

Abstract
Background
Pathogenic variants in KITLG, a crucial protein involved in pigmentation and neural crest cell migration, cause non‐syndromic hearing loss, Waardenburg syndrome type 2, familial progressive hyperpigmentation and familial progressive hyper‐ and hypopigmentation, all of which are inherited in an autosomal dominant manner.
Objectives
To describe the genotypic and clinical spectrum of biallelic KITLG‐variants.
Methods
We used a genotype‐first approach through the GeneMatcher data sharing platform to collect individuals with biallelic KITLG variants and reviewed the literature for overlapping reports.
Results
We describe the first case series with biallelic KITLG variants; we expand the known hypomelanosis spectrum to include a ‘sock‐and‐glove‐like’, symmetric distribution, progressive repigmentation and generalized hypomelanosis. We speculate that KITLG biallelic loss‐of‐function variants cause generalized hypomelanosis, whilst variants with residual function lead to a variable auditory‐pigmentary disorder mostly reminiscent of Waardenburg syndrome type 2 or piebaldism.
Conclusions
We provide consolidating evidence that biallelic KITLG variants cause a distinct auditory‐pigmentary disorder. We evidence a significant clinical variability, similar to the one previously observed in KIT‐related piebaldism.
Introduction
KITLG/c‐Kit and Ras/MAPK pathways are crucial for controlling pigmentation. 1 KITLG variants are a rare cause of autosomal dominant (AD), sensorineural hearing loss (HL) and Waardenburg syndrome type 2 (WS2). 2 , 3 Gain‐of‐function variants in KITLG are also associated with familial progressive hyperpigmentation (FPH) 4 and hyper‐ and hypopigmentation (FPHH). 5 Variants in KIT [OMIM:*164920], the KITLG receptor, cause AD piebaldism characterized by symmetric, (mainly) limb hypomelanosis, occasional repigmentation and rare HL. 6 , 7
Auditory‐pigmentary disorders are attributed to pathogenic variants in essential genes for differentiation and migration of melanocytes (Table S1). 8 The most common syndrome, Waardenburg syndrome (WS), is a neurocristopathy caused by migration defects of neural crest cells. WS is genetically and clinically heterogeneous. Type 1 (WS1, PAX3 [OMIM:*606597]) is distinguished by type 2 (WS2, MITF [OMIM:*156845], SNAI2 [OMIM:*602150], SOX10 [OMIM:*602229], KITLG [OMIM:*184745]) by the occurrence of dystopia canthorum; type 3 (WS3, PAX3) usually includes dystopia canthorum and upper limb abnormalities; and type 4 (WS4, EDN3 [OMIM:*131242], EDNRB [OMIM:*131244] and SOX10), known as Waardenburg‐Shah syndrome, includes Hirschsprung disease. 9 An allelic disorder to WS2 with AD‐transmission, Tietz albinism‐deafness syndrome, is caused by variants in MITF ([OMIM:*156845]), whereby heterochromia or pigmented patches are absent. 10
Here, we describe a case series with biallelic KITLG variants causing a variable yet distinct spectrum of hypomelanosis and sensorineural hearing loss.
Materials and methods
We used a genotype‐first approach through the GeneMatcher data sharing platform and data mining of aggregated DNA sequences from patients with ultra‐rare disorders across multiple research and diagnostic laboratories worldwide to collect individuals with biallelic KITLG variants. 11 This study was performed according to the Declaration of Helsinki and approved by the Instutional Review Board of University College London. Written informed consent for the publication of photographs and medical information was obtained from parents/guardians. Individuals were clinically characterized by consultants in clinical genetics. The genomic DNA of the proband in each family had been analysed by research exome (Individuals 1, 12 4 13 and 6 14 ), clinical exome (Individual 2 15 ) or genome (Individuals 3 16 , 17 and 5 16 , 17 ) sequencing, as previously described. 12 , 13 , 14 , 15 , 16 , 17 A review of previously reported and published pathogenic/likely pathogenic KITLG variants used ClinVar, LOVD and the HGMD Professional database version 2021.3. Additionally, querying KITLG in PubMed‐yielded publications that were reviewed for the reporting of familial data.
Results
We identified six unrelated individuals (two male, four female) with biallelic KITLG variants (Figs 1a, 2a–p) and a median age of 5 years at last observation. Individuals 1–4 presented with Waardenburg syndrome type 2‐like and individuals 5–6 showed generalized hypomenanosis. Detailed clinical descriptions are presented in Appendix S1 (Clinical Case Reports).
Figure 1.
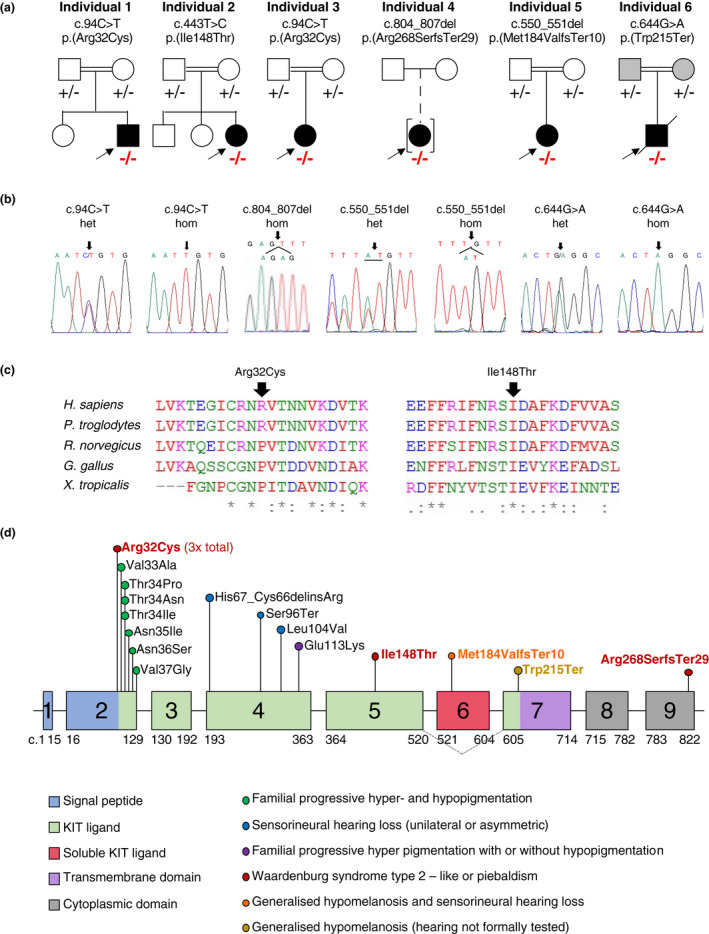
Molecular findings of individuals in our cohort. (a) Pedigree and segregation results (− represents the variant; homozygous variants are marked in red) for the six families with biallelic KITLG variants. (b) Available Sanger electropherograms showing heterozygous (het) or homozygous (hom) variants. (c) Interspecies alignment shows conservation of amino acids involved in non‐synonymous substitutions. (d) Schematic representation of the KITLG gene (NM_000889.4) with c. position, protein domains and features marked. An alternatively spliced isoform skips exon 6 and is represented with grey dotted lines (NM_003994.5). The phenotype of the KITLG variants is shown with coloured circles. The variants we describe are marked in red (WS), orange (generalized hypomelanosis and sensorineural hearing loss) or gold (generalized hypomelanosis with hearing not formally tested). [Colour figure can be viewed at wileyonlinelibrary.com]
Figure 2.
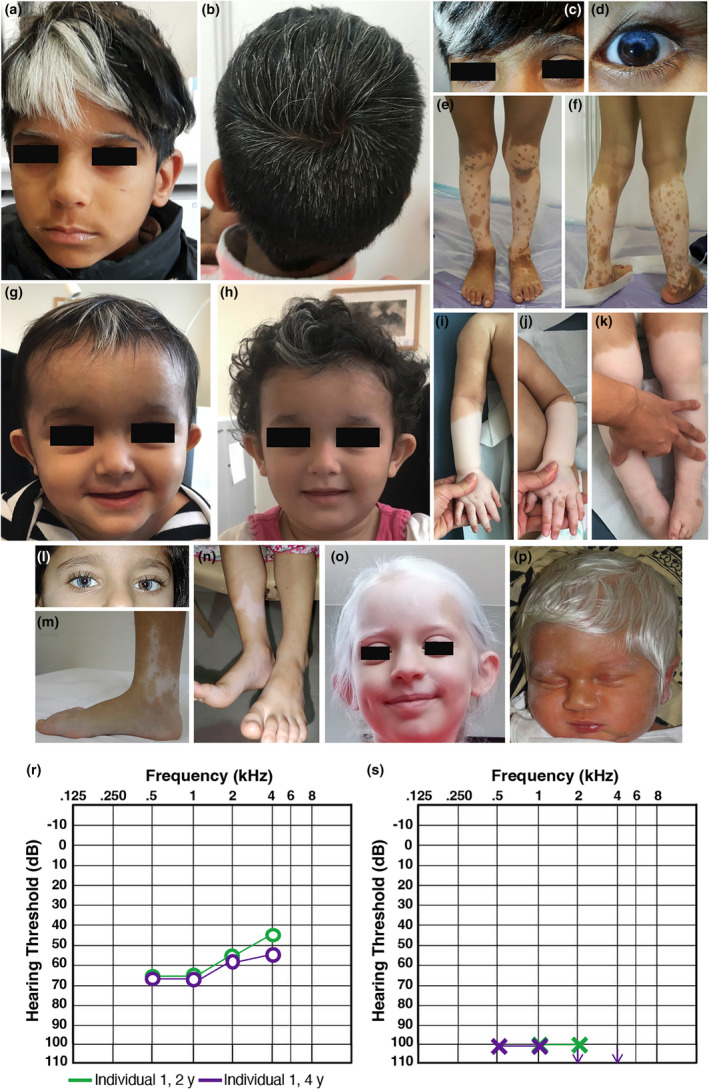
Clinical features of our cohort. (a–f) Individual 1 with partial heterochromia irides, hypomelanosis of hair including eyelashes and eyebrows and of the lower legs with multiple islands of pigmentation. (g–k) Individual 2 with blue irides, sparse white hair and hypopigmentation of the lower legs with a “sock‐and‐glove‐like” distribution and few islands of pigmentation on hands and feet. (l–m) Individual 3 with heterochromia irides, sparse white hair, and a hypopigmented area medially, on the right ankle with small islands of pigmentation. (n) Individual 4 with a hypopigmented area medially above the right ankle. (o) Individual 5 with generalized hypopigmentation of skin and hair. (p) Individual 6 with generalized hypopigmentation of skin and hair. (r, s) Auditory steady state responses from the right (r) and left (s) ears of Individual 1 at age 2 and 4 years of age showing asymmetric hearing loss. [Colour figure can be viewed at wileyonlinelibrary.com]
Hypomelanosis was present in all individuals and was either generalized (2/6) or affecting hair, irises and limbs (4/6). Islands of pigmentation within achromic areas on the limbs were present in 4/6 individuals and in two there was progressive repigmentation. Two individuals presented symmetric, lower limb hypomelanosis reminiscent of piebaldism, on one occasion with a ‘sock‐and‐glove‐like’ distribution. Heterochromia irises was present in 4/6 individuals. Hair hypomelanosis includes scattered leucotrichia including eyebrows and eyelashes (5/6), white forelock (3/6) and white‐silver hair reminiscent of albinism (2/6). Congenital/neonatal, sensorineural HL was identified in 5/5 individuals and appeared as asymmetric (2/5) (Figure 2r–s), bilateral, symmetric (2/5) or unilateral (1/5). Table S2 compares phenotypes in this case series with previously reported individuals with KITLG‐related WS2. Table S3 summarizes all individuals reporting KITLG variants, including those with FPHH and FPH.
The KITLG variants identified (GenBank accession: NM_000889.4) are summarized in Figure 1a and b. Table S4 shows evidence used to support variant interpretation. All variants are ultra rare or novel (Table S4).
Individuals 1 and 3 each presented with a homozygous c.94C>T, p.(Arg32Cys) variant that was previously reported in a 16‐year‐old Filipino WS2‐individual who had more extensive pigmentation anomalies compared to Individuals 1 and 3 (Figure 2a–f, l–m). 3 He had a reported, truncal, café‐au‐lait macule and a large well‐demarcated area of lower abdominal hyperpigmentation. Consistent with our individuals, he reported congenital, sensorineural HL and hypomelanotic areas with dappled pigmentation around the achromic patches. Melanotic skin was present on his back and buttocks, reportedly as post‐inflammatory hyperpigmentation due to psoriasis vulgaris. The authors noted that although the individual fulfilled criteria for WS, they could not exclude a diagnosis of FPHH with concomitant HL derived from a distinct genetic or non‐genetic origin. 3 These two individuals with identical, biallelic, pathogenic, variants provide strong evidence for c.94C>T, p.(Arg32Cys) as a causal allele for an auditory‐pigmentary disorder.
The homozygous likely pathogenic c.443T>C, p.(Ile148Thr) variant in individual 2 impacts a highly conserved amino acid (Figure 1c). The pattern of hypomelanosis affected hair (white forelock and scattered leucotrichia, eyebrows and eyelashes), eyes (blue irises) and limbs (symmetric, “sock‐and‐glove‐like” distribution). Within the depigmented areas on the limbs there were few pigmentation islands on fingers, toes and feet which developed progressively and with occasional symmetric distribution (lateral dorsal area of feet) (Figure 2g–k). Unilateral, sensorineural HL (left) was diagnosed at birth. The “sock‐and‐glove‐like” distribution of hypopigmentation is reminiscent of a peripheral demyelinating neuropathy but neurological examination was normal.
Individual 4 had a homozygous c.804_807delAGAG, p.(Arg268SerfsTer29) variant of uncertain significance that impacts the last six reference amino acids of the protein sequence. She has bilateral sensorineural HL and a hypomelanosis pattern that includes a fair skin complexion, patchy blue irises and two achromic patches over the lower third of the right shin and lateral aspect of right foot (Figure 2n). Due to the variant's C‐terminal location, we suspect an incomplete loss‐of‐function and preservation of protein expression.
Individual 5, with a homozygous c.550_551delAT, p.(Met184ValfsTer10) likely pathogenic variant, presented generalized hypomelanosis of the skin and hair (Figure 2o), as well as congenital, asymmetric, sensorineural HL. The frameshift variant introduces a premature stop codon in exon 6. However, alternative splicing of the shorter transcript variant “a”, that is also expressed in cochlear tissue, skips exon 6, leaving the shorter, primarily membrane‐bound transcript variant intact. 2
Individual 6, with a homozygous c.644G>A, p.(Trp215Ter) likely pathogenic variant, presented generalized hypomelanosis of skin, hair, and eyebrows but was lost to clinical follow‐up (Figure 2p). Hearing was not assessed before the individual's sudden death at 10 months, 22 days. He did not have heterochromia, amblyopia or nystagmus. His parents, who were confirmed carriers of the null variant, also exhibited lighter‐coloured skin than expected for their ethnic background.
In individuals with formal hearing assessments (5/5), the HL onset ranged from birth to neonatal period and the severity was moderate‐to‐profound. Laterality was variable: two individuals each had bilateral and asymmetric HL and one had unilateral HL, resembling the AD KITLG family with hereditary HL but no hypomelanosis. 2
Conclusions
We present four novel KITLG variants leading to a distinct spectrum of hypomelanosis and sensorineural hearing loss. Two out of four variants reported here (c.94C>T, p.(Arg32Cys) and c.443T>C, p.(Ile148Thr) map to the KIT ligand domain, where most FPHH and FPH variants also map (Figure 1d). We present the first variants in the following KITLG domains: the c.550_551del, p.(Met184ValfsTer10) frameshift variant affecting the soluble KIT ligand domain; the c.804_807del, p.(Arg268SerfsTer29) frameshift variant maps to the C‐terminal cytoplasmic domain, possibly contributing to a non‐generalized hypomelanosis; and the c.644G>A, p.(Trp215Ter) nonsense variant located at the beginning of the transmembrane domain in an individual with generalized hypomelanosis.
We expand the clinical spectrum caused by biallelic KITLG variants to include (a) “sock‐and‐glove‐like”, symmetric, hypomelanosis, (b) progressive re‐pigmentation and (c) generalized hypomelanosis. This significant clinical variability is similar to the one previously observed in individuals with piebaldism, in whom it correlates to the site and type of the KIT variant. 7 , 18 We hypothesize that (biallelic) loss‐of‐function variants cause generalized hypomelanosis while variants with residual protein function lead to a distinct spectrum of hypomelanosis and sensorineural hearing loss mostly reminiscent of WS2 or piebaldism. Functional studies are necessary to confirm this possible genotype–phenotype correlation.
Author contributions
RMaroofian was involved in conceptualization; BV, SD, DAS, AAR, MBT, PR, NB, RA, MD, RManju, DD, CJS, SN, EGK, HH, GB, MT, KMG and RMaroofian were involved in data curation; SD, DAS, AAR, SSL, PR, CJS and GB were involved in formal analysis; SSL and KMG were involved in funding acquisition; BV, SD, DAS, AAR, SSL, MBT, NB, RA, MD, CJS, SN, EGK, HH, GB, MT, KMG and RMaroofian were involved in investigation; BV, MT and RMaroofian were involved in project administration; MBT, NB, RA, MD, DD, SN, EGK, HH and MT provided key resources; SD and KMG were involved in supervision; SSL was involved in validation; BV, SD, SSL, MT and KMG were involved in visualization; BV, SD, MT, KMG and RMaroofian wrote the manuscript and prepared the original data; BV, SD, DAS, AAR, SSL, MBT, PR, NB, RA, MD, RManju, DD, CJS, SN, EGK, HH, GB, MT, KMG and RMaroofian wrote and edited the manuscript.
Supporting information
Appendix S1. Clinical Case Reports
Table S1. Differential diagnosis with known genetic syndromes causing hypomelanosis
Table S2. Prevalence of clinical features among Individuals with biallelic KITLG variants in the current case series and previously reported Individuals with KITLG‐related syndrome
Table S3. Summary of clinical features of patients with KITLG variants
Table S4. Evidence used to support variant interpretation
Acknowledgement
We are grateful to the participating families. Written informed consent for publication of photographs and medical information was obtained from parents/guardians.
Conflicts of Interest
None declared.
Funding source
The work was funded by a Bio‐CARe Woman Scientist Award, Department of Biotechnology, Ministry of Science and Technology, Government of India (grant no. BT/Bio‐CARe/07/ 9889/2013–14) (SSL and KMG) and by the John T. and Winifred M. Hayward Foundation (to MT). This work was supported by funding from the German Research Foundation through the Collaborative Research Center 889 (BV).
Data availability statement
Supporting data are available on request from the corresponding author. The genetic data are not publicly available due to privacy or ethical restrictions. Variants have been submitted to the Leiden Open Variation Database 3 (LOVD3) under variant accession IDs 0000386349–0000396354.
References
- 1. Grichnik JM, Burch JA, Burchette J, Shea CR. The SCF/KIT pathway plays a critical role in the control of normal human melanocyte homeostasis. J Invest Dermatol 1998; 111: 233–238. [DOI] [PubMed] [Google Scholar]
- 2. Zazo Seco C, Serrao de Castro L, van Nierop JW et al. Allelic mutations of KITLG, encoding KIT ligand, cause asymmetric and unilateral hearing loss and Waardenburg syndrome type 2. Am J Hum Genet 2015; 97: 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ogawa Y, Kono M, Akiyama M. Pigmented macules in Waardenburg syndrome type 2 due to KITLG mutation. Pigment Cell Melanoma Res 2017; 30: 501–504. [DOI] [PubMed] [Google Scholar]
- 4. Zhang C, Deng Y, Chen X et al. Linkage of a locus determining familial progressive hyperpigmentation (FPH) to chromosome 19p13.1‐pter in a Chinese family. Eur J Dermatol 2006; 16: 246–250. [PubMed] [Google Scholar]
- 5. Amyere M, Vogt T, Hoo J et al. KITLG mutations cause familial progressive hyper‐ and hypopigmentation. J Invest Dermatol 2011; 131: 1234–1239. [DOI] [PubMed] [Google Scholar]
- 6. Richards KA, Fukai K, Oiso N, Paller AS. A novel KIT mutation results in piebaldism with progressive depigmentation. J Am Acad Dermatol 2001; 44: 288–292. [DOI] [PubMed] [Google Scholar]
- 7. Hamadah I, Chisti M, Haider M et al. A novel KIT mutation in a family with expanded syndrome of piebaldism. JAAD Case Rep 2019; 5: 627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee TL, Lin PH, Chen PL, Hong JB, Wu CC. Hereditary hearing impairment with cutaneous abnormalities. Genes 2020; 12: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Read AP, Newton VE. Waardenburg syndrome. J Med Genet 1997; 34: 656–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet 2004; 38: 365–411. [DOI] [PubMed] [Google Scholar]
- 11. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015; 36: 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Makrythanasis P, Maroofian R, Stray‐Pedersen A et al. Biallelic variants in KIF14 cause intellectual disability with microcephaly. Eur J Hum Genet 2018; 26: 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kausthubham N, Shukla A, Gupta N et al. A data set of variants derived from 1455 clinical and research exomes is efficient in variant prioritization for early‐onset monogenic disorders in Indians. Hum Mutat 2021; 42: E15–E61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Girisha KM, von Elsner L, Neethukrishna K et al. The homozygous variant c.797G>a/p.(Cys266Tyr) in PISD is associated with a Spondyloepimetaphyseal dysplasia with large epiphyses and disturbed mitochondrial function. Hum Mutat 2019; 40: 299–309. [DOI] [PubMed] [Google Scholar]
- 15. Molina‐Ramírez LP, Kyle C, Ellingford JM et al. Personalised virtual gene panels reduce interpretation workload and maintain diagnostic rates of proband‐only clinical exome sequencing for rare disorders. J Med Genet 2022; 59: 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bademci G, Cengiz FB, Foster J et al. Variations in multiple syndromic deafness genes mimic non‐syndromic hearing loss. Sci Rep 2016; 6: 31622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Oiso N, Fukai K, Kawada A, Suzuki T. Piebaldism. J Dermatol 2013; 40: 330–335. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Clinical Case Reports
Table S1. Differential diagnosis with known genetic syndromes causing hypomelanosis
Table S2. Prevalence of clinical features among Individuals with biallelic KITLG variants in the current case series and previously reported Individuals with KITLG‐related syndrome
Table S3. Summary of clinical features of patients with KITLG variants
Table S4. Evidence used to support variant interpretation
Data Availability Statement
Supporting data are available on request from the corresponding author. The genetic data are not publicly available due to privacy or ethical restrictions. Variants have been submitted to the Leiden Open Variation Database 3 (LOVD3) under variant accession IDs 0000386349–0000396354.