

Abstract
Aim
The genetic relationship between schizophrenia (SCZ) and other nonpsychiatric disorders remains largely unknown. We examined the shared genetic components between these disorders based on multipopulation data sets.
Methods
We used two data sets for East Asian (EAS) and European (EUR) samples. SCZ data was based on the Psychiatric Genomics Consortium Asia with our own genome‐wide association study for EAS and Psychiatric Genomics Consortium for EUR. Nonpsychiatric data (20 binary traits [mainly nonpsychiatric complex disorders] and 34 quantitative traits [mainly laboratory examinations and physical characteristics]) were obtained from Biobank Japan and UK Biobank for EAS and EUR samples, respectively. To evaluate genetic correlation, linkage disequilibrium score regression analysis was utilized with further meta‐analysis for each result from EAS and EUR samples to obtain robust evidence. Subsequent mendelian randomization analysis was also included to examine the causal effect.
Results
A significant genetic correlation between SCZ and several metabolic syndrome (MetS) traits was detected in the combined samples (meta‐analysis between EAS and EUR data) (body mass index [rg = −0.10, q‐value = 1.0 × 10−9], high‐density‐lipoprotein cholesterol [rg = 0.072, q‐value = 2.9 × 10−3], blood sugar [rg = −0.068, q‐value = 1.4 × 10−2], triglycerides [rg = −0.052, q‐value = 2.4 × 10−2], systolic blood pressure [rg = −0.054, q‐value = 3.5 × 10−2], and C‐reactive protein [rg = −0.076, q‐value = 7.8 × 10−5]. However, no causal relationship on SCZ susceptibility was detected for these traits based on the mendelian randomization analysis.
Conclusion
Our results indicate shared genetic components between SCZ and MetS traits and C‐reactive protein. Specifically, we found it interesting that the correlation between MetS traits and SCZ was the opposite of that expected from clinical studies: this genetic study suggests that SCZ susceptibility was associated with reduced MetS. This implied that MetS in patients with SCZ was not associated with genetic components but with environmental factors, including antipsychotics, lifestyle changes, poor diet, lack of exercise, and living conditions.
Keywords: blood pressure, body mass index, cholesterol, C‐reactive protein, triglycerides
Schizophrenia (SCZ) is a common disorder with a complex pathophysiology. Although numerous studies have aimed to elucidate the biological causes, 1 , 2 , 3 , 4 , 5 , 6 , 7 little evidence has been available so far. One reason is the ambiguous diagnostic boundaries between SCZ and other psychiatric disorders 8 given that current diagnostic systems have mainly been based on descriptive criteria proposed by psychopathology. Although such criteria, e.g., the DSM or ICD systems, provide a consensus to some extent, classical epidemiological studies have estimated the shared genetic liability among psychiatric disorders. 9 , 10 , 11 , 12
To support this, recent advances in genomic analysis have provided important insight that would help address whether a genetic cause is shared between SCZ and other psychiatric disorders or whether it is unique to each disorder, based on data from association results of whole‐genome single nucleotide polymorphisms (SNPs). Specifically, the cross‐disorder group of the Psychiatric Genomics Consortium (PGC) reported a significant genetic overlap among them based on genetic correlation analysis and polygenic risk score analysis. 13 , 14 , 15 Interestingly, the genetic component of SCZ is close to that of bipolar disorder, of which contribution is larger than that of the relationship between bipolar disorder and major depressive disorder. 6 , 8 , 14 , 15
To determine the pathophysiology of SCZ and promote genome drug discovery, as well as prioritize the possible causality, understanding the genetic correlation between psychiatric disorders and/or other nonpsychiatric traits (disorders and quantitative traits) is crucial. 16 Specifically, nonpsychiatric traits can help uncover relationships that have yet to be reported or have been overlooked. For instance, several studies have reported that the genetic component for lower body mass index (BMI) correlated with that for susceptibility to SCZ. 16 , 17 , 18 Moreover, genetic variants associated with high C‐reactive protein (CRP) level (an instrumental variable) as a modifiable exposure to SCZ showed significant causality through mendelian randomization (MR) analysis, which is usually applied with genetic correlation analysis, 19 , 20 , 21 although some studies did not show a causal relationship. 22 , 23
Here, we examined the genetic relationship between SCZ and other nonpsychiatric disorders based on linkage disequilibrium score regression (LDSC) analysis, which is suitable for examining disorders with polygenic architecture. 16 Furthermore, we included subsequent analysis for the causal relationship between significant instrumental variants in nonpsychiatric traits and SCZ based on MR analysis. 24 The current study highlights the analyses using data from multiple populations. Although previous studies have mainly analyzed data based on European (EUR) populations, multipopulation analysis can provide more robust results. Therefore, we used data based on East Asian (EAS) populations in addition to that of EUR populations and finally assessed LDSC results using meta‐analysis to enhance statistical power.
Methods
Genome‐wide association study results in EAS and EUR samples
For EAS samples (a total of 24,504 patients with SCZ and 42,770 controls), we used SCZ results based on the meta‐analysis from our previous study (Japanese SCZ: 1,726 patients with SCZ and 7,408 controls) 6 and the PGC Asia (PGC EAS SCZ: 22,778 patients with SCZ and 35,362 controls), 25 as well as nonpsychiatric data from the JENGER database (40 binary traits and 63 quantitative traits: Supplementary Table 1), which is provided by Biobank Japan (http://jenger.riken.jp/result) (minimum of 12,302 and maximum of 212,453 samples). The nonpsychiatric traits were common diseases (e.g., type 2 diabetes), quantitative data of laboratory examinations (e.g., blood sugar [BS]), and physical characteristics (e.g., BMI). It should be noted that there were no overlapping samples between GWASs for SCZ and nonpsychiatric traits.
To enhance the statistical power during the primary meta‐analysis of SCZ results for EAS, the “metafor” package in R (https://www.metafor-project.org/doku.php) was used. We applied results from the fixed‐effects model with P heterogeneity ≥0.05 and the random‐effects model P heterogeneity <0.05.
For EUR samples, we used PGC results for SCZ (33,640 SCZ and 43,456 controls) 26 and UK Biobank data (http://www.nealelab.is/uk-biobank: a total of 4,236 phenotypes are listed) for nonpsychiatric traits (minimum of 51,453 and maximum of 361,194 samples). Among these nonpsychiatric traits, we used overlapped ones with the phenotypes listed in JENGER (20 binary traits and 34 quantitative traits).
LDSC analysis
All genetic correlation analyses were conducted using LDSC 16 software (https://github.com/bulik/ldsc). This software estimates the genetic correlation between two phenotypes based on the summary statistics. The genetic correlations between SCZ and nonpsychiatric traits were calculated in each population (EAS and EUR). However, specifically for EUR data, we only assessed the genetic correlation in identical pairs analyzed from EAS results, thereby excluding traits listed only in the UK Biobank, mentioned above.
For quality control, we used SNPs: (1) involved in the 1000 Genomes Project and HapMap Projects, (2) with a minor allele frequency ≥ 1%, and (3) INFO (imputation quality measures) ≥0.9.
Subsequent meta‐analysis was conducted by merging the “overlapped” traits (20 binary traits and 34 quantitative trait loci [QTL] traits) between results from the EAS and EUR samples. The “metafor” package in R was used based on the fixed‐effects model, because we assumed little differences of the genetic correlation between populations. Multiple comparisons were corrected using the false discovery rate (Benjamini–Hochberg method) to calculate q‐values (q‐values <0.05 were set as the significant threshold) in each trait (binary and QTL traits in the meta‐analysis).
MR analysis
For MR analysis, we only targeted traits that showed significant correlation during LDSC analysis. These nonpsychiatric traits were used as exposures, whereas SCZ was used as the outcome. The “exposure” SNPs, which surpassed the P‐value threshold of P < 5 × 10−8 in the results of the nonpsychiatric traits, were clamped to remove SNPs in linkage disequilibrium (r2 < 0.001 within a 10‐Mb window) and selected as instrumental variables. Our primary MR analysis method was inverse variance weighted (IVW) regression, particularly when P‐values of the Egger intercept exceeded 0.05. We also conducted: (1) weighted median analysis, (2) MR‐Egger regression analysis, and (3) MR pleiotropy residual sum and outlier (MR‐PRESSO) analysis as sensitivity analyses. During MR‐PRESSO, we checked the horizontal pleiotropy using the robust method and performed the global test to examine for outlier removal.
The aforementioned MR analyses were conducted using the “TwoSample MR” 27 package in R (https://mrcieu.github.io/TwoSampleMR/), with the default setting. The multiple comparisons were again corrected by false discovery rate and the significance level was set at q‐values <0.05 in each population (EAS and EUR).
Ethical statement
For the SCZ GWAS in the Japanese population, 6 written informed consent was obtained from all patients following a thorough explanation of the study. The study was approved by the ethics committees of Fujita Health University and other participating universities, which conform to the provisions of the Declaration of Helsinki.
Results
Initially, LDSC analysis was conducted between SCZ and nonpsychiatric traits in EAS samples (Tables 1 and Fig. 1; the full results can be seen in Table S1 and Figs. S1 and S2). Ten traits showed marginal significant correlation (P < 0.05) in this population (Table S1). Next, we conducted LDSC analysis for EUR samples and detected 8 of 54 “overlapped” traits (20 binary traits and 34 QTL traits) with P < 0.05 (Table S1). The subsequent meta‐analysis for the 54 “overlapped” traits in each population revealed no significant genetic correlation between SCZ and binary traits (q‐value <0.05); however, we detected six QTL traits that showed significant genetic correlation with SCZ (Table 1 and Fig. 1): BMI (rg = −0.101, q‐value = 1.01 × 10−9), CRP (rg = −0.0756, q‐value = 7.80 × 10−5), high‐density‐lipoprotein cholesterol (HDL‐C: rg = 0.0722, q‐value = 2.90 × 10−3), BS (rg = −0.0679, q‐value = 1.44 × 10−2), triglycerides (TGs: rg = −0.0524, q‐value = 2.44 × 10−2), and systolic blood pressure (SBP: rg = −0.0543, q‐value = 3.45 × 10−2). The direction of the genetic correlation for the six mentioned traits was identical between EAS and EUR results, suggesting consistent evidence between populations.
Table 1.
Results of genetic correlation analysis with P < 0.1
| EAS | EUR | Meta‐analysis (EAS + EUR) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Trait | Type | rg | SE | P‐value | rg | SE | P‐value | rg | SE | P‐value | q‐value |
| Osteoporosis | Binary | 0.107 | 0.0784 | 0.173 | 0.0789 | 0.0439 | 0.0725 | 0.0856 | 0.0383 | 0.0254 | 0.377 |
| Cirrhosis | Binary | 0.121 | 0.121 | 0.320 | 0.254 | 0.141 | 0.0719 | 0.178 | 0.0737 | 0.0538 | 0.377 |
| Epilepsy | Binary | 0.140 | 0.126 | 0.268 | 0.141 | 0.0907 | 0.121 | 0.141 | 0.0920 | 0.0565 | 0.377 |
| Prostate cancer | Binary | 0.0703 | 0.0510 | 0.168 | 0.0594 | 0.0614 | 0.333 | 0.0659 | 0.0392 | 0.0933 | 0.467 |
| BMI | QTL | −0.0713 | 0.0294 | 0.0155 | −0.112 | 0.0178 | 2.80E−10 | −0.101 | 0.0152 | 2.97E−11 | 1.01E−09 |
| CRP | QTL | −0.0363 | 0.0936 | 0.698 | −0.0772 | 0.0186 | 3.18E−05 | −0.0756 | 0.0182 | 3.40E−05 | 7.80E−05 |
| HDL‐C | QTL | 0.122 | 0.0467 | 9.00E−03 | 0.0614 | 0.0218 | 4.90E−03 | 0.0722 | 0.0198 | 2.55E−04 | 2.90E−03 |
| BS | QTL | −0.103 | 0.0475 | 0.0304 | −0.0588 | 0.0243 | 0.0154 | −0.0679 | 0.0216 | 1.70E−03 | 0.0144 |
| TGs | QTL | −0.0510 | 0.0344 | 0.139 | −0.0529 | 0.0211 | 0.0121 | −0.0524 | 0.0180 | 3.60E−03 | 0.0244 |
| SBP | QTL | −0.0694 | 0.0415 | 0.0943 | −0.0498 | 0.0225 | 0.0267 | −0.0543 | 0.0198 | 6.10E−03 | 0.0345 |
| GGT | QTL | −9.10E−03 | 0.0343 | 0.791 | −0.0465 | 0.0203 | 0.0219 | −0.0368 | 0.0175 | 0.0352 | 0.171 |
| DBP | QTL | −0.0345 | 0.0428 | 0.420 | −0.0385 | 0.0222 | 0.0834 | −0.0377 | 0.0197 | 0.0561 | 0.221 |
| Phosphorus | QTL | 1.40E−03 | 0.0758 | 0.985 | 0.0460 | 0.0233 | 0.0483 | 0.0422 | 0.0223 | 0.0584 | 0.221 |
| Lymphocyte count | QTL | −0.0545 | 0.0494 | 0.270 | 0.0449 | 0.0193 | 0.0198 | 0.0317 | 0.0180 | 0.0775 | 0.264 |
| MCV | QTL | 0.0190 | 0.0343 | 0.580 | 0.0279 | 0.0174 | 0.109 | 0.0261 | 0.0155 | 0.0928 | 0.287 |
Bold numbers represent a significant correlation after applying false discovery rate correction.
BMI, body mass index; BS, blood sugar; CRP, C‐reactive protein; DBP, diastolic blood pressure; EAS, East Asian; EUR, European; GGT, γ‐glutamyl transferase; HDL‐C, high‐density lipoprotein cholesterol; MCV, mean corpuscular volume; QTL, quantitative trait loci; rg, genetic correlation; SBP, systolic blood pressure; SE, standard error; TGs, triglycerides; QTL, quantitative trait loci.
Fig. 1.
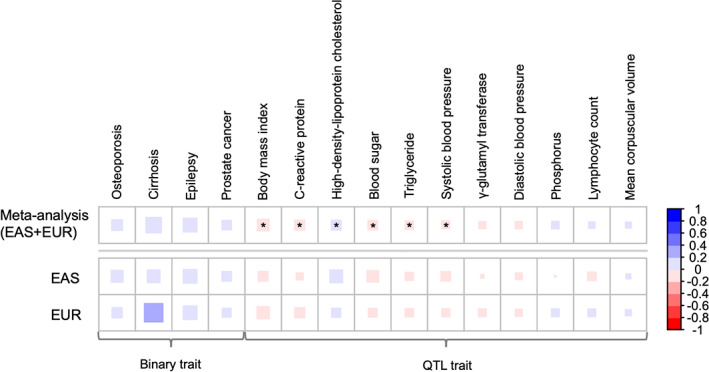
Heatmap of genetic correlation analysis with P‐values <0.1 (for overall samples). The colored box indicates the genetic correlation matrix (rg). Asterisk indicates q‐values <0.05. EAS, East Asian; EUR, European.
Table 2.
Results of mendelian randomization analysis
| EAS | EUR | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Trait | Method | No. of SNPs | beta | SE | P‐value | q‐value | No. of SNPs | beta | SE | P‐value | q‐value |
| BMI | Inverse variance weighted | 62 | −0.211 | 0.113 | 0.0617 | 0.434 | 298 | −0.165 | 0.0654 | 0.0116 | 0.0514 |
| Weighted median | 62 | −0.135 | 0.115 | 0.241 | 0.530 | 298 | −0.125 | 0.0727 | 0.0860 | 0.229 | |
| MR Egger | 62 | 0.178 | 0.364 | 0.625 | 0.772 | 298 | 0.280 | 0.191 | 0.142 | 0.341 | |
| MR‐PRESSO | 60 | −0.125 | 0.0949 | 0.192 | 0.530 | 278 | −0.140 | 0.0561 | 0.0134 | 0.0514 | |
| Egger intercept | 0.264 | 0.0135 | |||||||||
| CRP | Inverse variance weighted | 6 | −0.0530 | 0.141 | 0.706 | 0.795 | 183 | −0.0923 | 0.0378 | 0.0146 | 0.0514 |
| Weighted median | 6 | −0.0920 | 0.143 | 0.520 | 0.772 | 183 | −0.109 | 0.0422 | 0.0100 | 0.0514 | |
| MR Egger | 6 | 0.197 | 0.342 | 0.596 | 0.772 | 183 | −0.0602 | 0.0530 | 0.258 | 0.442 | |
| MR‐PRESSO | 6 | NA | NA | NA | 176 | −0.101 | 0.0332 | 2.79E−03 | 0.0514 | ||
| Egger intercept | 0.465 | 0.389 | |||||||||
| HDL‐C | Inverse variance weighted | 50 | 0.0756 | 0.0364 | 0.0379 | 0.431 | 247 | 0.0412 | 0.0387 | 0.287 | 0.459 |
| Weighted median | 50 | 0.0567 | 0.0543 | 0.296 | 0.530 | 247 | −0.0184 | 0.0439 | 0.674 | 0.887 | |
| MR Egger | 50 | −6.90E−03 | 0.0656 | 0.917 | 0.917 | 247 | −0.0786 | 0.0559 | 0.161 | 0.351 | |
| MR‐PRESSO | 50 | NA | NA | NA | 235 | 0.0406 | 0.0323 | 0.209 | 0.386 | ||
| Egger intercept | 0.139 | 2.00E−03 | 3.68E−03 | ||||||||
| BS | Inverse variance weighted | 15 | −0.117 | 0.105 | 0.265 | 0.530 | 93 | −0.135 | 0.0726 | 0.0629 | 0.189 |
| Weighted median | 15 | −0.145 | 0.141 | 0.303 | 0.530 | 93 | −0.0485 | 0.0694 | 0.485 | 0.728 | |
| MR Egger | 15 | −0.124 | 0.529 | 0.818 | 0.859 | 93 | −0.0421 | 0.118 | 0.723 | 0.887 | |
| MR‐PRESSO | 15 | NA | NA | NA | 86 | −0.126 | 0.0503 | 0.0144 | 0.0514 | ||
| Egger intercept | 0.989 | 0.321 | |||||||||
| TGs | Inverse variance weighted | 36 | −0.0469 | 0.0625 | 0.452 | 0.730 | 204 | −0.0143 | 0.0428 | 0.739 | 0.887 |
| Weighted median | 36 | −0.0960 | 0.0671 | 0.152 | 0.530 | 204 | −0.0208 | 0.0444 | 0.640 | 0.887 | |
| MR Egger | 36 | −0.207 | 0.0975 | 0.0409 | 0.431 | 204 | 0.0825 | 0.0616 | 0.182 | 0.364 | |
| MR‐PRESSO | 34 | −0.0626 | 0.0564 | 0.275 | 0.530 | 192 | 6.83E−03 | 0.0340 | 0.841 | 0.910 | |
| Egger intercept | 0.0452 | 0.0316 | |||||||||
| SBP | Inverse variance weighted | 22 | 0.207 | 0.164 | 0.206 | 0.530 | 158 | −0.0134 | 0.0831 | 0.872 | 0.910 |
| Weighted median | 22 | 0.0794 | 0.161 | 0.623 | 0.772 | 158 | 0.0212 | 0.0846 | 0.802 | 0.910 | |
| MR Egger | 22 | 0.987 | 0.574 | 0.101 | 0.530 | 158 | 0.702 | 0.268 | 9.56E−03 | 0.0514 | |
| MR‐PRESSO | 21 | 0.0416 | 0.114 | 0.719 | 0.795 | 153 | −5.01E−03 | 0.0687 | 0.942 | 0.942 | |
| Egger intercept | 0.172 | 5.65E−03 | |||||||||
Bold numbers represent results for prioritized analysis based on the significance of Egger intercept (if P > 0.05, we prioritized the “inverse variance–weighted” method, whereas if P < 0.05, we prioritized other methods).
Mendelian randomization (MR) pleiotropy residual sum and outlier (MR‐PRESSO) beta effects were calculated after removing the outliers, which were pleiotropic variants. If there was no outlier, we described “NA” in the column. The number of single nucleotide polymorphism (SNPs) in MR‐PRESSO was that after removing the outliers.
BMI, body mass index; BS, blood sugar; CRP, C‐reactive protein; EAS, East Asian; EUR, European; HDL‐C, high‐density lipoprotein cholesterol; SBP, systolic blood pressure; SE, standard error; TGs, triglycerides.
Further analysis on these “significant” traits was conducted through MR to examine causality. No significant causal relationship was obtained after correcting the multiple comparisons (Table 2). However, it is of note that we detected nonsignificant trend of the causal effects between CRP (beta = −0.0923, q‐value = 0.0514 in IVW, Egger intercept = 0.389) and SCZ, which is derived from EUR samples (Table 2). Whereas, the results with the significant P‐value of Egger intercept (P < 0.05, which means possible pleiotropy, thus prioritizing other “non‐IVW” methods) were not always consistent.
Discussion
In this genetic correlation analysis using multipopulation samples, we detected a significant correlation between several metabolic syndrome (MetS)–related traits/CRP and SCZ. Most of these have been previously reported mainly in EUR samples, 16 , 17 , 18 suggesting that our results replicated those presented in previous studies, albeit in a larger sample size with multipopulation samples. Nevertheless, we did not detect the significant causal relationship between these nonpsychiatric traits and SCZ after correcting the multiple comparisons.
MetS‐related traits and SCZ
The current study found a significant genetic correlation between MetS‐related traits, such as BMI, HDL‐C, BS, TGs, SBP, and SCZ. MetS is diagnosed based on various factors, namely large abdominal circumference, high TGs, low HDL‐C, high blood pressure, and high fasting blood glucose. 28 Most of our results implied shared genetic components between “non‐MetS”–related traits and SCZ; we found a negative rg between SCZ and BMI, TGs, SBP, and BS, and a positive rg between SCZ and HDL‐C, all of which showed the identical direction of the effect for EAS and EUR samples, suggesting robust results.
Nevertheless, in clinical practice, patients with SCZ generally demonstrate a higher prevalence of dyslipidemia, obesity, hypertension, and hyperglycemia than the general population and often have an increased risk of premature death caused by cardiovascular disease. 29 , 30 Some epidemiological studies targeting first episodes among drug‐naïve patients reported that such individuals had already exhibited high BMI or dyslipidemia. 31 , 32 , 33 This may imply that second‐generation antipsychotics, which is a major risk factor for obesity, are “independent” of MetS‐related traits in the early stages of SCZ.
On the contrary, longitudinal studies surveying BMI, one of the MetS‐related traits, during the adolescent period (e.g., aged 18 years) showed that patients with SCZ had a lower BMI than patients without SCZ. 34 , 35 This evidence is consistent with our current results and previous reports on genetic correlation, which supports the classical “somatotype” theory, suggesting that obesity in SCZ is an acquired/extrinsic effect from drug therapy (antipsychotics), lifestyle changes, poor diet, lack of exercise, and living conditions. 18
It is important to determine whether these MetS‐related traits in SCZ are derived from endogenous or extrinsic effects. 19 , 36 , 37 Our findings basically support the notion that extrinsic effects are the major cause for not only BMI but also other MetS‐related traits in SCZ. 28 In clinical settings, this speculation is encouraging for psychiatrists and patients because it implies that MetS‐related traits can be manageable. However, it is clear that MetS‐related traits in SCZ are caused by complex mechanisms and require further studies.
Causal effect of instrumental variables from nonpsychiatric traits on SCZ
Our MR analysis revealed no significant causal relationship between nonpsychiatric traits (MetS‐related traits and CRP) and SCZ after correcting the multiple comparisons (q‐value >0.05). This indicates that, at least, there is no “strong” causal effect based on these traits.
However, despite a nonsignificant trend, we stressed that one of the best P‐values (q‐value) was obtained in the instrumental variables from CRP of the EUR population (q‐valueIVW = 0.0514, Egger intercept = 0.389). This is not surprising because several studies have suggested that CRP is a potential biomarker for diagnosis, treatment response, and prevention of SCZ. 38 Specifically, previous clinical studies, including those with a cross‐sectional design, have shown that patients with SCZ had higher CRP values compared with the control group. 39 , 40 , 41 , 42 , 43 , 44 , 45 However, such a study design contains bias, which could be associated with the “status” of SCZ. 46
In contrast, recent evidence from MR analyses, which can theoretically reduce the bias caused by environmental factors, suggest that high CRP values had a protective effect against SCZ. 20 , 21 In the current study, a negative correlation was observed between CRP and SCZ in the LDSC analysis, while a negative beta was observed during MR analysis (exposure: CRP, outcome: SCZ [note: it was not “significant”]), although such observations were only observed in the EUR sample. Therefore, our results are at least in line with those presented in previous studies, which showed that higher CRP level had a protective effect against SCZ in EUR populations. 20 , 21 This is partially supported by evidence in EAS, which showed the same direction of causal effect, albeit not significantly. One possible reason for the nonsignificant trend of CRP on SCZ (in combined samples, EUR, and/or EAS samples) was that the sample size of EUR, as well as EAS, samples was not sufficient. We presume that the increased sample size may allow us to obtain “significant” causal effects; thus, further studies using multipopulation samples will be essential to obtain concrete results.
Limitations
There are several limitations to interpreting our current results. First, the sample size of GWASs, which were used in the LDSC and MR analyses, might not be sufficient for both the SCZ and nonpsychiatric traits. This means that insufficient statistical power overlooked the significant correlation and/or causal relationships. Specifically, MR analysis for EAS samples was typical because the numbers of instrumental variables were much lower than those for EUR samples. Therefore, further GWASs are required to deny the type II error. Second, in the LDSC and MR analyses for the EAS population, GWAS data for SCZ were mainly from Han Chinese and Japanese, whereas nonpsychiatric data were from Japanese. This might influence the results as a result of different ancestries (albeit not large compared with the difference between EAS and EUR). Therefore, we conducted further LDSC analyses to confirm whether our analysis in possible “stratified” EAS affects the result. We calculated the genetic correlation between nonpsychiatric traits (Biobank Japan: Japanese) and (1) SCZ in the Japanese population, 6 (2) SCZ in multiple EAS populations (PGC Asia 25 ), and (3) a “combined” sample of (1) and (2) (current results). However, based on these comparisons, we did not detect any clear difference in rg among these three subgroups (Table S2 and Figs. S3 and S4), specifically for the significant genetic correlation in six nonpsychiatric traits, except CRP (note: the genetic correlation of CRP was not significant in the EAS samples). This indicates that our comparison minimally affects the results.
Conclusion
Following LDSC analysis, the current study detected a correlation between MetS‐related traits and the inflammatory marker CRP and SCZ in multipopulation samples. Although most of the statistically significant data were derived from EUR samples, we believe that novel and/or replicable relationships will be discovered in EAS samples by increasing the sample size. Therefore, further GWAS with larger sample sizes from multiple populations should provide unexpected relationships between psychiatric disorders and other phenotypes, and hopefully clarify the pathophysiology of psychiatric disorders.
Disclosure statement
Dr N Iwata received research support or speakers' honoraria with Jansen, Takeda, Dainippon Sumitomo, Otsuka. The remaining authors have no conflicts of interest to declare.
Author contributions
R.A., T.S., M.I., and N.I. contributed to the conception and study design. R.A., M.I., T.S., K.N., A.S., T.A., K.I., and N.I. provide substantial contributions to the analysis and interpretation of clinical data. R.A., M.I., and T.S. wrote the first draft of the article. All authors have contributed to and approved the final version of the article.
Supporting information
Supplement Figure S1: Heat‐map of genetic correlation analysis of binary traits for overall samples
Supplement Figure S2: Heat‐map of genetic correlation analysis of quantitative trait loci (QTL) traits for overall samples
Supplement Figure S3: Heat‐map of genetic correlation analysis of binary traits within East Asian samples
Supplement Figure S4: Heat‐map of genetic correlation analysis of quantitative trait loci (QTL) traits within East Asian samples
Supplement Table S1: Results of genetic correlation analysis
Supplement Table S2: Results of genetic correlation analysis within East Asian samples
Acknowledgments
The authors thank all of the patients who participated in the present study. We also thank the members of the Strategic Research Program for Brain Sciences (SRPBS) team, staff at the Center for Research Promotion and Support in Fujita Health University for their assistance in sample collection, and MARUZEN‐YUSHODO Co., Ltd. (https://kw.maruzen.co.jp/kousei-honyaku/) for English‐language editing.
This study was supported by the SRPBS from the Japan Agency for Medical Research and Development (AMED) under grant numbers JP20dm0107097 (N.I., M.I., and T.S.), JP21wm0425008 (N.I. and T.S.), and JP21wm0525024 (M.I.); GRIFIN of P3GM from AMED under grant numbers JP20km0405201 (T.S. and N.I.), JP20km0405208 (M.I.), JP21tm0424220 (MI); JSPS Kakenhi grant numbers JP17H04251 (M.I.), JP21H02854 (M.I.), JP18K15497 (T.S.), and JP21K07490 (T.S.); and the Private University Research Branding Project from MEXT (N.I.). The funders played no role in the study design, data collection, and analysis; the decision to publish; or the preparation of the article.
References
- 1. Onitsuka T, Hirano Y, Nemoto K et al. Trends in big data analyses by multicenter collaborative translational research in psychiatry. Psychiatry Clin. Neurosci. 2022; 76: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Takahashi T, Suzuki M. Brain morphologic changes in early stages of psychosis: Implications for clinical application and early intervention. Psychiatry Clin. Neurosci. 2018; 72: 556–571. [DOI] [PubMed] [Google Scholar]
- 3. Yahata N, Kasai K, Kawato M. Computational neuroscience approach to biomarkers and treatments for mental disorders. Psychiatry Clin. Neurosci. 2017; 71: 215–237. [DOI] [PubMed] [Google Scholar]
- 4. Legge SE, Santoro ML, Periyasamy S, Okewole A, Arsalan A, Kowalec K. Genetic architecture of schizophrenia: A review of major advancements. Psychol. Med. 2021; 51: 2168–2177. [DOI] [PubMed] [Google Scholar]
- 5. Rees E, Kirov G. Copy number variation and neuropsychiatric illness. Curr. Opin. Genet. Dev. 2021; 68: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ikeda M, Takahashi A, Kamatani Y et al. Genome‐wide association study detected novel susceptibility genes for schizophrenia and shared trans‐populations/diseases genetic effect. Schizophr. Bull. 2019; 45: 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirano Y, Uhlhaas PJ. Current findings and perspectives on aberrant neural oscillations in schizophrenia. Psychiatry Clin. Neurosci. 2021; 75: 358–368. [DOI] [PubMed] [Google Scholar]
- 8. Brainstorm Consortium , Anttila V, Bulik‐Sullivan B et al. Analysis of shared heritability in common disorders of the brain. Science 2018; 360: eaap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jokiranta‐Olkoniemi E, Cheslack‐Postava K, Sucksdorff D et al. Risk of psychiatric and neurodevelopmental disorders among siblings of Probands with autism Spectrum disorders. JAMA Psychiat. 2016; 73: 622–629. [DOI] [PubMed] [Google Scholar]
- 10. Song J, Bergen SE, Kuja‐Halkola R, Larsson H, Landen M, Lichtenstein P. Bipolar disorder and its relation to major psychiatric disorders: A family‐based study in the Swedish population. Bipolar Disord. 2015; 17: 184–193. [DOI] [PubMed] [Google Scholar]
- 11. Van Snellenberg JX, de Candia T. Meta‐analytic evidence for familial coaggregation of schizophrenia and bipolar disorder. Arch. Gen. Psychiatry 2009; 66: 748–755. [DOI] [PubMed] [Google Scholar]
- 12. Mezey E, Kiss JZ. Coexpression of vasopressin and oxytocin in hypothalamic supraoptic neurons of lactating rats. Endocrinology 1991; 129: 1814–1820. [DOI] [PubMed] [Google Scholar]
- 13. Cross‐Disorder Group of the Psychiatric Genomics Consortium . Identification of risk loci with shared effects on five major psychiatric disorders: A genome‐wide analysis. Lancet 2013; 381: 1371–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cross‐Disorder Group of the Psychiatric Genomics Consortium , Lee SH, Ripke S et al. Genetic relationship between five psychiatric disorders estimated from genome‐wide SNPs. Nat. Genet. 2013; 45: 984–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cross‐Disorder Group of the Psychiatric Genomics Consortium . Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell 2019; 179: e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bulik‐Sullivan B, Finucane HK, Anttila V et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015; 47: 1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hubel C, Gaspar HA, Coleman JRI et al. Genetic correlations of psychiatric traits with body composition and glycemic traits are sex‐ and age‐dependent. Nat. Commun. 2019; 10: 5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ikeda M, Tanaka S, Saito T, Ozaki N, Kamatani Y, Iwata N. Re‐evaluating classical body type theories: Genetic correlation between psychiatric disorders and body mass index. Psychol. Med. 2018; 48: 1745–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ligthart S, Vaez A, Vosa U et al. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am. J. Hum. Genet. 2018; 103: 691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hartwig FP, Borges MC, Horta BL, Bowden J, Davey SG. Inflammatory biomarkers and risk of schizophrenia: A 2‐sample Mendelian randomization study. JAMA Psychiat. 2017; 74: 1226–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Prins BP, Abbasi A, Wong A et al. Investigating the causal relationship of C‐reactive protein with 32 complex somatic and psychiatric outcomes: A large‐scale cross‐consortium Mendelian randomization study. PLoS Med. 2016; 13: e1001976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perry BI, Upthegrove R, Kappelmann N, Jones PB, Burgess S, Khandaker GM. Associations of immunological proteins/traits with schizophrenia, major depression and bipolar disorder: A bi‐directional two‐sample mendelian randomization study. Brain Behav. Immun. 2021; 97: 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin BD, Alkema A, Peters T et al. Assessing causal links between metabolic traits, inflammation and schizophrenia: A univariable and multivariable, bidirectional Mendelian‐randomization study. Int. J. Epidemiol. 2019; 48: 1505–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ooi BNS, Loh H, Ho PJ et al. The genetic interplay between body mass index, breast size and breast cancer risk: A Mendelian randomization analysis. Int. J. Epidemiol. 2019; 48: 781–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lam M, Chen CY, Li Z et al. Comparative genetic architectures of schizophrenia in east Asian and European populations. Nat. Genet. 2019; 51: 1670–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schizophrenia Working Group of the Psychiatric Genomics Consortium . Biological insights from 108 schizophrenia‐associated genetic loci. Nature 2014; 511: 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hemani G, Zheng J, Elsworth B et al. The MR‐base platform supports systematic causal inference across the human phenome. Elife 2018; 7: e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mitchell AJ, Vancampfort D, Sweers K, van Winkel R, Yu W, De Hert M. Prevalence of metabolic syndrome and metabolic abnormalities in schizophrenia and related disorders‐‐a systematic review and meta‐analysis. Schizophr. Bull. 2013; 39: 306–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Manu P, Dima L, Shulman M, Vancampfort D, De Hert M, Correll CU. Weight gain and obesity in schizophrenia: Epidemiology, pathobiology, and management. Acta Psychiatr. Scand. 2015; 132: 97–108. [DOI] [PubMed] [Google Scholar]
- 30. Strassnig M, Kotov R, Cornaccio D, Fochtmann L, Harvey PD, Bromet EJ. Twenty‐year progression of body mass index in a county‐wide cohort of people with schizophrenia and bipolar disorder identified at their first episode of psychosis. Bipolar Disord. 2017; 19: 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lang X, Liu Q, Fang H et al. The prevalence and clinical correlates of metabolic syndrome and cardiometabolic alterations in 430 drug‐naive patients in their first episode of schizophrenia. Psychopharmacology (Berl) 2021; 238: 3643–3652. [DOI] [PubMed] [Google Scholar]
- 32. Zhang Q, He H, Bai X et al. Unveiling the metabolic profile of first‐episode drug‐naive schizophrenia patients: Baseline characteristics of a longitudinal study among Han Chinese. Front. Psych. 2021; 12: 702720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou Y, Song X, Guo Y, Lang X, Li Z, Zhang XY. Sex differences in metabolic disorder patterns of first‐episode drug‐naive patients with schizophrenia. Psychoneuroendocrinology 2021; 124: 105061. [DOI] [PubMed] [Google Scholar]
- 34. Sorensen HJ, Mortensen EL, Reinisch JM, Mednick SA. Height, weight and body mass index in early adulthood and risk of schizophrenia. Acta Psychiatr. Scand. 2006; 114: 49–54. [DOI] [PubMed] [Google Scholar]
- 35. Zammit S, Rasmussen F, Farahmand B et al. Height and body mass index in young adulthood and risk of schizophrenia: A longitudinal study of 1 347 520 Swedish men. Acta Psychiatr. Scand. 2007; 116: 378–385. [DOI] [PubMed] [Google Scholar]
- 36. Cao B, Jin M, Brietzke E et al. Serum metabolic profiling using small molecular water‐soluble metabolites in individuals with schizophrenia: A longitudinal study using a pre‐post‐treatment design. Psychiatry Clin. Neurosci. 2019; 73: 100–108. [DOI] [PubMed] [Google Scholar]
- 37. Annamalai A, Kosir U, Tek C. Prevalence of obesity and diabetes in patients with schizophrenia. World J. Diabetes 2017; 8: 390–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ullah I, Awan HA, Aamir A et al. Role and perspectives of inflammation and C‐reactive protein (CRP) in psychosis: An economic and widespread tool for assessing the disease. Int. J. Mol. Sci. 2021; 22: 13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brown AS, Sourander A, Hinkka‐Yli‐Salomaki S, McKeague IW, Sundvall J, Surcel HM. Elevated maternal C‐reactive protein and autism in a national birth cohort. Mol. Psychiatry 2014; 19: 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dickerson F, Stallings C, Origoni A et al. C‐reactive protein is elevated in schizophrenia. Schizophr. Res. 2013; 143: 198–202. [DOI] [PubMed] [Google Scholar]
- 41. Fernandes BS, Steiner J, Bernstein HG et al. C‐reactive protein is increased in schizophrenia but is not altered by antipsychotics: Meta‐analysis and implications. Mol. Psychiatry 2016; 21: 554–564. [DOI] [PubMed] [Google Scholar]
- 42. Metcalf SA, Jones PB, Nordstrom T et al. Serum C‐reactive protein in adolescence and risk of schizophrenia in adulthood: A prospective birth cohort study. Brain Behav. Immun. 2017; 59: 253–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wium‐Andersen MK, Orsted DD, Nordestgaard BG. Elevated C‐reactive protein associated with late‐ and very‐late‐onset schizophrenia in the general population: A prospective study. Schizophr. Bull. 2014; 40: 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhong X, Ao Q, Xing F. Serum levels of HCY, MIF, and hs‐CRP correlate with glycolipid metabolism in adults with never‐medicated first‐episode schizophrenia. Evid. Based Complement. Alternat. Med. 2021; 2021: 7394699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45. Li C, Shi Z, Ji J, Niu G, Liu Z. Associations of C‐reactive protein, free triiodothyronine, thyroid stimulating hormone and creatinine levels with agitation in patients with schizophrenia: A comparative cross‐sectional study. Neuropsychiatr. Dis. Treat. 2021; 17: 2575–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Orsolini L, Sarchione F, Vellante F et al. Protein‐C reactive as biomarker predictor of schizophrenia phases of illness? A systematic review. Curr. Neuropharmacol. 2018; 16: 583–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figure S1: Heat‐map of genetic correlation analysis of binary traits for overall samples
Supplement Figure S2: Heat‐map of genetic correlation analysis of quantitative trait loci (QTL) traits for overall samples
Supplement Figure S3: Heat‐map of genetic correlation analysis of binary traits within East Asian samples
Supplement Figure S4: Heat‐map of genetic correlation analysis of quantitative trait loci (QTL) traits within East Asian samples
Supplement Table S1: Results of genetic correlation analysis
Supplement Table S2: Results of genetic correlation analysis within East Asian samples