al. Targetable kinase gene fusions in high‐risk B‐ALL: a study from the Children's Oncology Group. Blood. 2017;129:3352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Roberts KG, Gu Z, Payne‐Turner D, McCastlain K, Harvey RC, Chen I‐M, et al. High frequency and poor outcome of Philadelphia chromosome‐like acute lymphoblastic leukemia in adults. J Clin Oncol. 2017;35:394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy‐cell leukemia. N Engl J Med. 2011;364:2305–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pettirossi V, Santi A, Imperi E, Russo G, Pucciarini A, Bigerna B, et al. BRAF inhibitors reverse the unique molecular signature and phenotype of hairy cell leukemia and exert potent antileukemic activity. Blood. 2015;125:1207–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Treon SP, Xu L, Yang G. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J Med. 2012;367:826–33. [DOI] [PubMed] [Google Scholar]
- 60. Treon SP, Xu L, Hunter Z. MYD88 mutations and response to ibrutinib in Waldenström's macroglobulinemia. N Engl J Med. 2015;373:584–6. [DOI] [PubMed] [Google Scholar]
- 61. Krzisch D, Guedes N, Boccon‐Gibod C. Cytogenetic and molecular abnormalities in Waldenström's macroglobulinemia patients: correlations and prognostic impact. Am J Hematol. 2021;96:1569–79. [DOI] [PubMed] [Google Scholar]
- 62. Johnson NA, Savage KJ, Ludkovski O, Ludkovski O, Ben‐Neriah S, Woods R, et al. Lymphomas with concurrent BCL2 and MYC translocations: the critical factors associated with survival. Blood. 2009;114:2273–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sarkozy C, Hung SS, Chavez EA, Duns G, Takata K, Chong LC, et al. Mutational landscape of gray zone lymphoma. Blood. 2021;137:1765–76. [DOI] [PubMed] [Google Scholar]
- 64. Griffin GK, Weirather JL, Roemer MGM, Lipschitz M, Kelley A, Chen P‐H, et al. Spatial signatures identify immune escape via PD‐1 as a defining feature of T‐cell/histiocyte‐rich large B‐cell lymphoma. Blood. 2021;137:1353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37:551–68.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lo‐Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–21. [DOI] [PubMed] [Google Scholar]
- 67. Hills RK, Castaigne S, Appelbaum FR, Delaunay J, Petersdorf S, Othus M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia:a meta‐analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014;15:986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377:454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S, et al. Gilteritinib or chemotherapy for relapsed or refractory. N Engl J Med. 2019;381:1728–40. [DOI] [PubMed] [Google Scholar]
- 70. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1‐mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–98. [DOI] [PubMed] [Google Scholar]
- 71. Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Roboz GJ, DiNardo CD, Stein EM, De Botton S, Mims AS, Prince GT, et al. Ivosidenib induces deep durable remissions in patients with newly diagnosed IDH1‐mutant acute myeloid leukemia. Blood. 2020;135:463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sekeres MA, Guyatt G, Abel G, Alibhai S, Altman JK, Buckstein R, et al. American Society of Hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020;4:3528–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, et al. CPX‐351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J Clin Oncol. 2018;36:2684–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL‐2 inhibition by ABT‐199 causes on‐target cell death in acute myeloid leukemia. Cancer Discov. 2014;4:362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383:617–29. [DOI] [PubMed] [Google Scholar]
- 77. Wei AH, Montesinos P, Ivanov V, Dinardo CD, Novak J, Laribi K, et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: a phase 3 randomized placebo‐controlled trial. Blood. 2020;135:2137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cortes JE, Heidel FH, Fiedler W, Smith BD, Robak T, Montesinos P, et al. Survival outcomes and clinical benefit in patients with acute myeloid leukemia treated with glasdegib and low‐dose cytarabine according to response to therapy. J Hematol Oncol. 2020;13:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 2021;137:751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Issa GC, Ravandi F, DiNardo CD, Jabbour E, Kantarjian HM, Andreeff M. Therapeutic implications of menin inhibition in acute leukemias. Leukemia. 2021;35:2482–95. [DOI] [PubMed] [Google Scholar]
- 81. Wei AH, Döhner H, Pocock C, Montesinos P, Afanasyev B, Dombret H, et al. Oral azacitidine maintenance therapy for acute myeloid leukemia in first remission. N Engl J Med. 2020;383:2526–37. [DOI] [PubMed] [Google Scholar]
- 82. Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy‐Durruthy R, et al. Single‐cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587:477–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Morita K, Wang F, Jahn K, Hu T, Tanaka T, Sasaki Y, et al. Clonal evolution of acute myeloid leukemia revealed by high‐throughput single‐cell genomics. Nat Commun. 2020;11:5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, et al. Three‐year follow‐up of treatment‐naïve and previously treated patients with CLL and SLL receiving single‐agent ibrutinib. Blood. 2015;125:2497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single‐arm trial. Lancet Oncol. 2015;16:169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Al‐Sawaf O, Zhang C, Tandon M, Sinha A, Fink A‐M, Robrecht S, et al. Venetoclax plus obinutuzumab versus chlorambucil plus obinutuzumab for previously untreated chronic lymphocytic leukaemia (CLL14): follow‐up results from a multicentre, open‐label, randomised, phase 3 trial. Lancet Oncol. 2020;21:1188–200. [DOI] [PubMed] [Google Scholar]
- 87. Yi S, Yan Y, Jin M, Bhattacharya S, Wang Y, Wu Y, et al. Genomic and transcriptomic profiling reveals distinct molecular subsets associated with outcomes in mantle cell lymphoma. J Clin Invest. 2022;135(5):e153283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first‐line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population‐based registry. Lancet Oncol. 2015;16:1111–22. [DOI] [PubMed] [Google Scholar]
- 89. Lockmer S, Ren W, Brodtkorb M, Østenstad B, Wahlin BE, Pan‐Hammarström Q, et al. M7‐FLIPI is not prognostic in follicular lymphoma patients with first‐line rituximab chemo‐free therapy. Br J Haematol. 2020;188:259–67. [DOI] [PubMed] [Google Scholar]
- 90. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B‐cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. [DOI] [PubMed] [Google Scholar]
- 91. Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B‐cell lymphoma. N Engl J Med. 2018;378:1396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24:679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wilson WH, Wright GW, Huang DW, Hodkinson B, Balasubramanian S, Fan Y, et al. Effect of ibrutinib with R‐CHOP chemotherapy in genetic subtypes of DLBCL. Cancer Cell. 2021;39:1643–53.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhang M, Xu P, Wang L, Cheng S, Zhao W. Genetic subtype guided rituximab‐based immunochemotherapy improves outcome in newly diagnosed diffuse large B‐cell lymphoma: first report of a randomized phase 2 study. Hematol Oncol. 2021;39. 10.1002/hon.26_2879 [DOI] [Google Scholar]
- 95. Spina V, Bruscaggin A, Cuccaro A, Martini M, Di Trani M, Forestieri G, et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood. 2018;131:2413–25. [DOI] [PubMed] [Google Scholar]
- 96. Rossi D, Diop F, Spaccarotella E, Monti S, Zanni M, Rasi S, et al. Diffuse large B‐cell lymphoma genotyping on the liquid biopsy. Blood. 2017;129:1947–57. [DOI] [PubMed] [Google Scholar]
- 97. Tamborero D, Dienstmann R, Rachid MH, Boekel J, Baird R, Braña I, et al. Support systems to guide clinical decision‐making in precision oncology: The Cancer Core Europe Molecular Tumor Board portal. Nat Med. 2020;26:992–4. [DOI] [PubMed] [Google Scholar]
- 98. Freeman SD, Hills RK, Virgo P, Khan N, Couzens S, Dillon R, et al. Measurable residual disease at induction redefines partial response in acute myeloid leukemia and stratifies outcomes in patients at standard risk without NPM1 mutations. J Clin Oncol. 2018;36:1486–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Heuser M, Freeman SD, Ossenkoppele GJ, Buccisano F, Hourigan CS, Ngai LL, et al. 2021 update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2021;138:2753–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kotrova M, Koopmann J, Trautmann H, Alakel N, Beck J, Nachtkamp K, et al. Prognostic value of low‐level MRD in adult acute lymphoblastic leukemia detected by low‐ and high‐throughput methods. Blood Adv. 2022;6:3006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dillon R, Potter N, Freeman S, Russell N. How we use molecular minimal residual disease (MRD) testing in acute myeloid leukaemia (AML). Br J Haematol. 2021;193:231–44. [DOI] [PubMed] [Google Scholar]
- 102. Fernando F, Robertson HF, El‐Zahab S, Pavlů J. How I use measurable residual disease in the clinical management of adult acute lymphoblastic Leukemia. Clin Hematol Int. 2021;3:130–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Paterno G, Del Principe MI, Venditti A. Detection and management of acute myeloid leukemia measurable residual disease: is it standard of care? Curr Opin Hematol. 2020;27:81–7. [DOI] [PubMed] [Google Scholar]
- 104. Bartram J, Patel B, Fielding AK. Monitoring MRD in ALL: methodologies, technical aspects and optimal time points for measurement. Semin Hematol. 2020;57:142–8. [DOI] [PubMed] [Google Scholar]
- 105. Zátopková M, Ševčíková T, Fanfani V, Chyra Z, Říhová L, Bezděková R, et al. Mutation landscape of multiple myeloma measurable residual disease: identification of targets for precision medicine. Blood Adv. 2022;6:368–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Riva G, Nasillo V, Ottomano AM, Bergonzini G, Paolini A, Forghieri F, et al. Multiparametric flow cytometry for MRD monitoring in hematologic malignancies: clinical applications and new challenges. Cancers (Basel). 2021;13:4582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Tettero JM, Freeman S, Buecklein V, Venditti A, Maurillo L, Kern W, et al. Technical aspects of flow cytometry‐based measurable residual disease quantification in acute myeloid leukemia: experience of the European LeukemiaNet MRD Working Party. Hemasphere. 2022;6:e676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Schuurhuis GJ, Heuser M, Freeman S, Béné M‐C, Buccisano F, Cloos J, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131:1275–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Della Starza I, Nunes V, Lovisa F, Silvestri D, Cavalli M, Garofalo A, et al. Droplet digital PCR improves IG‐/TR‐based MRD risk definition in Childhood B‐cell precursor acute lymphoblastic Leukemia. Hemasphere. 2021;5:e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ansuinelli M, Della Starza I, Lauretti A, Elia L, Siravo V, Messina M, et al. Applicability of droplet digital polymerase chain reaction for minimal residual disease monitoring in Philadelphia‐positive acute lymphoblastic leukaemia. Hematol Oncol. 2021;39:680–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Voso MT, Ottone T, Lavorgna S, Venditti A, Maurillo L, Lo‐Coco F, et al. MRD in AML: the role of new techniques. Front Oncol. 2019;9:655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Grassi S, Guerrini F, Ciabatti E, Puccetti R, Salehzadeh S, Metelli MR, et al. Digital droplet PCR is a specific and sensitive tool for detecting IDH2 mutations in acute myeloid leukemia patients. Cancers (Basel). 2020;12:1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vonk CM, Al Hinai ASA, Hanekamp D, Valk PJM. Molecular minimal residual disease detection in acute myeloid leukemia. Cancers (Basel). 2021;13:5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Patkar N, Kakirde C, Shaikh AF, Salve R, Bhanshe P, Chatterjee G, et al. Clinical impact of panel‐based error‐corrected next generation sequencing versus flow cytometry to detect measurable residual disease (MRD) in acute myeloid leukemia (AML). Leukemia. 2021;35:1392–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yoest JM, Shirai CL, Duncavage EJ. Sequencing‐based measurable residual disease testing in acute myeloid leukemia. Front Cell Dev Biol. 2020;8:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Yeom H, Lee Y, Ryu T, Noh J, Lee AC, Lee H‐B, et al. Barcode‐free next‐generation sequencing error validation for ultra‐rare variant detection. Nat Commun. 2019;10:977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pemovska T, Kontro M, Yadav B, Noh J, Lee AC, Lee H‐B, et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013;3:1416–29. [DOI] [PubMed] [Google Scholar]
- 118. Montero J, Sarosiek KA, DeAngelo JD, Edgren H, Eldfors S, Szwajda A, et al. Drug‐induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell. 2015;160:977–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Letai A. Functional precision cancer medicine‐moving beyond pure genomics. Nat Med. 2017;23:1028–35. [DOI] [PubMed] [Google Scholar]
- 120. Tognon CE, Sears RC, Mills GB, Gray JW, Tyner JW. Analysis of primary tumor specimens for evaluation of cancer therapeutics. Annu Rev Cancer Biol. 2021;5:39–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Yadav B, Pemovska T, Szwajda A, Kulesskiy E, Kontro M, Karjalainen R, et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep. 2014;4:5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Dietrich S, Oleś M, Lu J, Sellner L, Anders S, Velten B, et al. Drug‐perturbation‐based stratification of blood cancer. J Clin Invest. 2018;128:427–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Malani D, Kumar A, Bruck O, Kontro M, Yadav B, Hellesøy M, et al. Implementing a functional precision medicine tumor board for acute myeloid leukemia. Cancer Discov. 2022;12:388–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL‐2 family members. Cancer Cell. 2006;9:351–65. [DOI] [PubMed] [Google Scholar]
- 125. Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore VDG, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Bhola PD, Ahmed E, Guerriero JL, Sicinska E, Su E, Lavrova E, et al. High‐throughput dynamic BH3 profiling may quickly and accurately predict effective therapies in solid tumors. Sci Signal. 2020;13:eaay1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Snijder B, Vladimer GI, Krall N, Miura K, Schmolke A‐S, Kornauth C, et al. Image‐based ex‐vivo drug screening for patients with aggressive haematological malignancies: interim results from a single‐arm, open‐label, pilot study. Lancet Haematol. 2017;4:e595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kornauth C, Pemovska T, Vladimer GI, Bayer G, Bergmann M, Eder S, et al. Functional precision medicine provides clinical benefit in advanced aggressive hematological cancers and identifies exceptional responders. Cancer Discov. 2022;12:372–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Tseng YY, Boehm JS. From cell lines to living biosensors: new opportunities to prioritize cancer dependencies using ex vivo tumor cultures. Curr Opin Genet Dev. 2019;54:33–40. [DOI] [PubMed] [Google Scholar]
- 130. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54. [DOI] [PubMed] [Google Scholar]
- 131. Subbiah V, Puzanov I, Blay JY, Chau I, Lockhart AC, Raje NS, et al. Pan‐cancer efficacy of vemurafenib in BRAFV600‐mutant non‐melanoma cancers. Cancer Discov. 2020;10:657–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Di Nicolantonio F, Vitiello PP, Marsoni S, Siena S, Tabernero J, Trusolino L, et al. Precision oncology in metastatic colorectal cancer—from biology to medicine. Nat Rev Clin Oncol. 2021;18:506–25. [DOI] [PubMed] [Google Scholar]
- 133. Ray‐Coquard I, Pujade Lauraine E, Le Cesne A, Pautier P, Vacher Lavenue MC, Trama A, et al. Improving treatment results with reference centres for rare cancers: where do we stand? Eur J Cancer. 2017;77:90–8. [DOI] [PubMed] [Google Scholar]
- 134. van der Velden DL, Hoes LR, van der Wijngaart H, Van Berge Henegouwen JM, Van Werkhoven E, Roepman P, et al. The Drug Rediscovery protocol facilitates the expanded use of existing anticancer drugs. Nature. 2019;574:127–31. [DOI] [PubMed] [Google Scholar]
- 135. Horak P, Heining C, Kreutzfeldt S, Hutter B, Mock A, Hüllein J, et al. Comprehensive genomic and transcriptomic analysis for guiding therapeutic decisions in patients with rare cancers. Cancer Discov. 2021;11:2780–95. [DOI] [PubMed] [Google Scholar]
- 136. Sánchez R, Ayala R, Martínez‐López J. Minimal residual disease monitoring with next‐generation sequencing methodologies in hematological malignancies. Int J Mol Sci. 2019;20:2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Stuart T, Satija R. Integrative single‐cell analysis. Nat Rev Genet. 2019;20:257–72. [DOI] [PubMed] [Google Scholar]
- 138. Pfisterer U, Bräunig J, Brattås P, Heidenblad M, Karlsson G, Fioretos T. Single‐cell sequencing in translational cancer research and challenges to meet clinical diagnostic needs. Genes Chromosomes Cancer. 2021;60:504–24. [DOI] [PubMed] [Google Scholar]
- 139. Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AFM, Esfahani MS, et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8:364ra155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Camus V, Viennot M, Lequesne J, Viailly P‐J, Bohers E, Bessi L, et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: a prospective study. Haematologica. 2021;106:154–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Buedts L, Wlodarska I, Finalet‐Ferreiro J, Gheysens O, Dehaspe L, Tousseyn T, et al. The landscape of copy number variations in classical Hodgkin lymphoma: a joint KU Leuven and LYSA study on cell‐free DNA. Blood Adv. 2021;5:1991–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Steen CB, Luca BA, Esfahani MS, Azizi A, Sworder BJ, Nabet BY, et al. The landscape of tumor cell states and ecosystems in diffuse large B cell lymphoma. Cancer Cell. 2021;39:1422–37.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Reed SD, Anstrom KJ, Li Y, Schulman KA. Updated estimates of survival and cost effectiveness for imatinib versus interferon‐alpha plus low‐dose cytarabine for newly diagnosed chronic‐phase chronic myeloid leukaemia. Pharmacoeconomics. 2008;26:435–46. [DOI] [PubMed] [Google Scholar]
- 144. Kasztura M, Richard A, Bempong NE, Loncar D, Flahault A. Cost‐effectiveness of precision medicine: a scoping review. Int J Public Health. 2019;64:1261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Flowers CR, Link BK, Nastoupil LJ, Mcdonnell TJ, Kahl BS, Vij KR, et al. The Lymphoma Epidemiology of Outcomes (LEO) cohort study reflects the demographics and subtypes of patients diagnosed with non‐Hodgkin lymphoma in the United States. Blood. 2018;132:1702. [Google Scholar]
- 146. Ghesquières H, Rossi C, Cherblanc F, Le Guyader‐Peyrou S, Bijou F, Sujobert P, et al. A French multicentric prospective prognostic cohort with epidemiological, clinical, biological and treatment information to improve knowledge on lymphoma patients: study protocol of the “REal world dAta in LYmphoma and survival in adults” (REALYSA) cohort. BMC Public Health. 2021;21:432. [DOI] [PMC free article] [PubMed] [Google Scholar]