

Abstract
Objective
This study was undertaken to evaluate efficacy and long‐term safety of triheptanoin in patients >1 year old, not on a ketogenic diet, with drug‐resistant seizures associated with glucose transporter 1 deficiency syndrome (Glut1DS).
Methods
UX007G‐CL201 was a randomized, double‐blind, placebo‐controlled trial. Following a 6‐week baseline period, eligible patients were randomized 3:1 to triheptanoin or placebo. Dosing was titrated to 35% of total daily calories over 2 weeks. After an 8‐week placebo‐controlled period, all patients received open‐label triheptanoin through Week 52.
Results
The study included 36 patients (15 children, 13 adolescents, eight adults). A median 12.6% reduction in overall seizure frequency was observed in the triheptanoin arm relative to baseline, and a 13.5% difference was observed relative to placebo (p = .58). In patients with absence seizures only (n = 9), a median 62.2% reduction in seizure frequency was observed in the triheptanoin arm relative to baseline. Only one patient with absence seizures only was present in the control group, preventing comparison. No statistically significant differences in seizure frequency were observed. Common treatment‐emergent adverse events included diarrhea, vomiting, abdominal pain, and nausea, mostly mild or moderate in severity. No serious adverse events were considered to be treatment related. One patient discontinued due to status epilepticus.
Significance
Triheptanoin did not significantly reduce seizure frequency in patients with Glut1DS not on the ketogenic diet. Treatment was associated with mild to moderate gastrointestinal treatment‐related events; most resolved following dose reduction or interruption and/or medication for treatment. Triheptanoin was not associated with any long‐term safety concerns when administered at dose levels up to 35% of total daily caloric intake for up to 1 year.
Keywords: diet treatment, drug resistance, epilepsy, glucose transporter 1 deficiency syndrome, triheptanoin
Key Points.
Triheptanoin was tested in patients with Glut1DS not on the ketogenic diet
Triheptanoin did not significantly reduce overall seizure frequency; however, a clinically meaningful reduction in absence seizures was seen in some patients
There were no long‐term safety concerns when triheptanoin was administered at doses up to 35% of daily caloric intake
Triheptanoin was associated with mild/moderate gastrointestinal events; most resolved following dose modification and/or medication
Triheptanoin safety profile was consistent with other Glut1DS and LC‐FAOD studies
1. INTRODUCTION
Glucose transporter 1 deficiency syndrome (Glut1DS) is a severely debilitating disease characterized by seizures, developmental delay, and movement disorder. 1 , 2 , 3 , 4 Glut1DS is a rare disease, with an estimated birth incidence ranging from 1:24 000 to 1:90 000. 5 , 6 , 7 This birth incidence translates to an estimated point prevalence of ~8500–12 000 in the United States and Europe. Glut1DS is caused by a mutation in solute carrier family 2, member 1 gene SLC2A1, which encodes the GLUT1 protein responsible for transporting glucose across the blood–brain barrier. 3 , 8 Because glucose is the primary source of energy for the brain, this disorder results in a chronic state of energy deficiency in the brain.
Neurological symptoms in Glut1DS fall into three domains: epilepsy, cognitive/behavioral disturbances, and movement disorders. 4 The classical phenotype is a developmental and epileptic encephalopathy encompassing all three domains. 4 , 9 Seizures are present in approximately 90% of patients and usually present in early infancy. The laboratory hallmark of Glut1DS is a low cerebrospinal fluid glucose concentration (<60 mg/dl or 3.3 mmol·L–1 in all cases reported to date; <40 mg/dl or 2.2 mmmol·L–1 in the majority of cases). 4 , 10 Most patients (~90%) have a de novo heterozygous mutation in SLC2A1. 11 Approximately 10% of affected individuals have an affected parent (autosomal dominant inheritance pattern), 3 but autosomal recessive transmission has also been described in rare cases. 12
There are currently no approved drugs that specifically treat Glut1DS. Current treatment consists of a high‐fat ketogenic diet (KD) to provide an alternative energy source to glucose and antiseizure medications (ASMs). 4 , 13 However, seizures due to Glut1DS are typically refractory to conventional ASM treatment. 4 Reports suggest a KD is effective for the seizures of Glut1DS by generating ketone bodies that provide an alternative energy source to glucose. 4 , 14 , 15 , 16 , 17 However, the KD is difficult to tolerate, and patients may not be compliant with or refuse the diet. 17 , 18 These patients represent the subgroup of Glut1DS patients who are most in need of alternative therapy to the KD.
Triheptanoin (Dojolvi) is an odd‐carbon, medium‐chain triglyceride consisting of three 7‐carbon fatty acids on a glycerol backbone. Triheptanoin was approved in the United States (June 2020) as a source of calories and fatty acids for the treatment of pediatric and adult patients with molecularly confirmed long‐chain fatty acid oxidation disorders (LC‐FAODs). 19 , 20 , 21 Triheptanoin is metabolized to heptanoate and C4 and C5 ketone bodies, providing an alternative energy source to the brain. Furthermore, triheptanoin provides anaplerotic substrates to replenish intermediates of the tricarboxylic acid cycle, and triheptanoin can support gluconeogenesis in the brain. 22 , 23 , 24 , 25 A phase 2 study was designed to assess the safety and efficacy of triheptanoin in reducing the frequency of seizures in pediatric, adolescent, and adult populations with Glut1DS. The primary objectives of the trial were to evaluate the efficacy of triheptanoin compared to placebo, as measured by change in seizure frequency, and to evaluate the long‐term safety of triheptanoin in patients with Glut1DS not on a KD.
2. MATERIALS AND METHODS
2.1. Compliance, ethics, and informed consent
The trial (UX007G‐CL201, NCT01993186, EudraCT 2013–003771–35) was conducted over 3.5 years at 14 sites in Asia, Australia, Europe, and the United States.
The study was designed, conducted, recorded, and reported following the principles set forth in the Declaration of Helsinki. The investigators conducted the trial in conformance with those principles, current US Food and Drug Administration regulations, International Council for Harmonization (ICH) Good Clinical Practices (GCP) guidelines, EU Clinical Trials Directive 2001/20/EC, and local ethical and regulatory requirements. The institutional review board/independent ethics committee at each investigational site reviewed and approved the protocol, informed consent form, and any amendments thereof.
The method of obtaining and documenting informed consent complied with ICH GCP guidelines and all other applicable regulatory requirements. Written informed consent was obtained from the patient (or legal guardian in the case of minors or those with intellectual disability) before conducting any research‐related procedures; assent was obtained from minors as applicable.
2.2. Study design
UX007G‐CL201 was a randomized, double‐blind, placebo‐controlled, parallel‐group trial designed to assess the safety and efficacy of triheptanoin for the control of seizures in children and adults with Glut1DS not on a KD (Figure 1). Beginning with the screening visit, patients or their caregivers recorded seizure frequency during the 6‐week baseline period. At the end of the baseline period, eligible patients were randomized in a 3:1 ratio to either triheptanoin or placebo (safflower oil). Dosing was initiated using a 2‐week titration schedule until the patient reached 35% of total daily calories from the study drug. After an 8‐week placebo‐controlled, double‐blind treatment period, the open‐label extension period began, wherein all patients were treated with triheptanoin through Week 52 of the study.
FIGURE 1.
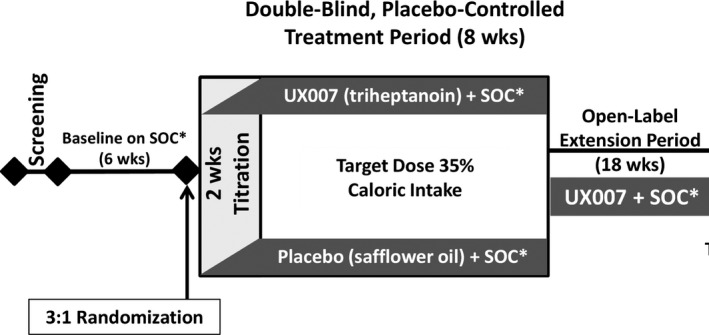
Study design of UX007G‐CL201, a randomized, double‐blind, placebo‐controlled, parallel‐group trial. *SOC, standard of care
2.3. Patients
Patients were at least 1 year of age with a diagnosis of Glut1DS confirmed by SLC2A1 mutation and were on a stable dose of ASM (up to three concomitant ASMs at stable doses were allowed) at study initiation. Inclusion into the study required an average of at least two observable seizures per 4‐week period in the past 6 months and at least two observable seizures during the baseline period. Eligible individuals were not on, or not fully compliant with, a prescribed diet plan (e.g., KD). Compliance was assessed by measuring plasma level of beta‐hydroxybutyrate (BHB). Patients with plasma BHB ≤ 1 mmol·L–1 (nonfasting) at screening could be included.
Key study‐specific exclusion criteria included serum alanine aminotransferase or aspartate aminotransferase levels exceeding 3 × upper limit of normal at screening; any known hypersensitivity to triheptanoin or safflower oil; prior use of triheptanoin within 30 days before screening; and a history of or current suicidal ideation, suicidal behavior, or suicide attempts. Prohibited medications included medium chain triglycerides, barbiturates, pancreatic lipase inhibitors, KetoCal, and other KD supplements.
2.4. Intervention
Patients and caregivers were required to record their daily diet for the 3 days before each visit. The diet history was reviewed by the site dietician to establish daily caloric intake.
Triheptanoin is a colorless to yellow oil for oral (PO) administration. Triheptanoin was initially titrated over 2 weeks to a dose of 35% of total daily caloric intake. Triheptanoin was mixed with food (or formula, as appropriate) and administered PO or by gastrostomy tube at least four times per day (breakfast, lunch, dinner, and before bed). The dose could be divided into smaller, more frequent doses with food as needed. Patients were instructed to mix the dose with small amounts of food and never administer triheptanoin without food.
Placebo consisted of safflower oil matching the appearance of triheptanoin. Dose level and mode of administration were identical to that of triheptanoin during the double‐blind treatment period. Both triheptanoin and placebo were manufactured, packaged, and labeled per Good Manufacturing Practice regulations.
2.5. Statistical analysis
A sample of approximately 40 patients was estimated to be adequate to detect a 50% between‐group difference in seizure frequency per 4 weeks with power of approximately 80% and population SD of 55% using two‐sample t test at one‐sided alpha level of .05.
The primary efficacy endpoint was reduction of seizure frequency from baseline to Week 8 (normalized to a 4‐week rate). Observable seizures were measured by a paper diary during the 6 weeks after the 2‐week titration period. An overnight electroencephalogram (EEG) was also recorded at baseline and Week 8, allowing an additional epilepsy outcome measure including absence seizures. The primary efficacy endpoint was derived for each patient based on patient seizure type at randomization. Observable seizure types included the following, as defined by the 2017 International League Against Epilepsy classification: generalized tonic–clonic, generalized tonic, generalized clonic, generalized atonic, focal (partial) to bilateral tonic–clonic, myoclonic, myoclonic–atonic, myoclonic–tonic, focal with awareness impairment, and focal motor without awareness impairment. At the randomization visit, each patient was classified as “observable seizures only,” “absence seizures only,” or “both” as determined by baseline seizure diary input and/or screening EEG.
The analysis plan specified the use of analysis of covariance (ANCOVA) for the primary analysis. However, seizure data were not normally distributed, rendering a priori analyses inappropriate for analysis by ANCOVA. Ad hoc, nonparametric analyses were conducted using the Hodges–Lehmann estimate of the location shift with 90% (or 95%) confidence interval for within‐group comparisons, and the Wilcoxon rank sum test for between‐group comparisons.
During the initial evaluation of the EEG data, there appeared to be a reduction in absence seizures. A post hoc EEG review was initiated to further explore this potential reduction in absence seizures. A total of 124 EEGs were reread; a total of six patients were “reclassified” either into or out of the “absence only” subgroup. Data from the post hoc EEG reread are presented in this article.
Secondary efficacy endpoints included changes from baseline in frequency of observable seizures and absence seizures and response rate. Other objectives included change from baseline to Week 8 in the Cambridge Neuropsychological Test Automated Battery (CANTAB), distance traveled in the 6‐min walk test (6MWT), time to onset of paroxysmal exercise‐induced dyskinesia (PED), and total score of the 88‐item Gross Motor Function Measure (GMFM‐88). Levels of triheptanoin metabolites (heptanoate, BHB, and beta‐hydroxypentanoate [BHP]) were periodically assessed using validated bioanalytical methods.
The incidence of adverse events and clinically significant changes in safety assessments were evaluated. Gastrointestinal (GI) treatment‐emergent adverse events (TEAEs) were categorized using the standardized MedDRA query. No inferential testing of safety endpoints was planned. An independent data safety monitoring committee with appropriate expertise in the conduct of clinical trials in children reviewed available safety information on a routine basis.
3. RESULTS
3.1. Patients
A total of 36 individuals were enrolled from 13 of the 14 sites and randomized to triheptanoin (25 patients) or placebo (11 patients; Figure 2). All enrolled patients (N = 36; 100%) received at least one dose of the study drug and were included in the efficacy and safety analysis sets. A total of 21 (58.3%) patients completed the study, including 16 (64.0%) patients initially randomized to triheptanoin. All but two (5.6%) patients (triheptanoin group) completed the double‐blind treatment period. The most common reason for discontinuation was consent withdrawal due to gastrointestinal discomfort (five [13.9%] patients) or compliance issues (five [13.9%] patients).
FIGURE 2.
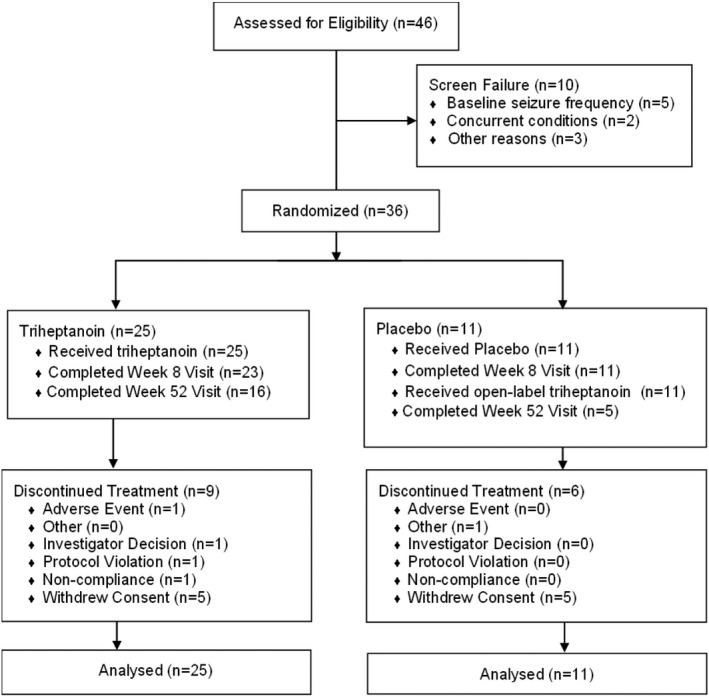
CONSORT (Consolidated Standards of Reporting Trials) diagram and patient disposition
Demographics and baseline characteristics are summarized in Table 1. The population consisted of 15 children (2 to <12 years: 41%), 13 adolescents (12 to <18 years: 36%), and eight adults (18 to <65 years: 22%) with genetically confirmed Glut1DS. The mean (SD) age was 14.3 (8.5) years (range = 4.3–53.8 years). A majority were female (22; 61.1%) and white (32; 88.9%), and their ethnicity was not Hispanic or Latino (30; 83.3%).
TABLE 1.
Demographic and baseline characteristics
| Characteristic | Triheptanoin, n = 25 | Placebo, n = 11 | Total, N = 36 |
|---|---|---|---|
| Age, years | |||
| Mean | 13.86 | 15.24 | 14.28 |
| Minimum, maximum | 4.3, 23.2 | 5.4, 53.8 | 4.3, 53.8 |
| Age group, n (%) | |||
| 2 to <12 years | 8 (32.0) | 7 (63.6) | 15 (41.7) |
| 12 to <18 years | 12 (48.0) | 1 (9.1) | 13 (36.1) |
| 18 to <65 years | 5 (20.0) | 3 (27.3) | 8 (22.2) |
| Sex, n (%) | |||
| Male | 10 (40.0) | 4 (36.4) | 14 (38.9) |
| Female | 15 (60.0) | 7 (63.6) | 22 (61.1) |
| Race, n (%) | |||
| American Indian or Alaska Native | 0 (.0) | 1 (9.1) | 1 (2.8) |
| Asian | 1 (4.0) | 0 (.0) | 1 (2.8) |
| Black or African American | 0 (.0) | 1 (9.1) | 1 (2.8) |
| White | 23 (92.0) | 9 (81.8) | 32 (88.9) |
| Other | 1 (4.0) | 0 (.0) | 1 (2.8) |
| Ethnicity, n (%) | |||
| Hispanic or Latino | 2 (8.0) | 1 (9.1) | 3 (8.3) |
| Not Hispanic or Latino | 21 (84.0) | 9 (81.8) | 30 (83.3) |
| Unknown | 2 (8.0) | 1 (9.1) | 3 (8.3) |
| Seizure history by type, n (%) | |||
| Generalized tonic–clonic | 12 (48.0) | 5 (45.5) | 17 (47.2) |
| Generalized tonic | 4 (16.0) | 3 (27.3) | 7 (19.4) |
| Generalized clonic | 1 (4.0) | 4 (36.4) | 5 (13.9) |
| Generalized atonic | 5 (20.0) | 3 (27.3) | 8 (22.2) |
| Focal [partial] to bilateral tonic–clonic | 6 (24.0) | 4 (36.4) | 10 (27.8) |
| Myoclonic | 9 (36.0) | 4 (36.4) | 13 (36.1) |
| Myoclonic–atonic | 4 (16.0) | 3 (27.3) | 7 (19.4) |
| Focal with impaired awareness | 8 (32.0) | 3 (27.3) | 11 (30.6) |
| Typical absence | 15 (60.0) | 4 (36.4) | 19 (52.8) |
| Atypical absence | 9 (36.0) | 6 (54.5) | 15 (41.7) |
| Myoclonic absence | 10 (40.0) | 1 (9.1) | 11 (30.6) |
| Eyelid myoclonia | 5 (20.0) | 1 (9.1) | 6 (16.7) |
| Glut1DS symptoms [ongoing], n (%) | |||
| Seizures | 25 (100) | 11 (100) | 36 (100) |
| Walking abnormality | 13 (52.0) | 10 (90.9) | 23 (63.9) |
| Coordination or motor abnormalities | 18 (72.0) | 10 (90.9) | 28 (77.8) |
| Paroxysmal exertional dyskinesia | 10 (40.0) | 6 (54.5) | 16 (44.4) |
| Movement disorder besides PED | 10 (40.0) | 3 (27.3) | 13 (36.1) |
| Cognitive abnormality | 20 (80.0) | 10 (90.9) | 30 (83.3) |
| Behavioral abnormality | 14 (56.0) | 5 (45.5) | 19 (52.8) |
| Developmental delay | 18 (72.0) | 10 (90.9) | 28 (77.8) |
Abbreviations: Glut1DS, glucose transporter 1 deficiency syndrome; PED, paroxysmal exercise‐induced dyskinesia.
All had a history of seizures; seizure types that occurred in at least 20% of patients in either arm are summarized in Table 1. Additional frequently reported symptoms included walking abnormality, coordination/motor abnormalities, PED, behavioral abnormality, developmental delay, and cognitive abnormality (Table 1). Most had reported a history of being on a prescribed high‐fat diet (16 patients [44.4%] on a KD and six patients [16.7%] on a modified Atkins diet). The most common reason for high‐fat diet discontinuation (13 patients) was noncompliance. Twelve patients (33%) had not tried a high‐fat diet.
3.2. Exposure and pharmacokinetics
Study drug was introduced over a 2‐week dose titration period to achieve study drug dose levels comprising up to 35% of total daily calories or the maximum tolerated dose. The target 35% dose level was achieved for 11 patients (44.0%) in the triheptanoin group and five patients (54.5%) in the placebo group. During the double‐blind treatment period, the median dose level was 33.36% of daily caloric intake for patients receiving triheptanoin (n = 25, range = 18.6%–62.4%) and 35.52% of daily caloric intake for patients receiving placebo (n = 11, range = 4.6%–69.7%). Among patients receiving triheptanoin and patients receiving placebo, the high end of the range was driven by a single patient in each group with unexplained low total calories for their 3‐day diet history.
The most common concomitant medications (i.e., used by ≥25% of patients) while exposed to triheptanoin were ASMs (83.3%; Table S1), vitamins (38.9%), analgesics (27.8%), anti‐inflammatory and antirheumatic products (25.0%), and psycholeptics (25.0%).
The concentration levels of triheptanoin were close to the lower limit of quantitation as anticipated due to rapid hydrolysis of triheptanoin in the intestine. The concentration levels of triheptanoin metabolites (heptanoate, BHB, and BHP) were greater than those of triheptanoin; the results were consistent with those observed in other clinical trials.
3.3. Efficacy
After 8 weeks of treatment, patients treated with triheptanoin (n = 25) demonstrated a median reduction of 12.6% in overall seizure frequency relative to baseline (Table 2). Patients in the placebo arm demonstrated no change in overall seizure frequency relative to baseline. Median difference in reduction from baseline for triheptanoin relative to placebo was 13.5% (p = .58). For the prespecified secondary analysis of the primary endpoint in patients with absence seizures only (n = 9), patients treated with triheptanoin (n = 8) demonstrated a median 62.2% reduction in seizure frequency relative to baseline. Median difference in reduction from baseline relative to placebo was −37.8% (p = .32); however, there was only one patient in the placebo group, so this finding was not statistically significant. There was no statistically significant difference in seizure frequency between any seizure subgroup during the double‐blind treatment period. Similar trends were seen at Week 31 and Week 52 (Table S2).
TABLE 2.
Seizure frequency (baseline to Week 8)
| Seizure frequency a | Triheptanoin, n = 25 | Placebo, n = 11 |
|---|---|---|
| Overall [diary and EEG], n | 25 | 11 |
| Median (range) at baseline | 96.6 (0–10 250) | 2.1 (0–1176) |
| Median (range) at Week 8 | 17.3 (0–5218) | 2.7 (0–576) |
| Median percent reduction from baseline | 12.6 | 0 |
| Median difference in percent reduction from baseline b | 13.45 | |
| p (95% CI) | .5812 (−38.63 to 80.95) | |
| Patients with any observable seizures [diary], n | 17 | 10 |
| Median (range) at baseline | 7.3 (0–1360) | 1.9 (0–248) |
| Median (range) at Week 8 | 8.0 (0–1341) | 5.4 (0–576) |
| Median percent reduction from baseline | 0 | 0 |
| Median difference in percent reduction from baseline b | 0 | |
| p (95% CI) | .8197 (−51.23 to 84.25) | |
| Patients with observable seizures only [diary], n | 6 | 5 |
| Median (range) at baseline | 38.3 (5–1360) | 8.2 (2–248) |
| Median (range) at Week 8 | 62.1 (3–1341) | 8.0 (2–576) |
| Median percent reduction from baseline | −3.8 | −62.6 |
| Median difference in percent reduction from baseline b | 38.92 | |
| p (95% CI) | .9273 (−84.09 to 192.46) | |
| Patients with any absence seizures [EEG], n | 17 | 6 |
| Median (range) at baseline | .0 (0–366) | .0 (0–42) |
| Median (range) at Week 8 | .0 (0–186) | .0 (0–0) |
| Median percent reduction from baseline | 0 | 0 |
| Median difference in percent reduction from baseline b | 0 | |
| p (95% CI) | .7276 (0–37.50) | |
| Patients with absence seizures only [EEG], n | 8 | 1 |
| Median (range) at baseline | 18.8 (6–366) | 42.0 (42–42) |
| Median (range) at Week 8 | 10.4 (0–186) | .0 (0–0) |
| Median percent reduction from baseline | 62.2 | 100 |
| Median difference in percent reduction from baseline b | −37.79 | |
| p (95% CI) | .3247 (−310.53 to .00) | |
Abbreviations: CI, confidence interval; EEG, electroencephalogram.
Each patient assigned to a “seizure type” population based on events during the randomization period.
Differences are expressed as triheptanoin – placebo.
Multiple secondary and exploratory objectives/endpoints were included to support efficacy evaluation. Secondary efficacy endpoints included changes in cognitive function (CANTAB), walking ability (6MWT), PED onset, and gross motor function (GMFM‐88). Overall, there were no statistically significant changes observed for secondary efficacy variables within and between treatment groups at Week 8.
3.4. Safety
TEAEs were reported for all 36 (100%) patients treated with triheptanoin throughout the double‐blind and extension periods and for nine of 11 (81.8%) patients receiving placebo in the double‐blind treatment period (Table 3). Investigators considered TEAEs to be related to the study drug for 31 of 36 (86.1%) patients in the triheptanoin group and five of 11 (45.5%) patients in the placebo group.
TABLE 3.
Common treatment‐related adverse events by study period
| System organ class, preferred term, patient incidence, n (%) | Double‐blind treatment period, Weeks 0–8 | Extension period, Weeks 9–52 | |
|---|---|---|---|
| Triheptanoin, n = 25 | Placebo, n = 11 | Triheptanoin [total], n = 34 a | |
| Patients with any related TEAE | 18 (72.0) | 5 (45.5) | 27 (79.4) |
| Gastrointestinal disorders | 17 (68.0) | 4 (36.4) | 21 (61.8) |
| Vomiting | 11 (44.0) | 1 (9.1) | 10 (29.4) |
| Diarrhea | 7 (28.0) | 3 (27.3) | 15 (44.1) |
| Abdominal pain upper | 6 (24.0) | 0 (.0) | 3 (8.8) |
| Abdominal pain | 5 (20.0) | 1 (9.1) | 3 (8.8) |
| Nausea | 5 (20.0) | 0 (.0) | 2 (5.9) |
| Investigations | 3 (12.0) | 1 (9.1) | 2 (5.9) |
| Weight increased | 3 (12.0) | 1 (9.1) | 1 (2.9) |
| Metabolism and nutrition disorders | 1 (4.0) | 1 (9.1) | 4 (11.8) |
| Decreased appetite | 1 (4.0) | 1 (9.1) | 4 (11.8) |
Abbreviation: TEAE, treatment‐emergent adverse event.
Triheptanoin includes all patients who received at least one dose of triheptanoin during the 8‐week double‐blind treatment period (n = 25) and/or the 44‐week extension period (N = 36). Placebo represents TEAEs reported during the 8‐week double‐blind treatment period only (n = 11).
Overall, most TEAEs were mild or moderate (Grade 1 or 2) in severity. Grade 3 (severe) TEAEs were reported for three (8.3%) patients while receiving triheptanoin; no Grade 4 (life‐threatening) or Grade 5 (death) TEAEs were reported. A total of three patients (8.3%) experienced serious TEAEs while receiving triheptanoin; none was assessed as related to the study drug by the investigator. One patient (2.8%; triheptanoin arm) had a serious TEAE of status epilepticus during the double‐blind treatment period that led to discontinuation. During the open‐label extension period, two (5.9%) patients experienced serious TEAEs (status epilepticus, seizure, and subcutaneous hematoma in one patient; status epilepticus in the second patient).
The most commonly (≥10%) reported TEAEs throughout the trial across triheptanoin and placebo groups were diarrhea (63.9% of patients), vomiting (55.6%), upper abdominal pain (27.8%), abdominal pain (22.2%), nausea (22.2%), constipation (19.4%), pyrexia (19.4%), decreased appetite (16.7%), viral upper respiratory tract infection (16.7%), headache (13.9%), and increased weight (11.1%).
During the double‐blind treatment period, GI TEAEs were reported for the triheptanoin group with a patient incidence approximately ≥2 times the patient incidence in the placebo group, included vomiting (triheptanoin, 44.0%; placebo, 18.2%) upper abdominal pain (triheptanoin, 28.0%; placebo, 0), abdominal pain (triheptanoin, 28.0%; placebo, 9.1%), and nausea (triheptanoin, 20.0%; placebo, 0). The overall incidence of related TEAE and GI TEAE was greater during the extension period and was primarily attributed to prolonged duration of exposure.
Throughout the trial, TEAEs led to dose modifications: dose reductions in 13 patients, dose increases in two patients, and dose interruptions in 18 patients. Most events leading to dose modifications were mild to moderate GI events.
3.5. Subgroup analysis of TEAEs
During the double‐blind treatment period, the reporting of TEAEs was less frequent in adults (Figure 3). The incidence of GI disorders was more common in children and adolescents (80% and 61.5%, respectively) compared to adults (37.5%). The incidence of nervous system disorders was more common in adults (37.5%) than in children (13.3%) or adolescents (15.4%).
FIGURE 3.
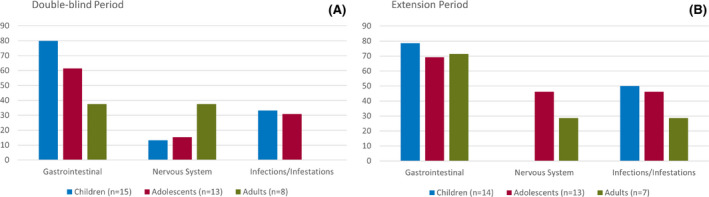
Treatment‐emergent adverse events by system organ class and age subgroup during the double‐blind period (placebo and triheptanoin treated) and extension period (open‐label triheptanoin). Children: 2 to <12 years old; adolescents: 12 to <18 years old; adults: ≥18 years old
Females experienced a higher incidence of nervous system disorders (females, 27.3%; males, 7.1%), whereas infections and infestations were higher in the male population (females, 13.6%; males, 42.9%).
A retrospective review of the TEAEs experienced by seven patients with Glut1DS who received triheptanoin and valproic acid concurrently showed all known adverse drug reactions previously described with valproic acid and triheptanoin.
3.6. Other safety assessments
Overall, safety assessments including routine laboratory testing, vital signs, physical examinations, and electrocardiograms did not reveal any clinically significant changes during the study; there were no serious TEAEs associated with clinically significant changes in any laboratory parameter. Throughout the trial, no suicidal ideation or behavior was reported. Self‐injurious behavior without suicidal intent was reported for one patient at the screening visit (before any study drug administration), and one patient (triheptanoin arm) at the Week 31 and Week 44 visits. This behavior was assessed by the investigator as not related to the study drug.
4. DISCUSSION
Results from previous clinical trials suggested triheptanoin could be of clinical benefit to patients with Glut1DS. 19 , 20 , 21 Based on those results, this randomized, double‐blind, placebo‐controlled, parallel‐group trial was designed to assess the safety and efficacy of triheptanoin in children, adolescents, and adults with seizures associated with Glut1DS. The prespecified primary endpoint was not achieved; overall, there was no reduction in the frequency (median %) of seizures (total or observable).
Most patients with drug‐resistant seizures associated with Glut1DS are treated with some type of prescribed high‐fat diet. 4 Because this study was designed to assess whether triheptanoin could be an alternative approach to seizure control in this population, eligibility was limited to patients who were not on a high‐fat diet, thereby complicating enrollment. Also, because enrollment was limited to patients who were not treated with a KD or modified Atkins diet, it is conceivable that the enrolled patients were inherently less responsive to any type of energy‐replacement therapy. The selection of safflower oil as the placebo also may have confounded the result. This metabolizable oil might have attenuated the difference between intervention and placebo, although the effect of the safflower oil is unknown. Furthermore, it is not known whether triheptanoin would influence ketosis in patients on and off a KD. Therefore, additional research would be necessary to determine whether triheptanoin may have an additive benefit when administered together with a KD.
Like all epilepsies, the development of new targeted pharmacological therapies for Glut1DS is complicated by its broad phenotypic spectrum, which is highlighted by the many epilepsy syndromes associated with Glut1DS. 13 , 26 , 27 , 28 In this trial, numerous seizure types were reported at baseline, including eight different seizure types exhibited by at least 20% of participants. The original study design specified a 1:1 randomization to triheptanoin or placebo, excluded absence seizures from contributing to the baseline seizure count for enrollment, and required a higher number of seizures during the baseline period. Enrollment, as a result, was extremely challenging with this design. The protocol was eventually amended to include patients with absence seizures, to decrease the number of seizures required during baseline, and to change to a 3:1 randomization schema. Although the changes to the protocol did facilitate enrollment, it also is likely that some of these changes may have affected the study outcome. The low number of observed seizures required for entry may have permitted selection of mildly affected patients, as more severely affected patients were unwilling to meet the criteria for screening. The low seizure frequency in many of the patients made it challenging to detect meaningful change during a short observation period, and the small number of placebo patients meant that few were available for comparison within the study subsets of observable, absence, or both seizure types.
The duration of the baseline treatment periods was designed to (1) minimize the likelihood of changes to concomitant ASM regimens, (2) minimize patient discontinuations due to lack of efficacy during the baseline period and/or placebo administration, and (3) be of sufficient length to allow for accurate determination of the seizure frequencies during those periods. Extending the placebo‐controlled treatment period was not deemed warranted in this population of predominantly young pediatric patients, who are especially vulnerable to the effects of seizures. All but two (5.6%) patients (triheptanoin group) completed the double‐blind treatment period.
Diaries were used by patients and caregivers to capture seizure events daily, thereby minimizing recall bias at site visits. As seen with most epilepsy trials, a limitation of the design for this trial was the use of an objective measure (diary) to track seizures as the primary efficacy variable. 29 Some sites reported that parents, who were responsible for diary completion, had a lack of clarity between clinical seizures and PEDs. Although EEG is a more objective measurement for the enumeration of absence seizures, the experience in this trial suggests clear criteria for detection and enumeration of events must be established a priori. With only one patient in the placebo arm, interpretation of the observed reduction in absence seizures is limited, but a reduction would be clinically relevant, warranting further investigation of triheptanoin in this population.
The secondary efficacy parameters encompassed the characteristic phenotype of individuals with Glut1DS, namely, seizures, delayed development, and complex movement disorders. Clinical assessments employed standard measures used in other diseases/conditions that affect the central nervous system and skeletal muscle. The 6MWT and GMFM‐88 were incorporated into the study design as a performance measure of muscle function to evaluate movement disorders typically seen in Glut1DS patients. The Columbia Neurological Score and CANTAB were included as measures of neurological involvement and cognitive function. Where possible, additional validated, age‐appropriate, patient‐reported outcomes were included to assess health‐related quality of life and activities of daily living (Short Form [SF]‐10/SF‐12 and Pediatric Evaluation of Disability Inventory Computer Adaptive Test).
Secondary efficacy variables did not show clinically meaningful differences in this study. The 6MWT was able to demonstrate the walking impairment of this study population at baseline, but it did not show any evidence of improvement in walking ability or fatigue over the course of the trial. The GMFM‐88 showed a ceiling effect for many of the patients at baseline, with minimal opportunity for improvement following randomization. Cognition as measured by the CANTAB may have been impacted by a study population with cognitive impairments at entry that were static in nature and not amenable to treatment. The randomization period/length of triheptanoin exposure may have been too short to measure cognitive changes.
After the initial 8‐week double‐blind treatment period, all patients were treated with triheptanoin through Week 52 during the open‐label extension period to assess long‐term safety and maintenance of effect. A majority of patients (58.3%) completed the study, including 16 (64%) patients initially randomized to triheptanoin, providing a reasonable assessment of long‐term safety in children, adolescents, and adults. Despite limitations in the characteristics of the study population and in trial enrollment and design, 94% of patients who participated in this study, and most patients who have participated in other triheptanoin studies, have chosen to continue in the extension/compassionate use program.
The safety profile observed throughout the trial is consistent with results of other trials with triheptanoin in Glut1DS and LC‐FAODs. 20 , 21 , 30 , 31 , 32 The most frequently reported TEAEs with the use of triheptanoin are mild‐to‐moderate GI events of vomiting, diarrhea, abdominal pain, and upper abdominal pain. Many of these events were managed with dose modification, temporary interruption, or symptomatic treatment of the TEAE. Tolerability improved for some patients when consuming triheptanoin in smaller, more frequent doses mixed with food throughout the day. Vomiting and abdominal pain were reported at a greater incidence in patients treated with triheptanoin than with placebo during the initial 8‐week double‐blind treatment period. The incidence of diarrhea was similar in both groups during the double‐blind treatment period, suggesting that this effect is related to the oil composition of triheptanoin and safflower oil. Weight increase was reported in five patients (four triheptanoin, one placebo) within the initial 60 days of study; contributing factors may have included a lack of diet adjustment for the increased caloric intake and normal growth in pediatric patients.
In conclusion, triheptanoin did not result in significant improvement in clinical outcomes associated with Glut1DS. The safety profile of triheptanoin was generally consistent with observations from other studies in Glut1DS and LC‐FAODs.
CONFLICT OF INTEREST
P.S. has received fees from Ultragenyx Pharmaceutical Inc, Zogenix, BioMarin, PTC Therapeutics, GW Pharma, and Neuraxpharm, and research grants from GW Pharma, PTC Therapeutics, Enecta, and Kolfarma. He has been an investigator for clinical trials for Ultragenyx Pharmaceutical Inc and Zogenix. He has served on a scientific advisory board for the Italian Medicines Agency; has received honoraria from GW Pharma, Kolfarma, and Eisai; and has received research support from the Italian Ministry of Health and San Paolo Foundation. S.A. has served as a consultant or received honoraria for lectures from Biocodex, BioMarin, Eisai, GW Pharma, Neuraxpharm, Nutricia, UCB Pharma, Ultragenyx Pharmaceutical Inc, and Zogenix. He has been an investigator for clinical trials for Eisai, UCB Pharma, Ultragenyx Pharmaceutical Inc, and Zogenix. I.E.S. has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, Chiesi, Encoded Therapeutics, Knopp Biosciences, and Xenon Pharmaceuticals; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, Chiesi, LivaNova, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, BioMarin, and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx Pharmaceutical Inc, GW Pharma, UCB, Eisai, Xenon Pharmaceuticals, Anavex Life Sciences, Ovid Therapeutics, Epygenix Therapeutics, Encoded Therapeutics, and Marinus; and has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, Care Beyond Diagnosis, Epilepsy Consortium, and UCB. She may accrue future revenue on pending patent WO61/010176 (filed 2008; Therapeutic Compound); has a patent for SCN1A testing held by Bionomics and licensed to various diagnostic companies; and has a patent "Molecular Diagnostic/Theranostic Target for Benign Familial Infantile Epilepsy (BFIE)" (PRRT2; 2011904493, 2012900190 and PCT/AU2012/001321; TECH ID: 2012–009). D.C.D. has served as advisor/consultant for AveXis, Biogen, Cytokinetics, Ionis Pharmaceuticals, Metafora Biosystems, Roche, Sanofi, Sarepta, and SMA Foundation; has received research grants from Hope for Children Research Foundation, National Institutes of Health, SMA Foundation, Cure SMA, Glut1 Deficiency Foundation, and US Department of Defense; has received clinical trial funding from Biogen, Mallinckrodt, PTC Therapeutics, Sarepta, Scholar Rock, and Ultragenyx Pharmaceutical Inc; and has received compensation as a member of a data and safety monitoring board (Canavan disease) for Aspa Therapeutics. M.T. has served as consultant for or received honoraria from Novartis and LivaNova; he has served on a scientific advisory board for Novartis and has been an investigator for clinical trials for LivaNova, Ovid Therapeutics, Novartis, GW Pharma, Ultragenyx Pharmaceutical Inc, and Pfizer. R.H. has served on a scientific advisory board for Zogenix. R.P.S. has received speaker honoraria from GW Pharma and has been an investigator for clinical trials for GW Pharma, Zogenix, Ultragenyx Pharmaceutical Inc, UCB Pharma, and Lundbeck. M.K.K. has served on advisory boards for Novartis and Taysha Gene Therapies; has received speaker honoraria from Novartis Pharmaceuticals, Greenwich Pharmaceuticals, and Lundbeck; serves on a data and safety monitoring board for Zogenix; and has received research funding from GW Research, Reneo Pharmaceuticals, Astellas Pharma, PTC Therapeutics, Marinus Pharmaceuticals, Stealth Biotherapeutics, EryDel, Ultragenyx Pharmaceutical Inc, Retrophin, LAM Therapeutics, Pfizer, Vtesse, Reata Pharmaceuticals, and Novartis Pharmaceuticals. None of the other authors has any conflict of interest to disclose. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Table S1‐S2
ACKNOWLEDGMENTS
We thank the patients and their families for participation in the trial. We would like to acknowledge the contributions of Jill Mayhew, Boglarka Bansagi, Giovanna Giudizioso, Maria Stella Vari, Genoa; Keren Proper, Kristin Engelstad, Ameneh Masud, Erin Stackowitz, Clinical Investigation Center, Robert Debré University Hospital; and LeiLei Dang. P.S. developed this work within the framework of the DINOGMI Department of Excellence of MIUR 2018‐2022 (legge 232 del 2016). Medical writing assistance was provided by Kimberly Denis‐Mize, PhD and Kerri Hebard‐Massey, PhD, and funded by Ultragenyx Pharmaceutical Inc. Open Access Funding provided by Universita degli Studi di Genova within the CRUI‐CARE Agreement. [Correction added on 06 July 2022, after first online publication: CRUI‐CARE funding statement has been added.]
Striano P, Auvin S, Collins A, Horvath R, Scheffer IE, Tzadok M, et al. A randomized, double‐blind trial of triheptanoin for drug‐resistant epilepsy in glucose transporter 1 deficiency syndrome. Epilepsia. 2022;63:1748–1760. 10.1111/epi.17263
Clinical trial registration: NCT01993186; EudraCT 2013–003771–35
Funding information
The study was funded by Ultragenyx Pharmaceutical Inc.
DATA AVAILABILITY STATEMENT
Requests for individual deidentified participant data and the clinical study report from this study will be available to researchers providing a methodologically sound proposal that is in accordance with the Ultragenyx Pharmaceutical Inc data sharing commitment. To gain access, data requestors will need to sign a data access and use agreement. Data will be shared via secured portal. The study protocol and statistical analysis plan for this study will be available on the relevant clinical trial registry websites with the tabulated results.
REFERENCES
- 1. Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep. 2013;13(4):342. [DOI] [PubMed] [Google Scholar]
- 2. De Vivo DC, Trifiletti RR, Jacobson RI, Ronen GM, Behmand RA, Harik SI. Defective glucose transport across the blood‐brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991;325(10):703–9. [DOI] [PubMed] [Google Scholar]
- 3. Koch H, Weber YG. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav. 2019;91:90–3. [DOI] [PubMed] [Google Scholar]
- 4. Klepper J, Akman C, Armeno M, Auvin S, Cervenka M, Cross HJ, et al. Glut1 deficiency syndrome (Glut1DS): state of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open. 2020;5(3):354–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coman DJ, Sinclair KG, Burke CJ, Appleton DB, Pelekanos JT, O'Neil CM, et al. Seizures, ataxia, developmental delay and the general paediatrician: glucose transporter 1 deficiency syndrome. J Paediatr Child Health. 2006;42(5):263–7. [DOI] [PubMed] [Google Scholar]
- 6. Larsen J, Johannesen KM, Ek J, Tang S, Marini C, Blichfeldt S, et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia. 2015;56(12):e203–8. [DOI] [PubMed] [Google Scholar]
- 7. Symonds JD, Zuberi SM, Stewart K, McLellan A, O'Regan M, MacLeod S, et al. Incidence and phenotypes of childhood‐onset genetic epilepsies: a prospective population‐based national cohort. Brain. 2019;142(8):2303–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castellotti B, Ragona F, Freri E, Solazzi R, Ciardullo S, Tricomi G, et al. Screening of SLC2A1 in a large cohort of patients suspected for Glut1 deficiency syndrome: identification of novel variants and associated phenotypes. J Neurol. 2019;266(6):1439–48. [DOI] [PubMed] [Google Scholar]
- 9. Verrotti A, D'Egidio C, Agostinelli S, Gobbi G. Glut1 deficiency: when to suspect and how to diagnose? Eur J Paediatr Neurol. 2012;16(1):3–9. [DOI] [PubMed] [Google Scholar]
- 10. Leen WG, Wevers RA, Kamsteeg EJ, Scheffer H, Verbeek MM, Willemsen MA. Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: a systematic review. JAMA Neurol. 2013;70(11):1440–4. [DOI] [PubMed] [Google Scholar]
- 11. Ebrahimi‐Fakhari D, Van Karnebeek C, Munchau A. Movement disorders in treatable inborn errors of metabolism. Mov Disord. 2019;34(5):598–613. [DOI] [PubMed] [Google Scholar]
- 12. Rotstein M, Engelstad K, Yang H, Wang D, Levy B, Chung WK, et al. Glut1 deficiency: inheritance pattern determined by haploinsufficiency. Ann Neurol. 2010;68(6):955–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arsov T, Mullen SA, Rogers S, Phillips AM, Lawrence KM, Damiano JA, et al. Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol. 2012;72(5):807–15. [DOI] [PubMed] [Google Scholar]
- 14. Veggiotti P, De Giorgis V. Dietary treatments and new therapeutic perspective in GLUT1 deficiency syndrome. Curr Treat Options Neurol. 2014;16(5):291. [DOI] [PubMed] [Google Scholar]
- 15. Gavrilovici C, Rho JM. Metabolic epilepsies amenable to ketogenic therapies: indications, contraindications, and underlying mechanisms. J Inherit Metab Dis. 2021;44(1):42–53. [DOI] [PubMed] [Google Scholar]
- 16. Pavon S, Lazaro E, Martinez O, Amayra I, Lopez‐Paz JF, Caballero P, et al. Ketogenic diet and cognition in neurological diseases: a systematic review. Nutr Rev. 2021;79(7):802–13. [DOI] [PubMed] [Google Scholar]
- 17. Schwantje M, Verhagen LM, van Hasselt PM, Fuchs SA. Glucose transporter type 1 deficiency syndrome and the ketogenic diet. J Inherit Metab Dis. 2020;43(2):216–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Varesio C, Pasca L, Parravicini S, Zanaboni MP, Ballante E, Masnada S, et al. Quality of life in chronic ketogenic diet treatment: the GLUT1DS population perspective. Nutrients. 2019;11(7):1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vockley J, Burton B, Berry G, Longo N, Phillips J, Sanchez‐Valle A, et al. Effects of triheptanoin (UX007) in patients with long‐chain fatty acid oxidation disorders: results from an open‐label, long‐term extension study. J Inherit Metab Dis. 2021;44(1):253–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vockley J, Burton B, Berry GT, Longo N, Phillips J, Sanchez‐Valle A, et al. Results from a 78‐week, single‐arm, open‐label phase 2 study to evaluate UX007 in pediatric and adult patients with severe long‐chain fatty acid oxidation disorders (LC‐FAOD). J Inherit Metab Dis. 2019;42(1):169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vockley J, Burton B, Berry GT, Longo N, Phillips J, Sanchez‐Valle A, et al. UX007 for the treatment of long chain‐fatty acid oxidation disorders: safety and efficacy in children and adults following 24 weeks of treatment. Mol Genet Metab. 2017;120(4):370–7. [DOI] [PubMed] [Google Scholar]
- 22. Gu L, Zhang GF, Kombu RS, Allen F, Kutz G, Brewer WU, et al. Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats. II. Effects on lipolysis, glucose production, and liver acyl‐CoA profile. Am J Physiol Endocrinol Metab. 2010;298(2):E362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Karall D, Brunner‐Krainz M, Kogelnig K, Konstantopoulou V, Maier EM, Moslinger D, et al. Clinical outcome, biochemical and therapeutic follow‐up in 14 Austrian patients with long‐chain 3‐hydroxy acyl CoA dehydrogenase deficiency (LCHADD). Orphanet J Rare Dis. 2015;10:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Karall D, Mair G, Albrecht U, Niedermayr K, Karall T, Schobersberger W, et al. Sports in LCHAD deficiency: maximal incremental and endurance exercise tests in a 13‐year‐old patient with long‐chain 3‐hydroxy acyl‐CoA dehydrogenase deficiency (LCHADD) and heptanoate treatment. JIMD Rep. 2014;17:7–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marin‐Valencia I, Good LB, Ma Q, Malloy CR, Pascual JM. Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I‐deficient (G1D) brain. J Cereb Blood Flow Metab. 2013;33(2):175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mullen SA, Marini C, Suls A, Mei D, Della Giustina E, Buti D, et al. Glucose transporter 1 deficiency as a treatable cause of myoclonic astatic epilepsy. Arch Neurol. 2011;68(9):1152–5. [DOI] [PubMed] [Google Scholar]
- 27. Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE. Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology. 2010;75(5):432–40. [DOI] [PubMed] [Google Scholar]
- 28. Suls A, Mullen SA, Weber YG, Verhaert K, Ceulemans B, Guerrini R, et al. Early‐onset absence epilepsy caused by mutations in the glucose transporter GLUT1. Ann Neurol. 2009;66(3):415–9. [DOI] [PubMed] [Google Scholar]
- 29. Karoly P, Goldenholz DM, Cook M. Are the days of counting seizures numbered? Curr Opin Neurol. 2018;31(2):162–8. [DOI] [PubMed] [Google Scholar]
- 30. Hainque E, Gras D, Meneret A, Atencio M, Luton MP, Barbier M, et al. Long‐term follow‐up in an open‐label trial of triheptanoin in GLUT1 deficiency syndrome: a sustained dramatic effect. J Neurol Neurosurg Psychiatry. 2019;90(11):1291–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mochel F, Hainque E, Gras D, Adanyeguh IM, Caillet S, Heron B, et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J Neurol Neurosurg Psychiatry. 2016;87(5):550–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pascual JM, Liu P, Mao D, Kelly DI, Hernandez A, Sheng M, et al. Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. 2014;71(10):1255–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1‐S2
Data Availability Statement
Requests for individual deidentified participant data and the clinical study report from this study will be available to researchers providing a methodologically sound proposal that is in accordance with the Ultragenyx Pharmaceutical Inc data sharing commitment. To gain access, data requestors will need to sign a data access and use agreement. Data will be shared via secured portal. The study protocol and statistical analysis plan for this study will be available on the relevant clinical trial registry websites with the tabulated results.