

Abstract
Migraine is a highly prevalent neurovascular disorder afflicting more than 15% of the global population. Nearly three times more females are afflicted by migraine in the 18–50 years age group, compared to males. Migraine attacks are most often sporadic, but a subgroup of individuals experience a gradual increase in frequency over time; among these, up to 1%–2% of the global population develop chronic migraine. Although migraine symptoms have been known for centuries, the underlying mechanisms remain largely unknown. Two theories have dominated the current thinking—a neurovascular theory and a central neuronal theory with the origin of the attacks in the hypothalamus. During the last decades, the understanding of migraine has markedly advanced. This is supported by the early seminal demonstration of the trigeminovascular reflex 35 years ago and the insight that calcitonin gene‐related peptide (CGRP) is a key molecule released in acute migraine attacks. The more recent findings that gepants, small molecule CGRP receptor blockers, and monoclonal antibodies generated against CGRP, or its canonical receptor are useful for the treatment of migraine, are other important issues. CGRP has been established as a key molecule in the neurobiology of migraine. Moreover, monoclonal antibodies to CGRP or the CGRP receptor represent a breakthrough in the understanding of migraine pathophysiology and have emerged as an efficacious prophylactic treatment for patients with severe migraine with excellent tolerability. This review describes the progression of research to reach the clinical usefulness of a large group of molecules that have in common the interaction with CGRP mechanisms in the trigeminal system to alleviate the burden for individuals afflicted by migraine.
Keywords: CGRP, CGRP receptor, gepants, migraine, monoclonal antibodies

Introduction
Migraine is established as the most prevalent and disabling neurovascular brain disorder with severe socioeconomic impact, affecting females to a higher degree than males (Fig. 1). It currently ranks as the sixth most prevalent disorder worldwide. It is a major cause of disability, thus posing a heavy burden on individuals and society [1]. Diagnostically, it is characterized by moderate to severe headache attacks, often unilateral, and accompanied by nausea, vomiting, photophobia, and phonophobia [2]. In many cases, it is initially associated with an aura phenomenon, lasting for 20–60 min, and often of visual nature. Experimental and clinical translational research has provided key observations adding to the understanding of the underlying neurobiology and as a stimulus for the development of novel therapies. There is consensus on a genetic background of migraine clinically based on interviews with patients; however, genome‐wide association screening (GWAS) studies have failed to produce data to pinpoint one specific locus [3]. Thus, migraine is likely a polygenic phenotype as GWAS studies initially reported 38 independent loci associated with the risk of migraine. More recent work reported that 123 loci are associated with the risk of migraine with links to all chromosomes [4]. It is therefore improbable that a single gene can be responsible for the origin of common forms of migraine.
Fig. 1.
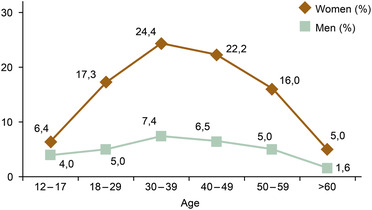
Global age‐standardized prevalence of migraine in males and females [1].
Studies of a rare condition, familial hemiplegic migraine, however, has been more successful in pointing towards a more specific single mechanism—increased sensitivity of central nervour system (CNS) glutaminergic signaling [5]. The debate is still ongoing regarding the importance of peripheral sites as the origin of migraine attacks, posing the question: does migraine start in the dura mater and/or in extracerebral arteries [6]? Do migraine attacks require a peripheral sensory input to be activated [7]? Recent imaging studies suggest that midbrain and brainstem structures are the drivers of migraine attacks [8]. Today, much evidence suggests that migraine attacks may start in the hypothalamus, sometimes already on the day before the headache (the prodromal phase), continue with the activation of the thalamus and the brainstem, and then the trigeminal system. The trigeminovascular system is likely necessary for the characteristic headache (the core of a migraine attack). Finally, after cessation of the headache phase there are imaging data showing that there are still alterations in the brain that correlate with CNS symptoms, such as tiredness (the postdrome) [9, 10]. Current debate favors the view that migraine is a CNS disorder, in which attacks starts in subcortical regions, exemplified by studies of premonitory symptoms [11] and supported by a series of elegant studies during continuous scanning of patients during the migraine cycle [12]. The imaging studies collectively point towards a hypothalamus–thalamus–brainstem pathway as a putative driver of the migraine biology [10]. Thus, the functional brain imaging studies have demonstrated brainstem areas to be specifically activated in migraine attacks, sometimes referred to as the “migraine generator” [13]. The link from the hypothalamus with the dorsal rostral pons, the spinal trigeminal nuclei, and sensory trigeminovascular system, are key parts in understanding the transmission of headache pain.
Historical background on the treatment of migraine
The American neurologist Harald Wolff [14] observed a widened temporal artery with pulsating quality in migraine patients that responded to administration of ergotamine with vessel constriction and reduced headache. Therefore, a vascular target seemed at this time a likely explanation of migraine and served as inspiration in the development of migraine therapies related to the role of 5‐hydroxytryptamine (5‐HT) and vasoconstriction [15]. The work ultimately resulted in the successful development of the triptan class of drugs, 5‐HT1B/1D receptor agonists for acute treatment of migraine attacks introduced during the early 1980s [16]. The role of intracranial and extracranial vasculature was still in focus and postulated to be a key part of the migraine attack [17]. Studies of perivascular innervation of the intracranial circulation, first with a focus on the autonomic nerves and later with the immunohistochemistry of neuropeptides, revealed numerous colocalizations [18]. The first sensory neuropeptide in the intracranial arteries is substance P (SP), originating in the trigeminal ganglion [19, 20, 21]. It was soon proposed to be a key messenger molecule responsible for neurogenic inflammation and sensitization of nociceptors located in, for example, the dura mater [22]. This was soon followed by other neurokinins and calcitonin gene‐related peptide (CGRP) that also originated in the trigeminal ganglion and colocalized with SP [23]. However, CGRP was, for nearly two decades, deemed not interesting, because it did not induce neurogenic inflammation [24] or did not activate the trigeminal neurons and only resulted in vasodilatation of meningeal arteries [24]. Thus, much effort focused on SP, and specific neurokinin blockers were developed that were effective in the inhibition of neurogenic inflammation, the proposed decisive model [25]. However, when tested in randomized clinical trials they were all without effect on migraine [26, 27, 28], and hence abandoned as antimigraine treatment candidates. Recent detailed work has shed new light on SP and the related neurokinins, as well as on their receptors in different parts of the trigeminovascular system [29].
What inspired the continued work on CGRP mechanisms in migraine?
Soon after the discovery of CGRP [30], work was initiated to develop the immunohistochemistry and specific radioimmunoassay (for quantification) for the study of neuropeptides associated with the intracranial vasculature [18]. CGRP is a 37‐amino acid peptide produced by neurons both in the CNS and in the peripheral nervous system [31]. The peptide exists in two forms, αCGRP and βCGRP, that differ by three amino acids in humans and are encoded by two different genes, CALCA and CALCB. CGRP was earlier shown to be a potent vasodilator in various parts of the vascular system [32, 33, 34]; it relaxed the cerebral arteries in association with increased levels of cyclic adenosine 3′5′‐monophosphate (cAMP), and the response was unrelated to the patency of the endothelium; later, CGRP was found to have effects throughout the body and in numerous organs [35]. The demonstration of CGRP in the trigeminal system verified its colocalization with SP, and surgical denervation removed these peptides from the intracranial perivascular innervation [23], while retrograde tracing verified its origin in neurons of the trigeminal ganglion [36].
In addition, CGRP was earlier found in the CNS; however, recent in‐depth mapping revealed an astonishing abundance of CGRP and its canonical receptor in the brain and in particular in migraine‐related regions and the trigeminovascular system [37, 38]. The CGRP expressing regions are linked with many cerebral systems, some of which are related to those involved in migraine attacks.
From basic research to clinical proof
Initial reports describing the presence of CGRP in trigeminal neurons and its potent vasodilator effects mediated via adenylyl cyclase resulted in the demonstration of the trigeminovascular reflex (cerebral artery constriction activates the sensory perivascular nerve fibers to release CGRP to maintain the resting artery diameter and blood flow) [39]. This inspired me to suggest that CGRP is involved in the migraine pathophysiology [23, 32, 33, 39]. The first clinical observations by Goadsby and Edvinsson linked the release of CGRP to primary headaches. The initial study in patients with severe trigeminal neuralgia showed elevated levels of CGRP in the jugular vein (but CGRP elevation was not seen in the peripheral cubital fossa vein) and this was associated with facial flushing symptoms [40]. Subsequently, two studies were performed on migraine patients with acute severe attacks showing up in the emergency room because of the severity of the migraine pain. These studies revealed that only CGRP and not SP, neuropeptide Y, or vasoactive intestinal peptide were released during these acute attacks. Subcutaneous sumatriptan aborted the headache and normalized the CGRP levels [41, 42]. Subsequent mechanistic and immunohistochemical studies on patients and human tissues firmly established the importance of CGRP in migraine pathophysiology [31, 43]. Once the peptide has been released from the sensory nerve, it is only slowly removed from the extracellular space due to the lack of specific reuptake machinery. Another interesting aspect of neuropeptides in general is their volume transmission, diffusion‐driven distribution into extracellular fluid over a relatively large distance [44]. Another feature of neuropeptides is the fact that they can be released at both synapses and nonsynaptic sites such as neurons and axons [45].
It was reported that the CGRP receptor is a G protein–coupled receptor that consists of a seven‐transmembrane (7‐TM) part, the calcitonin receptor‐like receptor (CLR), and a 1‐TM part, the receptor amplifying membrane protein 1 (RAMP1) (Fig. 2). When activated, it couples with a receptor component protein (RCP) and adenylyl cyclase [46, 47]. RCP has been suggested to be essential for CLR signaling [48]. Another important aspect of CGRP receptor signaling and regulation is the removal from the cell surface by internalization. Studies have examined this process, and evidence has revealed that tagged CLR/RAMP1 (the canonical CGRP receptor) is internalized into endosomes in response to CGRP [49].
Fig. 2.
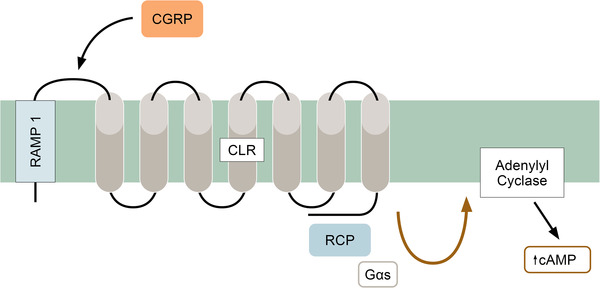
The components of the calcitonin gene‐related peptide (CGRP) receptor (CLR and RAMP1) and the signal pathway from RCP (receptor component protein) to the adenylyl cyclase system. Upon release of CGRP, it docks into the space between CLR and RAMP1, causing its activation [31]. This complex is then internalized into the cell.
In parallel, the industry worked to develop small CGRP blocking drugs—the first of these was olcegepant, which soon paved the way for several others in the new drug class “gepants” (Fig. 3). The developed small molecule receptor antagonists were shown to be competitive antagonists at the human CLR/RAMP1 receptor [50, 51]. After additional basic studies [51, 52], olcegepant was given intravenously to patients with acute migraine attacks [53]. Olcegepant not only aborted the attack rapidly, but pain freedom persisted in some patients for up to 24 h. Further proof of the importance of CGRP came, when intravenous (iv) CGRP resulted in migraine‐like attacks more frequently in migraine patients than in healthy subjects [54].
Fig. 3.
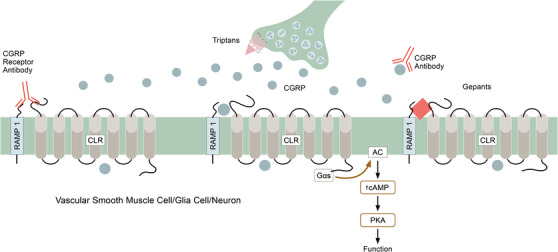
Released calcitonin gene‐related peptide (CGRP) is prevented from activating the CGRP receptor in several ways. (i) Presynaptic triptans may reduce the release of CGRP. (ii) Small molecule gepants may compete with CGRP at the receptor site. (iii). Monoclonal antibodies may adhere to CGRP, forming a large complex that does not fit at the receptor site. (iv) Specific monoclonal antibodies directed towards the N‐terminals of CLR and of RAMP1, which effectively blocks CGRP from reaching the active receptor site [31].
The recognition of the important role of CGRP in migraine triggered further efforts to target CGRP and its receptor as a therapeutic approach for migraine treatment. Since the dipeptide olcegepant was not suited for oral use, other pharmaceutical companies explored this target in more detail and developed their own gepants. The second gepant to appear clinically was telcagepant. The subsequent trials met the primary as well as the secondary endpoints and were well tolerated [55, 56]. However, in prophylaxis studies, reversible liver toxicity was observed, resulting in halting of its further development. The liver issue has now been solved with a new group of gepants. Their chemical structures and the developmental process of novel gepants have now been revealed in some detail [57]. At present, we are seeing the appearance of a further series of gepants for both acute and prophylactic use (ubrogepant, aterogepant, and rimegepant). These gepants have successfully passed the required randomized clinical trials for the treatment of migraine and are available in the United States [58, 59].
After 2 decades of basic research on the CGRP family of peptides, the focus now aimed at finding other means or molecules that can be used to block the CGRP responses [31]. A different approach was to examine whether specific antibodies could be designed toward CGRP. Two lines of research appeared; one was the further development of monoclonal antibodies (mAbs) binding CGRP, where we now have three different molecules on the market (eptinezumab, fremanezumab, and galcanezumab). The understanding of the uniqueness of the CGRP receptor led AMGEN to construct an mAb recognizing the N‐terminal of the two components of the canonical CGRP receptor: CLR and RAMP1. Erenumab is a highly specific antagonist against the CGRP receptor. The research behind it provided tools that could assist in further understanding of migraine pathophysiology. CGRP‐related therapies offer considerable improvements over existing drugs; as they are designed selectively to act on the trigeminal pain system, they are more specific and have few or mild adverse effects [60]. The development of the small molecules, gepants, resurfaced with CGRP receptor antagonists such as atogepant, rimegepant, and ubrogepant; they are effective for acute relief from migraine headache attacks and can also be useful as prophylactics.
Monoclonal antibodies
Basic research in the field of neuropeptides relied on methods to produce reagents for immunohistochemistry and radioimmunoassay for their quantification of the production of specific and selective antibodies directed towards various parts of the molecule under study [61]. Usually, these antibodies were polyclonal and therefore not suited for therapy. By using humanized or human antibodies, side effects were reduced and target specificity improved. Thus, the mAbs have high specificity towards the target and long half‐lives; they are metabolized by the reticuloendothelial system and not by the liver enzymes and have, therefore, a low potential for liver or renal toxicity. Due to their large molecular size, the mAbs cross the blood–brain barrier (BBB) only in very low concentrations [62], which limits possible CNS side effects that would be expected due to the rich expression of CGRP and CGRP receptors in numerous regions within the brain [38].
Patients with multiple attacks per month, as commonly seen in frequent episodic migraine (EM) or chronic migraine (CM), need prophylactic drugs. Those that are available were found by serendipity and include β‐adrenoceptor blockers, antidepressants, antiepileptics, and botulinum toxin [63]. The first three of these drugs have widespread effects in the CNS, are not specific for the migraine targets, and are often accompanied by side effects that limit their usage [63]. More than 90% of patients treated with these drugs have, after 1 year, stopped taking them due to either low efficacy or unwanted side effects.
The first published results with mAbs aimed at the trigeminovascular system showed a somewhat different pharmacological profile compared to the gepants [64, 65]. Although the CGRP‐directed antibody reduced the vasodilatory effect of CGRP, the inhibition was not competitive. Today, four mAbs are available: erenumab is a human mAb designed to bind the N‐terminals of CLR and RAMP1 of the CGRP receptor, thereby stopping CGRP from activating the receptor [66]. The other three mAbs (fremanezumab, galcanezumab, and eptinezumab) are directed towards the 37‐amino acid molecule, acting equally well at α‐ and β‐CGRP [67]. Collectively, these molecules have all emerged as effective and well tolerated for the preventive treatment of migraine. They are much larger than other preventive medications, which, as said, limits their ability to pass the BBB [68, 69]. The tolerability profile is excellent and does not differ to any major extent from their control groups.
Because CGRP is widely expressed in the body, discussion is ongoing regarding possible cardiovascular or intestinal side effects [70]. A common question is why are there so few peripheral effects, and would there not be interactions with pain systems generally [71]? The mAbs have now been approved by regulatory authorities FDA and EMA, and greater than 500,000 patients are on these treatments. Numerous patients have reported that “the CGRP directed medications have transformed their lives” [60]. Amazingly, the mAbs have effects both in EM and CM patients who have not responded well to other available preventive treatments [72, 73]. The American Headache Society has provided a statement paper for which patients should have the mAbs for migraine prophylaxis [74].
A site of antimigraine effect
The debate regarding the site of action of gepants and mAbs is ongoing. Currently, there is consensus that the mAbs cannot pass the BBB, hence the place for relieving the migraine pain is peripheral [62, 69, 75], suggesting action on the trigeminovascular system [31]. Interestingly, studies of the trigeminal system using quantitative and semiquantitative methods have revealed that the trigeminal ganglion (TG) is freely accessed by molecules in the blood, molecules that can have effects on its cellular structures [75, 76]. There is no alteration in the integrity of the BBB during migraine attacks [77]. Thus, reportedly, there was no alteration in the BBB (interictal and ictal) in acute glyceryl trinitrate–induced migraine attacks [78]. Furthermore, magnetic resonance imaging performed during the aura phase reported no evidence of increased BBB permeability [79]. In addition, the BBB was intact in spontaneous attacks of migraine without aura [80]. Furthermore, induced dural inflammation in a preclinical model did not show effects on BBB integrity calculated as the permeability surface area [75].
Where could the CGRP directed medications act to produce their effects? The studies have shown that the brain is protected from passage of the gepants and triptans (less than 2–3% passes the BBB), and for the mAbs less than 0,01% passes the BBB. Then the trigeminal system offers the most logical site of action of these acute and prophylacitc drugs [68, 75]. From available data, it is obvious that mAbs act on sites outside the BBB, putatively within the trigeminovascular system, the central or peripheral aspects of the sensory C‐ and Aδ‐fibers [81, 82]. Given this suggestion, there are at least four tentative places for the action of the anti‐CGRP drugs: (i) the most peripheral ends of the C‐ and Aδ‐fibers, in part located in the adventitia of intracranial vasculatures and on the dura mater with mast cells [83], (ii) the trigeminal ganglion with neurons and satellite glial cells [84, 85], (iii) the trigeminal nucleus caudalis, though it is limited by the BBB [11, 86], and recently (iv) the nodes of Ranvier, which offers a novel target site [87]. The debate is ongoing and possibly several of these sites may be involved.
The details of the molecular interaction of mAbs have been studied. Manoukian et al. showed in a cell model that CGRP induces a concentration‐dependent increase in cAMP and CGRP receptor internalization at different concentrations (EC50 8.4 pM vs. 7.9 nM, respectively). The lack of effect of the antibodies and gepants at the resting stage agrees with human data, where the small molecule CGRP antagonist telcagepant [88, 89] and the CGRP antibody fremanezumab or the CGRP receptor antibody erenumab did not have vasomotor effects by themselves [90, 91]. An experimental anti‐CGRP molecule (8E11) and an anti‐CGRP receptor antibody (AA58) blocked both effects, but the cAMP effect occurred at lower concentrations [92]. Using a combination of flow cytometry and confocal microscopy, the study showed that CGRP and the CGRP receptor were internalized and localized to the endosomes. It has been suggested that the endosomes and not the plasma membrane is the site of pain transmission [93]. Hypothetically, the increase in CGRP may contribute to migraine pain via CGRP receptor internalization and endosomal signaling [92].
Clinical effects of the CGRP antibodies
The mAbs either bind to the CGRP receptor, or the α and β isoforms of CGRP. They are not broken down by liver enzymes, which adds to their long half‐life in plasma, varying between 3 and 5 weeks. The mAbs are effective in EM and CM and have few side effects. The mAbs have, during the last few years, passed phase III, some even phase IV, and are now on the market in numerous countries [60, 94]. Since there are no direct comparative trials, we have to rely on meta‐analysis papers; the overall conclusion is that their efficacy is good with few and mild side effects [95, 96]. The mAbs have been proposed as disease‐modifying drugs since they can help to slow down the natural progression of migraine [97]. All mAbs studies showed a significant reduction in their primary endpoints, either meaning change from baseline in monthly migraine days (MMD) or a change in headache hours from baseline [98].
Erenumab is a humanized IgG2A mAb that targets the CGRP receptor [66]. Early onset of its efficacy has been documented in many trials. In the first week, 43% of EM and 26% of CM patients receiving erenumab 140 mg had a ≥50% decrease in weekly migraine days [99]. Furthermore, CM patients treated with erenumab for a month disclosed a reduction by 12.2 MMD, along with a reduction in the use of medication, the intensity of pain, and disability. Subcutaneous erenumab, 70 mg and 140 mg monthly, has been evaluated in phase III randomized controlled trials (RCTs) (STRIVE and ARISE) for the prevention of EM [99, 100]. Besides significant reduction in the use of antimigraine drugs, erenumab led to substantial improvements. The most common side effects noted were upper respiratory tract infection and pain at the injection site. In the STRIVE trial, besides a drop in the number of MMD, statistically significant reductions in the number of days requiring the use of antimigraine drugs and improvement in physical functioning scores and daily activities were observed [100]. Phase IIIb LIBERTY trial examining EM patients with a history of 2–4 preventive drug failures verified the supremacy of erenumab 140 mg and established that erenumab worked well for patients with refractory migraine [73]. Recently, a 5‐year open‐label study validated that erenumab is a safe drug with effects that remain over this period [101, 102]. Lipton et al. reported that CM can reverse to EM within a year of erenumab treatment [103].
Eptinezumab is the only CGRP mAb designed for iv use quarterly. It is a fully humanized IgG1λ mAb targeting both α and β isoforms of CGRP [104]. Phase III RCTs have assessed eptinezumab for prophylaxis of EM (PROMISE‐1) and CM (PROMISE‐2) [105]. There was ≥75% reduction in MMD observed in 24.7%, 22.2%, and 29.7% of patients medicated with eptinezumab 30, 100, and 300 mg, respectively, over 12 weeks. Interestingly, the occurrence of migraine on the first day after the infusion was significantly reduced by approximately half. In the PROMISE‐2 trial, the doses of 100 and 300 mg showed significant reductions in MMDs over the 6‐month trial period. The ≥75% migraine responder rates (RRs) were up to 38.5% (100 mg) and 42.3% (300 mg dose) and 22.7% (placebo) (months 4–6). The ≥50% migraine RRs were 60.7% (100 mg), 63.4% (300 mg dose), and 44.5% (placebo) (months 4–6). A 1‐year open‐label safety study of 300 mg eptinezumab in CM (PREVAIL) reported a reduction in migraine‐associated disability and improvement in patient functioning [106]. In addition, besides effective prevention, eptinezumab was found to achieve headache pain freedom after 4 h and absence of most bothersome symptoms after 2 h as compared to placebo [107].
Fremanezumab binds equally well with α and β isoforms of CGRP. It is a humanized IgG2κ mAb evaluated at doses of 225 mg administered monthly and 675 mg quarterly for the prevention of EM (HALO‐EM) [108]. The phase III study, HALO‐CM trial assessed the doses of 675 mg quarterly and 225 mg monthly for the prevention of CM [109]. Patients treated with fremanezumab with concurrent medication overuse headache (overuse of antimigraine drugs) had reported a statistically significant reduction of monthly medication use days compared to placebo [110]. A long‐term study (52 weeks) from the HALO trials showed that fremanezumab reduced MMD in patients with EM and CM by −5.1 and −8.0 days, respectively [111]. Side effects observed were redness at the site of injection, induration at the site of injection, diarrhea, anxiety, and depression [108]. The FOCUS trial was a phase III trial in EM and CM patients who had failed 2–4 preventive medications [72]. Fremanezumab was effective with MMD reduction in both patient populations. Interestingly, a post hoc analysis stratified the results by age and sex and reported that fremanezumab was effective in all age groups and equally between men and women [112].
Galcanezumab is a humanized IgG4 mAb acting well at both α and β forms of CGRP. Subcutaneous monthly doses of 120 mg or 240 mg have been investigated for the prophylaxis of EM (EVOLVE‐1 and EVOLVE‐2) and CM (REGAIN) [113, 114, 115, 116]. Analysis of the three trials revealed a greater number of EM or CM patients treated with galcanezumab achieved ≥50% reduction in MMD compared to placebo, establishing the efficacy of this antibody [114]. The EVOLVE‐1 trial on the prevention of EM compared galcanezumab (120 mg and 240 mg) with a placebo. Galcanezumab displayed fast onset starting at month 1 that lasted through month 6. Patients had lesser MMDs needing acute treatment. Galcanezumab improved both Migraine Disability Assessment (MIDAS) and daily functioning scores compared to placebo [116]. In the EVOLVE‐2 trial, the side effects seen were pain at the local injection site, local reactions, and itching [115]. REGAIN (evaluation of galcanezumab in the prevention of CM) comprised a 3‐month double‐blind, placebo‐controlled treatment phase and a 9‐month open‐label extension phase [113]. Common side effects of galcanezumab were pain at the site of injection, upper respiratory tract infection, reactions at the local site of administration, backache, and sinusitis [113]. In a 1‐year open‐label study of self‐administered subcutaneous monthly injections as prophylactic therapy, both 120 mg and 240 mg doses were found to be safe and associated with a reduction in MMD [117]. Long‐term galcanezumab lead to improvement in functional impairment and disability [118].
The overall results from the clinical trials of mAbs against CGRP and the CGRP receptor have collectively demonstrated stable effects in EM and CM. The response remained for more than 5 years of therapy, with remaining efficacy and with few side effects. These novel treatments designed to target the specific pathophysiology of migraine, where CGRP plays a key role, already have an important place in the therapy of severe migraine. Long‐term risks, especially in comorbid conditions, have so far not disclosed severe side effects. The role and interaction of these mAbs are now monitored in special subsets of the population, such as pregnant and lactating women and in children, and in other conditions where the CGRP family of peptides may have a role.
Conclusion
The current view suggests that migraine has an elusive genetic background. Various triggers may elicit attacks, whether due to stress or fluctuations in hormones, which trigger cells in the hypothalamus to initiate the attack. The connectivity in the brain implies the involvement of the thalamus and brainstem regions, ultimately resulting in enhanced activity in the trigeminal system and sensitization at both central and peripheral sites. The new specific CGRP‐directed medications aim to play down this activity.
CGRP was earlier proposed to be involved in migraine pathophysiology [32] and later basic and clinical research formed the foundation for a new group of specific remedies, mAbs directed towards CGRP or the CGRP receptor, and small molecule drugs (gepants) acting on the CGRP receptor [31]. The mAbs are now approved for prophylaxis of migraine, and the first of the gepants are in clinical use. The experience has been reviewed by several groups and is very encouraging [60]. It took basic research more than 3 decades to be translated to clinical practice, but the result for the patients has been extraordinary [31]. Ongoing work aims to unravel the biology of CGRP signaling, expand the clinical evidence for the role of CGRP in migraine headache, and potentially find other ways to treat the different patient groups of primary headache disorders, like cluster headache, inter alia. All in all, the latest findings have provided new insights into the central role of the trigeminal system in the pathophysiology of migraine pain.
Conflict of interest
The authors declare no conflict of interest in this work.
Acknowledgments
The work reviewed has been partially supported by grants from the Lundbeck Foundation, Denmark, the Swedish Research Council (grant 5958), and the Heart–Lung Foundation, Sweden.
Edvinsson L. Calcitonin gene‐related peptide (CGRP) is a key molecule released in acute migraine attacks—Successful translation of basic science to clinical practice. J Intern Med. 2022;292:575–586.
From the symposium Neuropeptides: The diverse dialects of the nervous system.
References
- 1. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1211–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 2018;38:1–211. [DOI] [PubMed] [Google Scholar]
- 3. Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH, et al. Meta‐analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet. 2016;48:856–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hautakangas H, Winsvold BS, Ruotsalainen SE, Bjornsdottir G, Harder AVE, Kogelman LJA, et al. Genome‐wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype‐specific risk alleles. Nat Genet. 2022;54:152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pelzer N, Louter MA, van Zwet EW, Nyholt DR, Ferrari MD, Van den Maagdenberg AM, et al. Linking migraine frequency with family history of migraine. Cephalalgia. 2019;39:229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levy D, Labastida‐Ramirez A, MaassenVanDenBrink A. Current understanding of meningeal and cerebral vascular function underlying migraine headache. Cephalalgia. 2019;39:1606–22. [DOI] [PubMed] [Google Scholar]
- 7. Goadsby PJ, Akerman S. The trigeminovascular system does not require a peripheral sensory input to be activated—migraine is a central disorder. Focus on ‘Effect of cortical spreading depression on basal and evoked traffic in the trigeminovascular sensory system’. Cephalalgia. 2012;32:3–5. [DOI] [PubMed] [Google Scholar]
- 8. May A, Burstein R. Hypothalamic regulation of headache and migraine. Cephalalgia. 2019;39:1710–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moller M, Mehnert J, May A. Hypothalamic activation discriminates painful and non‐painful initiation of the trigeminal autonomic reflex—an fMRI study. Cephalalgia. 2020;40:79–87. [DOI] [PubMed] [Google Scholar]
- 10. Schulte LH, May A. The migraine generator revisited: continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain. 2016;139:1987–93. [DOI] [PubMed] [Google Scholar]
- 11. Goadsby PJ, Holland PR, Martins‐Oliveira M, Hoffmann J, Schankin C, Akerman S. Pathophysiology of migraine: a disorder of sensory processing. Physiol Rev. 2017;97:553–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Charles A. The pathophysiology of migraine: implications for clinical management. Lancet Neurol. 2018;17:174–82. [DOI] [PubMed] [Google Scholar]
- 13. Weiller C, May A, Limmroth V, Kaube H, Schayck RV, Coenen HH, et al. Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995;1:658–60. [DOI] [PubMed] [Google Scholar]
- 14. Dalessio DJ, editor. Wolff´s headache and other head pain. New York: Oxford University Press; 1980. [Google Scholar]
- 15. den Boer MO, Villalon CM, Heiligers JP, Humphrey PP, Saxena PR. Role of 5‐HT1‐like receptors in the reduction of porcine cranial arteriovenous anastomotic shunting by sumatriptan. Br J Pharmacol. 1991;102:323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferrari MD, Roon KI, Lipton RB, Goadsby PJ. Oral triptans (serotonin 5‐HT(1B/1D) agonists) in acute migraine treatment: a meta‐analysis of 53 trials. Lancet. 2001;358:1668–75. [DOI] [PubMed] [Google Scholar]
- 17. Levy D, Labastida‐Ramirez A, MaassenVanDenBrink A. Current understanding of meningeal and cerebral vascular function underlying migraine headache. Cephalalgia. 2019;39:1606–22. [DOI] [PubMed] [Google Scholar]
- 18. Edvinsson L, Uddman R. Neurobiology in primary headaches. Brain Res Brain Res Rev. 2005;48:438–56. [DOI] [PubMed] [Google Scholar]
- 19. Tsuda M, Mizokoshi A, Shigemoto‐Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen‐activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. [DOI] [PubMed] [Google Scholar]
- 20. Edvinsson L, McCulloch J, Uddman R. Substance P: immunohistochemical localization and effect upon cat pial arteries in vitro and in situ. J Physiol. 1981;318:251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Edvinsson L, Uddman R. Adrenergic, cholinergic and peptidergic nerve fibres in dura mater–involvement in headache? Cephalalgia. 1981;1:175–9. [DOI] [PubMed] [Google Scholar]
- 22. Moskowitz MA. The neurobiology of vascular head pain. Ann Neurol. 1984;16:157–68. [DOI] [PubMed] [Google Scholar]
- 23. Uddman R, Edvinsson L, Ekman R, Kingman T, McCulloch J. Innervation of the feline cerebral vasculature by nerve fibers containing calcitonin gene‐related peptide: trigeminal origin and co‐existence with substance P. Neurosci Lett. 1985;62:131–6. [DOI] [PubMed] [Google Scholar]
- 24. Levy D, Burstein R, Strassman AM. Calcitonin gene‐related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol. 2005;58:698–705. [DOI] [PubMed] [Google Scholar]
- 25. Buzzi MG, Moskowitz MA. The antimigraine drug, sumatriptan (GR43175), selectively blocks neurogenic plasma extravasation from blood vessels in dura mater. Br J Pharmacol. 1990;99:202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goldstein DJ, Offen WW, Klein EG, Phebus LA, Hipskind P, Johnson KW, et al. Lanepitant, an NK‐1 antagonist, in migraine prevention. Cephalalgia. 2001;21:102–6. [DOI] [PubMed] [Google Scholar]
- 27. Goldstein DJ, Wang O, Saper JR, Stoltz R, Silberstein SD, Mathew NT. Ineffectiveness of neurokinin‐1 antagonist in acute migraine: a crossover study. Cephalalgia. 1997;17:785–90. [DOI] [PubMed] [Google Scholar]
- 28. Diener HC, Group R. RPR100893, a substance‐P antagonist, is not effective in the treatment of migraine attacks. Cephalalgia. 2003;23:183–5. [DOI] [PubMed] [Google Scholar]
- 29. Edvinsson JC, Reducha PV, Sheykhzade M, Warfvinge K, Haanes KA, Edvinsson L. Neurokinins and their receptors in the rat trigeminal system: differential localization and release with implications for migraine pain. Mol Pain. 2021;17:17448069211059400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rosenfeld MG, Mermod JJ, Amara SG, Swansong LW, Sawchenko PE, Rivier J, et al. Production of a novel neuropeptide encoded by the calcitonin gene via tissue‐specific RNA processing. Nature. 1983;304:129–35. [DOI] [PubMed] [Google Scholar]
- 31. Edvinsson L, Haanes KA, Warfvinge K, Krause DN. CGRP as the target of new migraine therapies—successful translation from bench to clinic. Nat Rev Neurol. 2018;14:338–50. [DOI] [PubMed] [Google Scholar]
- 32. Edvinsson L. Functional‐role of perivascular peptides in the control of cerebral‐circulation. Trends Neurosci. 1985;8:126–31. [Google Scholar]
- 33. Edvinsson L, Fredholm BB, Hamel E, Jansen I, Verrecchia C. Perivascular peptides relax cerebral arteries concomitant with stimulation of cyclic adenosine monophosphate accumulation or release of an endothelium‐derived relaxing factor in the cat. Neurosci Lett. 1985;58:213–7. [DOI] [PubMed] [Google Scholar]
- 34. Uddman R, Edvinsson L, Ekblad E, Hakanson R, Sundler F. Calcitonin gene‐related peptide (CGRP): perivascular distribution and vasodilatory effects. Regul Pept. 1986;15:1–23. [DOI] [PubMed] [Google Scholar]
- 35. Russell FA, King R, Smillie SJ, Kodji X, Brain SD. Calcitonin gene‐related peptide: physiology and pathophysiology. Physiol Rev. 2014;94:1099–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Edvinsson L, Hara H, Uddman R. Retrograde tracing of nerve fibers to the rat middle cerebral artery with true blue: colocalization with different peptides. J Cereb Blood Flow Metab. 1989;9:212–8. [DOI] [PubMed] [Google Scholar]
- 37. Edvinsson L, Warfvinge K. Recognizing the role of CGRP and CGRP receptors in migraine and its treatment. Cephalalgia. 2019;39:366–73. [DOI] [PubMed] [Google Scholar]
- 38. Warfvinge K, Edvinsson L. Distribution of CGRP and CGRP receptor components in the rat brain. Cephalalgia. 2019;39:342–53. [DOI] [PubMed] [Google Scholar]
- 39. McCulloch J, Uddman R, Kingman TA, Edvinsson L. Calcitonin gene‐related peptide: functional role in cerebrovascular regulation. Proc Natl Acad Sci USA. 1986;83:5731–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goadsby PJ, Edvinsson L, Ekman R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol. 1988;23:193–6. [DOI] [PubMed] [Google Scholar]
- 41. Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48–56. [DOI] [PubMed] [Google Scholar]
- 42. Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28:183–7. [DOI] [PubMed] [Google Scholar]
- 43. Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol. 2010;6:573–82. [DOI] [PubMed] [Google Scholar]
- 44. van den Pol AN. Neuropeptide transmission in brain circuits. Neuron. 2012;76:98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bost A, Shaib AH, Schwarz Y, Niemeyer BA, Becherer U. Large dense‐core vesicle exocytosis from mouse dorsal root ganglion neurons is regulated by neuropeptide Y. Neuroscience. 2017;346:1–13. [DOI] [PubMed] [Google Scholar]
- 46. Hay DL, Pioszak AA. Receptor activity‐modifying proteins (RAMPs): new insights and roles. Annu Rev Pharmacol Toxicol. 2016;56:469–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McLatchie LM, Fraser NJ, Main MJ, Wise A, Brown J, Thompson N, et al. RAMPs regulate the transport and ligand specificity of the calcitonin‐receptor‐like receptor. Nature. 1998;393:333–9. [DOI] [PubMed] [Google Scholar]
- 48. Evans BN, Rosenblatt MI, Mnayer LO, Oliver KR, Dickerson IM. CGRP‐RCP, a novel protein required for signal transduction at calcitonin gene‐related peptide and adrenomedullin receptors. J Biol Chem. 2000;275:31438–43. [DOI] [PubMed] [Google Scholar]
- 49. Gingell JJ, Hendrikse ER, Hay DL. New insights into the regulation of CGRP‐family receptors. Trends Pharmacol Sci. 2019;40:71–83. [DOI] [PubMed] [Google Scholar]
- 50. Doods H, Arndt K, Rudolf K, Just S. CGRP antagonists: unravelling the role of CGRP in migraine. Trends Pharmacol Sci. 2007;28:580–7. [DOI] [PubMed] [Google Scholar]
- 51. Doods H, Hallermayer G, Wu D, Entzeroth M, Rudolf K, Engel W, et al. Pharmacological profile of BIBN4096BS, the first selective small molecule CGRP antagonist. Br J Pharmacol. 2000;129:420–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Edvinsson L, Alm R, Shaw D, Rutledge RZ, Koblan KS, Longmore J, et al. Effect of the CGRP receptor antagonist BIBN4096BS in human cerebral, coronary and omental arteries and in SK‐N‐MC cells. Eur J Pharmacol. 2002;434:49–53. [DOI] [PubMed] [Google Scholar]
- 53. Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, et al. Calcitonin gene‐related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104–10. [DOI] [PubMed] [Google Scholar]
- 54. Ashina M, Hansen JM, Á Dunga BO, Olesen J. Human models of migraine—short‐term pain for long‐term gain. Nat Rev Neurol. 2017;13:713–24. [DOI] [PubMed] [Google Scholar]
- 55. Edvinsson L, Linde M. New drugs in migraine treatment and prophylaxis: telcagepant and topiramate. Lancet. 2010;376:645–55. [DOI] [PubMed] [Google Scholar]
- 56. Edvinsson L, Ho TW. CGRP receptor antagonism and migraine. Neurotherapeutics. 2010;7:164–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dubowchik GM, Conway CM, Xin AW. Blocking the CGRP pathway for acute and preventive treatment of migraine: the evolution of success. J Med Chem. 2020;63:6600–23. [DOI] [PubMed] [Google Scholar]
- 58. Dodick DW, Lipton RB, Ailani J, Lu K, Finnegan M, Trugman JM, et al. Ubrogepant for the treatment of migraine. N Engl J Med. 2019;381:2230–41. [DOI] [PubMed] [Google Scholar]
- 59. Lipton RB, Dodick DW, Ailani J, Lu K, Finnegan M, Szegedi A, et al. Effect of ubrogepant vs placebo on pain and the most bothersome associated symptom in the acute treatment of migraine: the ACHIEVE II randomized clinical trial. JAMA. 2019;322:1887–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Charles A, Pozo‐Rosich P. Targeting calcitonin gene‐related peptide: a new era in migraine therapy. Lancet. 2019;394:1765–74. [DOI] [PubMed] [Google Scholar]
- 61. Hokfelt T, Bartfai T, Bloom F. Neuropeptides: opportunities for drug discovery. Lancet Neurol. 2003;2:463–72. [DOI] [PubMed] [Google Scholar]
- 62. Johnson KW, Morin SM, Wroblewski VJ, Johnson MP. Peripheral and central nervous system distribution of the CGRP neutralizing antibody [(125)I] galcanezumab in male rats. Cephalalgia. 2019;39:1241–8. [DOI] [PubMed] [Google Scholar]
- 63. Hepp Z, Dodick DW, Varon SF, Gillard P, Hansen RN, Devine EB. Adherence to oral migraine‐preventive medications among patients with chronic migraine. Cephalalgia. 2015;35:478–88. [DOI] [PubMed] [Google Scholar]
- 64. Edvinsson L, Nilsson E, Jansen‐Olesen I. Inhibitory effect of BIBN4096BS, CGRP(8‐37), a CGRP antibody and an RNA‐Spiegelmer on CGRP induced vasodilatation in the perfused and non‐perfused rat middle cerebral artery. Br J Pharmacol. 2007;150:633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Juhl L, Edvinsson L, Olesen J, Jansen‐Olesen I. Effect of two novel CGRP‐binding compounds in a closed cranial window rat model. Eur J Pharmacol. 2007;567:117–24. [DOI] [PubMed] [Google Scholar]
- 66. Shi L, Lehto SG, Zhu DX, Sun H, Zhang J, Smith BP, et al. Pharmacologic characterization of AMG 334, a potent and selective human monoclonal antibody against the calcitonin gene‐related peptide receptor. J Pharmacol Exp Ther. 2016;356:223–31. [DOI] [PubMed] [Google Scholar]
- 67. Dodick DW. CGRP ligand and receptor monoclonal antibodies for migraine prevention: evidence review and clinical implications. Cephalalgia. 2019;39:445–58. [DOI] [PubMed] [Google Scholar]
- 68. Edvinsson L. CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br J Clin Pharmacol. 2015;80:193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Noseda R, Schain AJ, Melo‐Carrillo A, Tien J, Stratton J, Mai F, et al. Fluorescently‐labeled fremanezumab is distributed to sensory and autonomic ganglia and the dura but not to the brain of rats with uncompromised blood brain barrier. Cephalalgia. 2020;40:229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Messlinger K, MaassenVanDenBrink A. Cardio‐ and cerebrovascular safety of erenumab, a monoclonal antibody targeting CGRP receptors—important studies on human isolated arteries. Cephalalgia. 2019;39:1731–4. [DOI] [PubMed] [Google Scholar]
- 71. Tan KK, Brown MJ, Hargreaves RJ, Shepheard SL, Cook DA, Hill RG. Calcitonin gene‐related peptide as an endogenous vasodilator: immunoblockade studies in vivo with an anti‐calcitonin gene‐related peptide monoclonal antibody and its Fab' fragment. Clin Sci (Lond). 1995;89:565–73. [DOI] [PubMed] [Google Scholar]
- 72. Ferrari MD, Diener HC, Ning X, Galic M, Cohen JM, Yang R, et al. Fremanezumab versus placebo for migraine prevention in patients with documented failure to up to four migraine preventive medication classes (FOCUS): a randomised, double‐blind, placebo‐controlled, phase 3b trial. Lancet. 2019;394:1030–40. [DOI] [PubMed] [Google Scholar]
- 73. Reuter U, Goadsby PJ, Lanteri‐Minet M, Wen S, Hours‐Zesiger P, Ferrari MD, et al. Efficacy and tolerability of erenumab in patients with episodic migraine in whom two‐to‐four previous preventive treatments were unsuccessful: a randomised, double‐blind, placebo‐controlled, phase 3b study. Lancet. 2018;392:2280–7. [DOI] [PubMed] [Google Scholar]
- 74. American Headache Society . The American Headache Society Position statement on integrating new migraine treatments into clinical practice. Headache. 2019;59:1–18. [DOI] [PubMed] [Google Scholar]
- 75. Lundblad C, Haanes KA, Grande G, Edvinsson L. Experimental inflammation following dural application of complete Freund's adjuvant or inflammatory soup does not alter brain and trigeminal microvascular passage. J Headache Pain. 2015;16:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Eftekhari S, Salvatore CA, Johansson S, Chen TB, Zeng Z, Edvinsson L. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood–brain barrier. Brain Res. 2015;1600:93–109. [DOI] [PubMed] [Google Scholar]
- 77. Edvinsson L, Tfelt‐Hansen P. The blood–brain barrier in migraine treatment. Cephalalgia. 2008;28:1245–58. [DOI] [PubMed] [Google Scholar]
- 78. Schankin CJ, Maniyar FH, Seo Y, Kori S, Eller M, Chou DE, et al. Ictal lack of binding to brain parenchyma suggests integrity of the blood–brain barrier for 11C‐dihydroergotamine during glyceryl trinitrate‐induced migraine. Brain. 2016;139:1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hougaard A, Amin FM, Christensen CE, Younis S, Wolfram F, Cramer SP, et al. Increased brainstem perfusion, but no blood–brain barrier disruption, during attacks of migraine with aura. Brain. 2017;140:1633–42. [DOI] [PubMed] [Google Scholar]
- 80. Amin FM, Hougaard A, Cramer SP, Christensen CE, Wolfram F, Larsson HBW, et al. Intact blood–brain barrier during spontaneous attacks of migraine without aura: a 3T DCE‐MRI study. Eur J Neurol. 2017;24:1116–24. [DOI] [PubMed] [Google Scholar]
- 81. Melo‐Carrillo A, Noseda R, Nir RR, Schain AJ, Stratton J, Strassman AM, et al. Selective inhibition of trigeminovascular neurons by fremanezumab: a humanized monoclonal anti‐CGRP antibody. J Neurosci. 2017;37:7149–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Melo‐Carrillo A, Strassman AM, Nir RR, Schain AJ, Noseda R, Stratton J, et al. Fremanezumab—a humanized monoclonal anti‐CGRP antibody‐inhibits thinly myelinated (Adelta) but not unmyelinated (C) meningeal nociceptors. J Neurosci. 2017;37:10587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Melo‐Carrillo A, Schain AJ, Stratton J, Strassman AM, Burstein R. Fremanezumab and its isotype slow propagation rate and shorten cortical recovery period but do not prevent occurrence of cortical spreading depression in rats with compromised blood brain barrier. Pain. 2020;161:1037–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Eftekhari S, Salvatore CA, Calamari A, Kane SA, Tajti J, Edvinsson L. Differential distribution of calcitonin gene‐related peptide and its receptor components in the human trigeminal ganglion. Neuroscience. 2010;169:683–96. [DOI] [PubMed] [Google Scholar]
- 85. Frederiksen SD, Warfvinge K, Ohlsson L, Edvinsson L. Expression of pituitary adenylate cyclase‐activating peptide, calcitonin gene‐related peptide and headache targets in the trigeminal ganglia of rats and humans. Neuroscience. 2018;393:319–32. [DOI] [PubMed] [Google Scholar]
- 86. Akerman S, Holland PR, Goadsby PJ. Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci. 2011;12:570–84. [DOI] [PubMed] [Google Scholar]
- 87. Edvinsson JCA, Warfvinge K, Krause DN, Blixt FW, Sheykhzade M, Edvinsson L, et al. C‐fibers may modulate adjacent Adelta‐fibers through axon‐axon CGRP signaling at nodes of Ranvier in the trigeminal system. J Headache Pain. 2019;20:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Edvinsson L, Chan KY, Eftekhari S, Nilsson E, de Vries R, Saveland H, et al. Effect of the calcitonin gene‐related peptide (CGRP) receptor antagonist telcagepant in human cranial arteries. Cephalalgia. 2010;30:1233–40. [DOI] [PubMed] [Google Scholar]
- 89. Chan KY, Edvinsson L, Eftekhari S, Kane SA, Lynch J, Hargreaves RJ, et al. Characterization of the calcitonin gene‐related peptide receptor antagonist telcagepant (MK‐0974) in human isolated coronary arteries. J Pharmacol Exp Ther. 2010;334:746–52. [DOI] [PubMed] [Google Scholar]
- 90. Ohlsson L, Haanes KA, Kronvall E, Xu C, Snellman J, Edvinsson L. Erenumab (AMG 334), a monoclonal antagonist antibody against the canonical CGRP receptor, does not impair vasodilatory or contractile responses to other vasoactive agents in human isolated cranial arteries. Cephalalgia. 2019;39:1745–52. [DOI] [PubMed] [Google Scholar]
- 91. Ohlsson L, Kronvall E, Stratton J, Edvinsson L. Fremanezumab blocks CGRP induced dilatation in human cerebral, middle meningeal and abdominal arteries. J Headache Pain. 2018;19:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Manoukian R, Sun H, Miller S, Shi D, Chan B, Xu C. Effects of monoclonal antagonist antibodies on calcitonin gene‐related peptide receptor function and trafficking. J Headache Pain. 2019;20:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Yarwood RE, Imlach WL, Lieu T, Veldhuis NA, Jensen DD, Herenbrink CK, et al. Endosomal signaling of the receptor for calcitonin gene‐related peptide mediates pain transmission. Proc Natl Acad Sci USA. 2017;114:12309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Ashina M. Migraine. N Engl J Med. 2020;383:1866–76. [DOI] [PubMed] [Google Scholar]
- 95. McGinley JS, Houts CR, Nishida TK, Buse DC, Lipton RB, Goadsby RJ, et al. Systematic review of outcomes and endpoints in preventive migraine clinical trials. Headache. 2021;61:253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang X, Chen Y, Song J, You C. Efficacy and safety of monoclonal antibody against calcitonin gene‐related peptide or its receptor for migraine: a systematic review and network meta‐analysis. Front Pharmacol. 2021;12:649143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Martelletti P, Edvinsson L, Ashina M. Shaping the future of migraine targeting calcitonin‐gene‐related‐peptide with the disease‐modifying migraine drugs (DMMDs). J Headache Pain. 2019;20:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tepper S, Ashina M, Reuter U, Brandes JL, Doležil D, Silberstein S, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Neurol. 2017;16:425–34. [DOI] [PubMed] [Google Scholar]
- 99. Goadsby PJ, Reuter U, Hallstrom Y, Broessner G, Bonner JH, Zhang F, et al. A controlled trial of erenumab for episodic migraine. N Engl J Med. 2017;377:2123–32. [DOI] [PubMed] [Google Scholar]
- 100. Dodick DW, Ashina M, Brandes JL, Kudrow D, Lanteri‐Minet M, Osipova V, et al. ARISE: a phase 3 randomized trial of erenumab for episodic migraine. Cephalalgia. 2018;38:1026–37. [DOI] [PubMed] [Google Scholar]
- 101. Ashina M, Kudrow D, Reuter U, Dolezil D, Silberstein S, Tepper SJ, et al. Long‐term tolerability and nonvascular safety of erenumab, a novel calcitonin gene‐related peptide receptor antagonist for prevention of migraine: a pooled analysis of four placebo‐controlled trials with long‐term extensions. Cephalalgia. 2019;39:1798–808. [DOI] [PubMed] [Google Scholar]
- 102. Ashina M, Goadsby PJ, Reuter U, Silberstein S, Dodick DW, Xue F, et al. Long‐term efficacy and safety of erenumab in migraine prevention: results from a 5‐year, open‐label treatment phase of a randomized clinical trial. Eur J Neurol. 2021;28:1716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lipton RB, Tepper SJ, Silberstein SD, Kudrow D, Ashina M, Reuter U, et al. Reversion from chronic migraine to episodic migraine following treatment with erenumab: results of a post‐hoc analysis of a randomized, 12‐week, double‐blind study and a 52‐week, open‐label extension. Cephalalgia. 2021;41:6–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Baker B, Schaeffler B, Beliveau M, Rubets I, Pederson S, Trinh M, et al. Population pharmacokinetic and exposure‐response analysis of eptinezumab in the treatment of episodic and chronic migraine. Pharmacol Res Perspect. 2020;8:e00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ashina M, Saper J, Cady R, Schaeffler BA, Biondi DM, Hirman J, et al. Eptinezumab in episodic migraine: a randomized, double‐blind, placebo‐controlled study (PROMISE‐1). Cephalalgia. 2020;40:241–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, et al. Safety and efficacy of ALD403, an antibody to calcitonin gene‐related peptide, for the prevention of frequent episodic migraine: a randomised, double‐blind, placebo‐controlled, exploratory phase 2 trial. Lancet Neurol. 2014;13:1100–7. [DOI] [PubMed] [Google Scholar]
- 107. Winner PK, McAllister P, Chakhava G, Ailani J, Ettrup A, Krog M, et al. Effects of intravenous eptinezumab vs placebo on headache pain and most bothersome symptom when initiated during a migraine attack: a randomized clinical trial. JAMA. 2021;325:2348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dodick DW, Silberstein SD, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, et al. Effect of fremanezumab compared with placebo for prevention of episodic migraine: a randomized clinical trial. JAMA. 2018;319:1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Silberstein SD, Dodick DW, Bigal ME. Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med. 2017;377:2113–22. [DOI] [PubMed] [Google Scholar]
- 110. Silberstein SD, Cohen JM, Seminerio MJ, Yang R, Ashina S, Katsarava Z. The impact of fremanezumab on medication overuse in patients with chronic migraine: subgroup analysis of the HALO CM study. J Headache Pain. 2020;21:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Goadsby PJ, Silberstein SD, Yeung PP, Cohen JM, Ning X, Yang R, et al. Long‐term safety, tolerability, and efficacy of fremanezumab in migraine: a randomized study. Neurology. 2020;95:e2487–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. MaassenVanDenBrink A, Terwindt GM, Cohen JM, Cohen JM, Barash S, Ramirez V, et al. Impact of age and sex on the efficacy of fremanezumab in patients with difficult‐to‐treat migraine: results of the randomized, placebo‐controlled, phase 3b FOCUS study. J Headache Pain. 2021;22:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Detke HC, Goadsby PJ, Wang S, Friedman DI, Selzler KJ, Aurora SK. Galcanezumab in chronic migraine: the randomized, double‐blind, placebo‐controlled REGAIN study. Neurology. 2018;91:e2211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Forderreuther S, Zhang Q, Stauffer VL, Aurora SK, Lainez MJA. Preventive effects of galcanezumab in adult patients with episodic or chronic migraine are persistent: data from the phase 3, randomized, double‐blind, placebo‐controlled EVOLVE‐1, EVOLVE‐2, and REGAIN studies. J Headache Pain. 2018;19:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Skljarevski V, Matharu M, Millen BA, Ossipov MH, Kim BK, Yang JY. Efficacy and safety of galcanezumab for the prevention of episodic migraine: results of the EVOLVE‐2 phase 3 randomized controlled clinical trial. Cephalalgia. 2018;38:1442–54. [DOI] [PubMed] [Google Scholar]
- 116. Stauffer VL, Dodick DW, Zhang Q, Carter JN, Ailani J, Conley RR. Evaluation of galcanezumab for the prevention of episodic migraine: the EVOLVE‐1 randomized clinical Trial. JAMA Neurol. 2018;75:1080–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ford JH, Foster SA, Stauffer VL, Ruff DD, Aurora SK, Versijpt J. Patient satisfaction, health care resource utilization, and acute headache medication use with galcanezumab: results from a 12‐month open‐label study in patients with migraine. Patient Prefer Adherence. 2018;12:2413–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Camporeale A, Kudrow D, Sides R, Wang S, Van Dycke A, Selzler KJ, et al. A phase 3, long‐term, open‐label safety study of galcanezumab in patients with migraine. BMC Neurol. 2018;18:188. [DOI] [PMC free article] [PubMed] [Google Scholar]