

Abstract
The GC reaction results in the selection of B cells acquiring effector Ig secreting ability by progressing toward plasmablastic differentiation. This transition is associated with exclusion from the GC microenvironment. The aberrant expansion of plasmablastic elements within the GC fringes configures an atypical condition, the biological characteristics of which have not been defined yet. We investigated the in situ immunophenotypical and transcriptional characteristics of a nonclonal germinotropic expansion of plasmablastic elements (GEx) occurring in the tonsil of a young patient. Compared to neighboring GC and perifollicular regions, the GEx showed a distinctive signature featuring key regulators of plasmacytic differentiation, cytokine signaling, and cell metabolism. The GEx signature was tested in the setting of diffuse large B‐cell lymphoma (DLBCL) as a prototypical model of lymphomagenesis encompassing transformation at different stages of GC and post‐GC functional differentiation. The signature outlined DLBCL clusters with different immune microenvironment composition and enrichment in genetic subtypes. This report represents the first insight into the transcriptional features of a germinotropic plasmablastic burst, shedding light into the molecular hallmarks of B cells undergoing plasmablastic differentiation and aberrant expansion within the noncanonical setting of the GC microenvironment.
Keywords: Diffuse large B‐cell lymphoma ⋅ Digital spatial profiling ⋅ Germinal center ⋅ Plasmablast
Through Digital Spatial Profiling, we explored the in situ transcriptome of reactive follicle germinal center dark and light zones, peri‐follicularregions, and of an atypical germinal center plasmablastic expansion(GEx). The GEx distinctive signature applied to diffuse large B‐celllymphomas (DLBCL) hinted at subgroups with positive enrichment in MYD88/CD79B mutant cases.
Introduction
Within secondary lymphoid organs, immune cells display topographic compartmentalization underlying functional commitment toward different stages of immune response induction and regulation. In lymphoid follicles, GCs represent a complex specialized microenvironment sustaining B‐cell proliferative bursts underlying somatic hypermutation and class‐switch recombination of Ig genes, communication with T‐cell subsets with helping function, and interplay with specialized mesenchymal scaffolds (i.e. FDCs) [1]. These events play through the dynamical iteration of elements between the dark zone (DZ), intermediate zone, and light zone (LZ) of the GC [2], eventually resulting in the differentiation and displacement from the GC of cells acquiring effector capabilities through the synthesis and secretion of Igs (i.e. plasmablasts and plasma cells) [3]. Alterations in the topographic compartmentalization of GC and extra‐GC populations in lymphoid tissues are commonly observed in the setting of lymphoproliferative diseases, where the accumulation of cells with morphological or immunophenotypical features conflicting with their topographic localization represents a hallmark of histopathological analyses. This assumption reached its highest expression in diffuse large B‐cell lymphoma (DLBCL), where prognostic subclassification is based on the presumed derivation from elements transforming at different functional stages of their GC reaction trajectories, which is recapitulated by the cell of origin (COO), as determined by gene expression profiling [4, 5, 6].
We have investigated here an atypical germinotropic expansion of nonclonal B cells with plasmablastic features confined to a single enlarged GC structure in the tonsil of a young patient, through in‐situ immunolocalization analyses and high throughput digital spatial profiling. Comparing the features of the atypical germinotropic expansion (GEx) with those of topographically preserved DZ, LZ, and peri‐follicular (Peri) regions of interest (ROIs), we identified transcriptional programs discriminating the different microenvironments and a unique transcriptomic profile of the GEx ROIs featuring the overexpression of transcripts involved in plasmacytoid differentiation, cytokine signaling, and cell metabolism.
To probe the reflection of the identified transcriptional signature in a setting of B‐cell lymphomatous transformation encompassing the full spectrum of GC‐ and post‐GC differentiation, the 20 GEx hallmark genes were used to cluster a large transcriptomic dataset of DLBCL. The GEx signature highlighted two clusters with positive enrichment in genetic subtypes associated with a non‐GC COO, and consistent immune microenvironment composition.
Results
Pathology of the tonsil
Histopathological analysis of the left tonsil from a young patient with clinical bilateral hypertrophy revealed, in a background of lymphoid follicles with hyperplastic features and preserved GC DZ, LZ, and mantles, an isolated abnormal follicle with flattened mantle zone, enlarged GC without evident DZ/LZ polarization, preservation of rare tingible body macrophages, and populated by a predominance of monomorphic plasmacytoid cells with immature morphology (Fig. 1A, inset). Quantitative immunophenotypical characterization of the reactive follicular (DZ and LZ) and Peri regions, and of the atypical germinotropic plasmablastic expansion (GEx), highlighted differences in the immune profile of resident elements (Fig. 1B and C). The reactive preserved GC DZ and LZ regions were characterized by B cells with strong CD20 expression, dense Ki‐67 immunoreactivity (higher in the DZ), negativity for IRF4 (except for scattered cells), in a background of CD10‐expressing cells, Bcl‐2 negativity, slight T‐cell infiltration (denser in the LZ), and no evidence of light‐chain restriction (Fig. 1B and C). At contrast, the composition of the GEx displayed a CD20+ B‐cell phenotype, high Ki‐67+ proliferative fraction, diffuse IRF4 positivity with a fraction of IRF4+ cells coexpressing CD10, negativity for Bcl‐2, and immunophenotypical restriction for lambda light chain (Fig. 1B and C). Most of the cells populating the GEx also expressed CD138 (in the absence of CD30), indicating partial acquisition of a plasmablastic phenotype. Immunohistochemistry for HHV8 and in situ hybridization for EBER (EBV) proved negative (Supporting information Fig. S1). On the basis of the GEx lambda light‐chain restriction, analysis of the Ig light and heavy chain genes rearrangement, as well as analysis of incomplete rearrangements, were performed on DNA extracted by laser microdissection of the GEx, which revealed a polyclonal profile (Supporting information Fig. S2A–C). The strong and diffuse immunoreactivity of IRF4 and the co‐occurrence of IRF4/CD10 double‐expressing elements also prompted the analysis of IRF4 gene rearrangement by fluorescence in situ hybridization (FISH), which did not reveal any abnormality (Supporting information Fig. S3), allowing to exclude an IRF4‐rearranged lymphoma.
Figure 1.
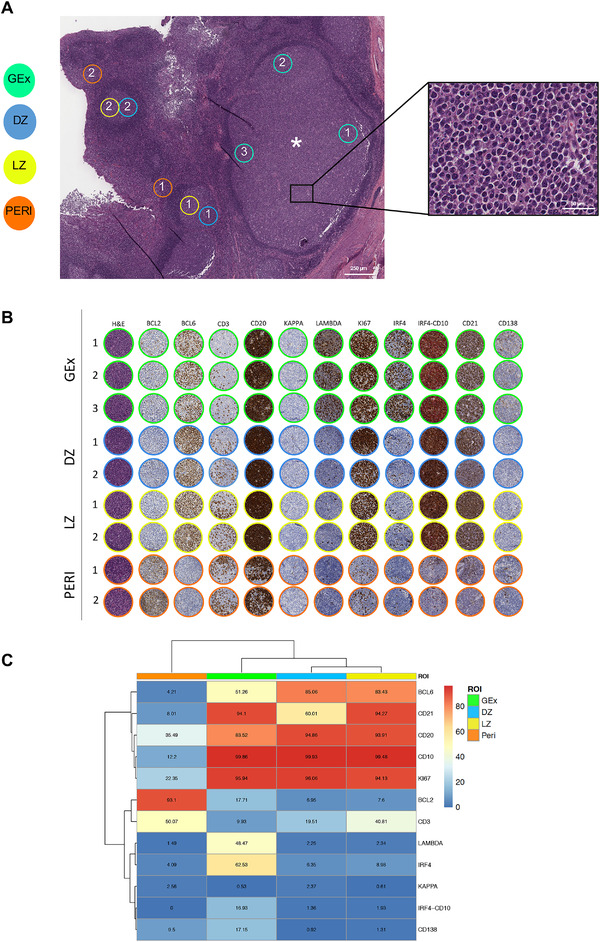
(A) Digitalized slide selection of the H&E‐stained section of the tonsil highlighting the presence of an aberrantly expanded GC (asterisk) characterized by the presence of elements with plasmacytoid morphology (inset). On the H&E, representative regions relative to GC dark zone (DZ) and light zone (LZ) areas, perifollicular (Peri) areas, and germinotropic plasmablastic expansion (GEx) areas, are highlighted. Original magnification ×50. (B) Comparative analysis of H&E and IHC for Bcl‐2, Bcl‐6, CD3, CD20, Kappa and Lambda light chain, Ki67, IRF4, IRF4/CD10, CD2, CD138 in the DZ, LZ, Peri, and GEx areas highlighted in (A). (C) Heatmap of the average expression of the quantitative immunohistochemical analysis of the markers evaluated in the DZ (n = 5), LZ (n = 5), Peri (n = 5), and GEx (n = 9) areas highlighted in (B).
To further investigate the nature of the GEx from a genetic perspective, a targeted NGS experiment was performed on the microdissected DNA, probing the mutational status of 54 genes recurrently mutated in lymphomas (Supporting information Table S1). NGS did not highlight pathogenic mutations associated with B‐cell lymphomas (Supporting information Table S2), rather it detected polymorphisms and missense single nucleotide variants with benign, uncertain, or not reported clinical significance, including a SNP in the coding sequence of the KMT2D gene (c.14077A>G; variant allele frequency [VAF] 47.5%) [7]. Such KMT2D variant in the setting of a polyclonal expansion and in the absence of other relevant genetic lesions did not substantiate GEx transformed nature.
Digital Spatial Profiling of the GEx regions reveals a distinctive profile
We subsequently explored the in situ transcriptional profile of the GC plasmablastic burst through the Nanostring GeoMx Digital Spatial Profiling technology (NanoString, Seattle WA). The expression of 1824 genes from key cancer‐associated transcriptional programs (Supporting information Table S3) was determined on five DZ, five LZ, and five Peri ROIs selected from morphologically or phenotypically preserved follicles or perifollicular areas, and on nine GEx ROIs. Principal component analysis (PCA) and unsupervised hierarchical clustering were analyzed to investigate the differences in the transcriptional profile of the 24 ROIs. PCA revealed that ROIs segregated according to their spatial classification, with Peri regions showing neatly separated profiles from GC ROIs, including DZ, LZ, and GEx, which clustered together with other GC regions, showing some degree of intermixing with LZ ROIs (Fig. 2A). Consistently, clustering analysis confirmed the same degree of relationship between the different ROIs (Fig. 2B). The different spatial regions could be discriminated according to the differential expression of 474 genes (Fig. 2C, Supporting information Table S4), which were enriched in biological pathways including cell cycle and mitotic checkpoints, PI3K‐Akt, NF‐kappa B, and B‐cell receptor signaling (Fig. 2D, KEGG, Reactome, and GO‐BP libraries have been considered, Supporting information Table S4). mRNA expression of the transcripts relative to the IHC markers evaluated for quantitative immunopehnotypical analyses showed consistency with the protein expression pattern (Supporting information Fig. S4).
Figure 2.
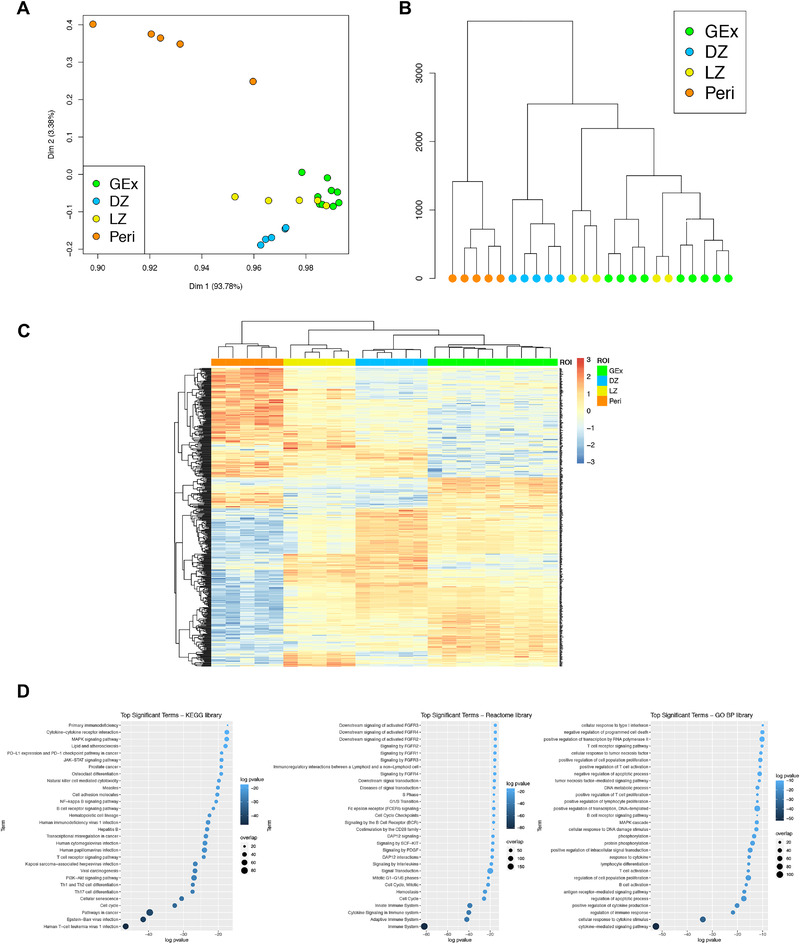
(A) Two‐dimensional principal component reduction of the DZ (n = 5), LZ (n = 5), Peri (n = 5), and GEx (n = 9) regions of interest (ROIs) profiles according to Digital Spatial Profiling of 1824 genes. (B) Unsupervised hierarchical clustering of the 24 ROIs. (C) Heatmap of differentially expressed genes among GEx, DZ, LZ, and Peri ROIs. Kruskal–Wallis test has been applied to compare gene expression among ROI groups. (D) Top significant terms from gene set enrichment analysis on genes differentially expressed among DZ (n = 5), LZ (n = 5), Peri (n = 5), and GEx (n = 9) ROIs (Gene Ontology Biological Process, KEGG, and Reactome Pathway libraries).
We then investigated whether GEx ROIs could be defined by a specific gene signature. Pairwise differential expression analysis performed on the different ROIs allowed to identify candidate genes reflecting the distinctive profiles between the GEx ROIs in comparison with DZ LZ and Peri ROIs (Fig. 3A–D). Among the 20 differentially expressed genes, 17 were significantly upregulated in GEx ROIs, while three were downmodulated (Fig. 3A‐D). GEx hallmark genes are included, along with the plasma cell differentiation markers PRDM1, IRF4, TNFRSF17 (BCMA), and CD9, genes which are involved in 2‐oxoglutarate metabolism (GOT2, IDH2), in IL17 pathway (IL17RB, HSP90B1), and cytokine signaling (RASAL1, LTB), in PI3K‐Akt pathway (SGK1, BCL2L1), in lymphocyte activation (ADA, SCL7A5, FCRL2), and cell‐surface regulation of immune activation (CD24, LILRB1), in cell adhesion (ANKRD28) and response to abiotic (i.e. osmotic) stress (SLK1). Moreover, the long noncoding RNA FAM30 was also listed among the GEx hallmarks.
Figure 3.
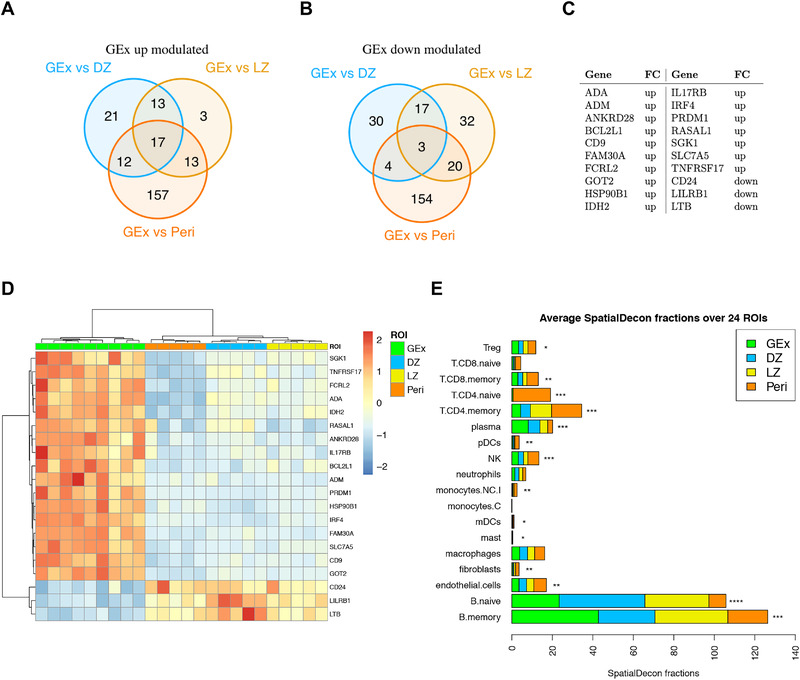
(A) Venn diagram of upmodulated genes from three different comparisons (i.e. GEx vs. DZ, GEx vs. LZ, and GEx vs. Peri). (B) Venn diagram of downmodulated genes from three different comparisons (i.e. GEx vs. DZ, GEx vs. LZ, and GEx vs. Peri). (C) GEx signature genes. These genes are significantly differentially expressed in GEx in each pairwise comparison. (D) Heatmap of differentially expressed genes in GEx as compared to DZ, LZ, and Peri ROIs. The GEx signature shows a high discriminatory capacity between GEx ROIs and the other regions. (E) Average Spatial Decon fractions of cell types in the four ROI subgroups; DZ (n = 5), LZ (n = 5), Peri (n = 5), and GEx (n = 9) ROIs. Kruskal‐Wallis test has been applied to compare faction distributions among groups (Supporting information Table S5).
Spatial immune deconvolution of GEx ROIs shows enrichment in memory B cells and plasma cells
Based on the evidence of a distinct transcriptional profile of GEx regions in comparison with other GC and P regions, we investigated the immune composition of the ROIs according to transcriptional deconvolution. A SpatialDecon [8] approach using the safeTME matrix was adopted, which highlighted that GEx ROIs displayed a different microenvironment composition as compared with canonical LZ and DZ GC ROIs, also differing from Peri ROIs (Fig. 3E, Supporting information Table S5), further indicating that a perturbation of the normal GC milieu. Specifically, GEx ROIs were positively enriched in plasma cells and memory B cells as compared with other ROIs, while being poorly infiltrated by T cells (Kruskal–Wallis p‐values < 0.001) (Fig. 3E). To validate the transcriptional immune deconvolution output, software‐based segmentation and quantitative evaluation of the IRF4 (MUM1 epitope) and CD138 IHC markers highlighting cells undergoing plasma cell differentiation, and of CD3 T‐cell marker were performed in 24 ROIs corresponding to the profiled nine GEx, five DZ, five LZ, and five Peri regions (Supporting information Fig. S5A and B). Analysis of the three protein markers supported a higher frequency of plasma cells and a decreased infiltration of T cells in the GEx ROIs, in line with the transcriptional deconvolution analysis.
The GEx signature outlines DLBCL clusters with different enrichments in genetic subtypes and microenvironment composition
The molecular profiles of nonmalignant GC compartments can be exploited to probe GC microenvironment imprints in B‐cell lymphomas with different degrees of relationship with GC subpopulations, such as DLBCL, in which the COO has shown prognostic significance in the setting of standard chemoimmunotherapy regimens [9]. To investigate whether the transcriptional hallmarks identified in the GEx ROIs could be traced in the heterogeneous spectrum of B‐cell malignant transformation recapitulated by DLBCL, we applied the GEx gene signature to a dataset of 481 DLBCL cases relative to Schmitz et al. [6]. Based on the expression of the 20 genes of the GEx signature, DLBCL clustered into two main groups (Fig. 4A), with the cluster 1 characterized by the overexpression of 17 genes (Supporting information Fig. S6A) and by a trend toward a worse prognosis (Supporting information Fig. S6B and C). We subsequently investigated the distribution of the major DLBCL genetic subtypes according to Schmitz and colleagues [6] and found that the relative frequency of the subtypes was significantly different in the two clusters identified by the GEx signature (Fig. 4B and C, Supporting information Tables S6 and S7). Specifically, cluster 1, which was characterized by the general overexpression of hallmark genes of the GEx ROIs, showed a neat enrichment in cases with MYD88 and CD79B mutation co‐occurrence (MCD, Fisher p‐value < 10e‐05), a higher frequency of cases with BCL6 fusions and NOTCH2 mutations (BN2, Fisher p‐value = 0.02), and a markedly lower frequency of cases with EZH2 and BCL2 lesions (EZB, Fisher p‐value < 10e‐9) (Fig. 4C, Supporting information Table S7) suggesting that the genes positively characterizing GEx ROIs underlie a specific biology related with MCD genetics, known to be enriched in ABC clones undergoing plasmablastic or plasmacytic commitment [6]. The analysis of the expression of GEx hallmark genes across different DLBCL genetic subtypes was further extended by including the dataset reported by Chapuy et al. [5]. Consistent with what observed in the Schmitz's genetic groups (Supporting information Fig. S6D), the GEx gene signature showed higher expression in the Chapuy's C5 cluster, positively enriched in MYD88 mutant cases, and lower expression in the C3 cluster characterized by EZH2 mutations (Supporting information Figs. S6E, Table S8).
Figure 4.
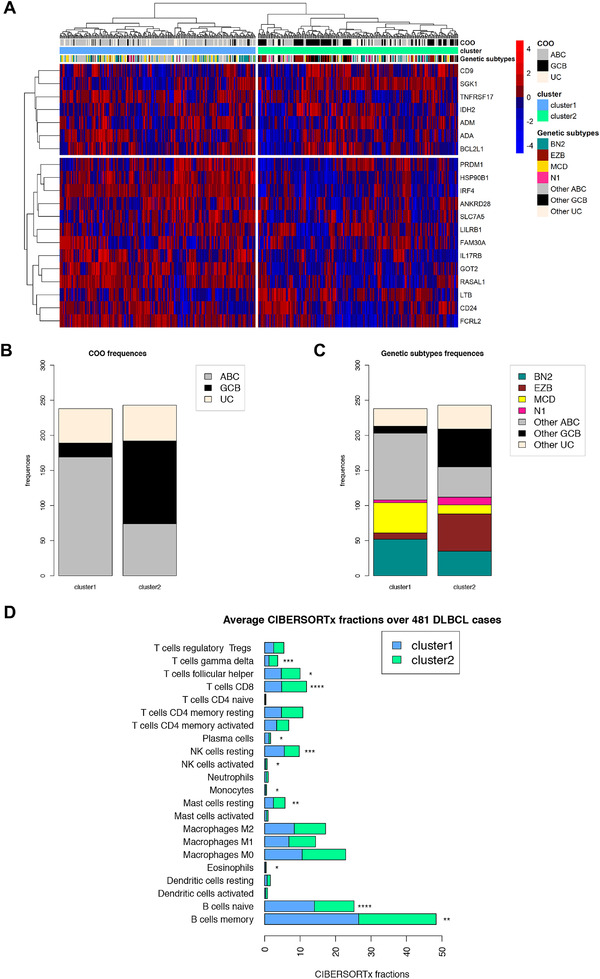
(A) Unsupervised clustering analysis of the 481 DLBCL cases relative to Schmitz et al. dataset [6] based on the GEx signature. Two distinct clusters are identified; the cluster 1 (blue) is characterized by a higher gene expression, while a lower gene expression characterizes the cluster 2 (green). (B) Barplot of cell of origin (COO) absolute frequencies observed among clusters. Cluster 1 is enriched by ABC cases, whereas cluster 2 is enriched by GCB cases (Fisher p‐values < 10e‐15). (C) Barplot of genetic subtypes absolute frequencies observed among clusters (Fisher p‐values reported in Supporting information Table S8). (D) Average CIBERSORTx fractions of cell types in the two clusters relative to Schmitz et al. dataset [6]. Wilcoxion‐Mann‐Withney test has been applied to compare faction distributions among groups (Supporting information Table S9).
Prompted by the finding of a different microenvironment composition of GEx ROIs in comparison with other GC and Peri ROIs, we estimated the microenvironment of the DLBCL clusters identified in Shmitz's cohort according to GEx signature expression, through CIBERSORTx deconvolution. Consistently with the deconvolution of in situ transcriptional profiles of GEx ROIs, cluster 1, which was characterized by upregulation of GEx hallmark genes, showed a significant enrichment in memory B cells and plasma cells (Mann–Whitney p‐values < 0.001, Supporting information Table S9), and a decrease in CD8 T cells and follicular T helper cells (Fig. 4D), indicating that a link between the GEx that is a GC‐related lesion, and lymphomatous clones enriched in specific non‐GC genetics, cell‐of‐origin and microenvironment, may exist.
Discussion
The transition of B cells undergoing selection and refinement of their IG receptor in the GC reaction toward effectors capable of Ig secretion implies the acquisition of plasmablastic/plasmacytoid features within the GC microenvironment. The spatial localization of these functional and phenotypical intermediates is still poorly characterized and depends on the dynamical modulation of chemotactic receptor or ligand axes interweaving with BCR‐controlled programs [10]. Proliferating cells with plasmablastic or plasmacytoid features accumulating within the GC, therefore, represent an element of atypia even in the setting of nonclonal events, and little is known about the molecular signature characterizing their transient state [11]. In this report, we genetically, transcriptionally, and phenotypically characterized a nonclonal atypical germinotropic expansion of plasmablastic cells, investigating differential features emerging from the comparison with neighboring GC and extra‐GC regions. The GEx ROIs were characterized by the unique co‐occurrence of IRF4 (MUM1) and CD10 expression, which highlighted a transitory state engendered by IRF4 control of GC exit [12] and CD10 ectopeptidase retention that can be observed in DLBCL with plasmablastic differentiation [13]. The finding of a KMT2D variant in the absence of clonal restriction and of other cooperating genetic lesions prompts the question whether an altered GC B‐cell homeostasis could underlie focal GEx occurrence. Kmt2d conditional deletion in early murine B cells induces the atypical oligoclonal expansion of CD19+B220+ B cells with variable degree of plasmacytic differentiation (i.e. CD138+); moreover, shRNA interference of Kmt2d expression in normal mouse hematopoietic precursors followed by immunization results in the aberrant persistence of GCs [14]. Consistently, in the setting of Kmt2d deficiency an increase in the frequency of transitional B cells and GC B cells is observed in response to immunization [14]. These models underscore the influence of Kmt2d competence in the physiological regulation of GC B‐cell dynamics. However, there is no evidence linking the reported KMT2D variant with defective protein activity or supporting its involvement either direct or through the cooperation with biased GC microenvironmental programs, to the transcriptional and phenotypical changes observed in the GEx.
On digital spatial profiling, we identified genes allowing the neat discrimination of the different ROIs according to their topography (DZ, LZ, GEx, Peri). providing an unprecedented near‐transcriptome characterization of these lymphoid tissue spatial compartments in their native state in situ. A set of 20 genes were found differentially expressed in GEx ROIs as compared with neighboring DZ, LZ, and Peri ROIs. The discriminating signature resulted in positively enriched in the key transcription factors driving plasma cell differentiation, IRF4 and PRDM1, and included the B‐cell differentiation receptor BCMA involved in the transduction of trophic signals from APRIL and BAFF TNF superfamily ligands [15]. Such molecular features supportive of a plasmablastic phenotype were also supported by the downregulation of CD24, a signal transducer negatively modulated in response to BCR activation and along plasmablastic transition [16]. The GC localization of the plasmablastic expansion found resonance in the overexpression of CD9. The tetraspanin CD9 has been reported to mark a subset of B cells in the human GC characterized by plasmablastic differentiation and Blimp1 (PRDM1) expression [17]. These CD9+ GC B cells more efficiently give rise to CD20‐CD38+ plasmablasts as compared with their CD9‐ counterpart. Moreover, in the murine setting, the efficient plasmablastic or plasmacytic differentiation of CD9+ B cells is shared by non‐GC B cells endowed with prompt commitment to Ig‐secreting effectors such as B1 B cells and marginal zone B‐cell subsets [18]. By applying the GEx differentially expressed genes signature to a well‐characterized (both transcriptionally and genetically) DLBCL dataset, we aimed at investigating whether the atypical status of nonclonal Gex could be represented in the transcriptional signature of a subset of DLBCL of either ABC or GCB COO. Previous reports described a subset of ABC‐DLBCL expressing PRDM1/BLIMP1 and demonstrated that loss of function of this antigen is harbinger of a poor prognosis [19]. Expression of IRF4/MUM1 is routinely employed in the diagnostic setting for the distinction of non‐GC subtypes based on immunohistochemical algorithms [20]. Moreover, specific subtypes of large B‐cell lymphomas characterized by IRF4 rearrangement have been recently described and recognized as independent entities in the most recent WHO classification [21]. We report additional molecular markers potentially associated with plasmablastic commitment in the GC, including the receptor of IL17B/IL25 IL17RB, the overexpression of which marks lymphoplasmacytic lymphomas with mutant MYD88 L265P and CXCR4 WT [22]. In the GC setting, IL17B/IL25 signaling could enforce NF‐κB activity [23] through TRAF6, its cooperation with CD40 signaling being required for B‐cell affinity maturation and plasma cell differentiation [24]. From the genes positively and negatively characterizing GEx ROIs, no relevant clues emerge about the mechanisms leading to the atypical GC retention and expansion of the plasmablastic elements. Under normal conditions, the suppression of Bach2 and Pax5 transcripts, along with the downregulation of Bcl‐6, IRF8, and PU‐1 and the activation of BLIMP1 and MUM1/IRF4, drive the development of PCs resulting from the GC reaction (1). Once their effector or memory fate is established, B cells escape from the GCs through the suppression of the BCL‐6‐induced “confinement factor” S1PR2 and the expression of promigratory receptors that are likely to be involved in GC exit such as EBI2 and S1PR1 [25]. The downmodulation of lymphotoxin beta transcript emerging from the GEx ROIs profiling can imply an impaired activation of the FDC meshwork by resident elements [26], which would in turn impact the maintenance of a functional GC microenvironment licensing atypical plasmablastic expansion within the GC contexture. Indeed, some cases of abrupt or florid follicular hyperplasia have been described in which activated B cells are mainly localized in the GCs, partly twisting the normal follicular architecture and even showing immunohistochemical light‐chain (oligoclonal) restriction, leading to diagnostic concern for neoplasia [27]. The perturbation of normal GC dynamics in GEx was also indicated by an overall depletion in the T‐cell compartment on transcriptional deconvolution analysis, which was similar to that of the DZ, conflicting with the memory B and plasma‐cell enrichment. In the analyzed DLBCL datasets, GEx hallmark genes characterized subgroups with positive enrichment in MCD genetics and ABC COO, indicating that GC‐centered proliferations of plasmablastic elements may be transcriptionally linked with non‐GC DLBCL. These cases characterized by a dyscrasia between cytoarchitectural, phenotypical, and topographic profile of a B‐cell expansion with plasmablastic or plasmacytic features may help in finding a link with the pathogenesis of specific DLBCL subsets [28] and reconcile an ABC‐COO with the GC microenvironment [29].
Materials and methods
Clinical setting
This study started from the incidental finding of the reactive germinotropic plasmablastic expansion, described in Results section, in the tonsil of a young patient who underwent tonsillectomy for clinical hypertrophy. Sample was obtained and handled according to the Declaration of Helsinki. Informed consent for surgery and histopathological studies was obtained from the legal representatives. The case was included in the study 05/2018 approved by the University of Palermo Institutional Review Board.
Histological, immunohistochemical, and molecular analyses
Tonsillar tissue has been formalin‐fixed and paraffin‐embedded and 3‐μm thick sections have been stained with H&E. IHC has been performed at the Pathology Department of ASST Monza, San Gerardo Hospital, Monza, Italy using a Dako Omnis platform (Dako, Denmark) using antibodies directed against CD20 (L26), CD3 (Polyclonal), Bcl2 (124), Bcl6 (PG‐B6p), CD21, ki‐67 (Mib‐1), IRF4 (MUM1), CD10, CD30 (Ber‐H2), CD138 (Mi15), HHV8 (13B10), kappa and lambda light chains. Double immunohistochemistry has been performed for MUM1 and CD10 using 3‐3'‐diaminobenzidine and 3‐amino‐9‐ethylcarbazole as chromogens, respectively. FISH study has been performed using a IRF4/DUSP22 (6p25) Break Apart kit (Kreatech, Leica Biosystem, Germany) at the Pathology Department of Spedali Riuniti di Brescia. Quantitative evaluation of immunophenotypical markers was performed by applying the HALO image analysis software (v3.2.1851.229, Indica Labs) to regions selected on whole slide digital scans acquired using an Aperio CS2 slide scanner with the ImageScope software (v12.3.28013, Leica Biosystems, Germany).
Quantitative PCR to detect clonal immunoglobulin genes rearrangement was performed after laser microdissection on H&E‐stained slides, using an LMD6 platform (Leica Microsystems, Germany).
Immunoglobulin heavy‐ and light‐chain genes rearrangement analysis
Immunoglobulin heavy‐ (IGH) and light‐chain (IGK) genes rearrangements were assessed by gene scan PCR assays (IdentiClone, InVivoScribe Technologies, San Diego, California, USA) following manufacture protocols. Briefly, PCR amplifications of DNA fragments corresponding to the VH‐JH and DH‐JH segments of the variable region of the IGH gene and to the VK‐JK and VK/intron‐Kde segments of the variable region of the IGK gene were conducted using a multiplex PCR, followed by size analysis of fluorescently labeled PCR fragments separated by capillary electrophoresis using a SeqStudio Genetic Analyzer (Applied Biosystems, Waltham, MA, USA). Fragments analysis was conducted using the Genemapper software (Life Technologies, Carlsbad, CA, USA) according to BIOMED‐2 recommendations. A monoclonal rearrangement is identified as prominent products of the same size within the expected reference size ranges, whereas polyclonal PCR products show a Gaussian size distribution. IGH and IGK clonal and polyclonal controls were included in the analyses.
Targeted next‐generation sequencing
To investigate the presence of gene mutations, the Lymphoma Solution targeted panel (Sophia Genetics, Switzerland), that covers 54 relevant genes associated with many B‐ and T‐cell lymphomas (193 kb) (Supporting information Table S1) was used on the DNA extracted from the microdissected GEx GC. Library preparation was conducted by initial DNA enzymatic fragmentation, end repair, A‐tailing, and adapter ligation, according to the manufacturer's protocol. Purified amplified libraries were captured by hybridization with the capture probes. Last, captured pools were amplified and sequenced on a MiniSeq platform (Illumina, USA; paired‐end 2 × 150 bp; high‐output kit). The percentage of target regions with coverage of 500× and 1000× was 100 and 99.99%, respectively. Single nucleotide variants and small insertions and deletions were analyzed using Sophia DDMTM software version 5.10.13 ‐ p5.5.55 (Sophia Genetics). FASTQ files were uploaded to the data portal and aligned with the human reference genome (GRCh37/hg19). After annotation in DDM, nonsynonymous variants located in exonic or ±1.2 intronic splice regions were retained, and common variants (>1% frequency) present in the 1000Genomes, ExAC, and GnomAD databases were removed. To avoid false‐positive results, mutations with a VAF lower than 10% were not considered. Integrative Genomics Viewer (Broad Institute, USA) was used for visualizing the variants aligned against the reference genome to confirm the accuracy of the variant calls by checking for possible strand biases and sequencing errors. Variants were checked in the databases ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), Varsome (https://varsome.com), and literature, and further classified using two different prediction algorithms (SIFT http://sift.bii.a‐star.edu.sg and Polyphen‐2 [PP2] http://genetics.bwh.harvard.edu/pph2/) (Supporting information Table S2).
Digital spatial profiling
The transcriptional landscape of 15 different spatially resolved ROIs of the tonsil (five peri/interfollicular ROIs, five DZ, and five LZ ROIs from morphologically normal follicles) and nine ROIs from the GEx was determined by Digital Spatial Profiling on slides stained with CD271/NGFR (as an FDC marker to highlight the LZ) and CD20 (as a B‐cell marker). The 24 selected and segmented ROIs were profiled using a GeoMx Digital Spatial Profiler (NanoString) as previously described (9), applying the Cancer Transcriptome Atlas panel (https://www.nanostring.com/products/geomx‐digital‐spatial‐profiler/geomx‐rna‐assays/geomx‐cancer‐transcriptome‐atlas/) (Supporting information Table S3).
Bioinformatics and data analysis
NanoString Technologies performed the quality control (QC) analyses and the gene expression normalization. First, sequencing QC analysis was performed on counts from each ROI, to allow identification of undersequenced samples. Biological Probe QC analysis was performed to identify outlier probes (five probes/target) to be removed from the downstream data analysis. Finally, QC analysis on raw data was performed before Q3 normalization, which highlighted adequate sequencing depth and coverage. Raw counts were normalized against the 75th percentile of signal from their own ROI and normalized data were used to perform PCA using FactoMine R package. For hierarchical clustering analysis of the ROIs, the Euclidean distance metric across samples was considered and complete aggregation method was used for building tree within the R package hclust.
The genes differentially expressed among the ROI types have been identified through the Kruskal–Wallis test (adjusted p‐values < 0.05). The gene set enrichment analysis has been carried out through the “enrichR” R package [30] considering the KEGG, Reactome Pathway, and Gene Ontology Biological Process libraries.
Differential expression analyses were carried out by applying the moderated t‐test, using the limma package [31]; pairwise comparisons between GEx and DZ/LZ/Peri ROIs were considered. Upregulated or downregulated genes were selected for subsequent analysis if their expression values were found to exceed the threshold of 0.05 FWER (Bonferroni correction).
The spatial GEx signature was assessed in the dataset published by Schmitz et al. [6] and the clinical information was downloaded from the Supporting information.
After centering and scaling expression values, unsupervised hierarchical clustering analysis based on the GEx signature was performed to identify potential discriminative clusters based on Ward.D2 method on the Euclidean distance. Survival analysis was performed using log‐rank test implemented in “survival” R package. Differences in patient characteristics among groups were analyzed with the Fisher's exact test.
CIBERSORTx (http://cibersortx.stanford.edu) [32] has been used to calculate the proportions of microenvironment cell included in the LM22 signature matrix on Schmitz et al. [6] RNA‐seq data. This dataset was downloaded and analyzed using the authors’ normalization setting which included fragments per kilobase of transcript per million space. Bulk‐mode batch correction (B‐mode) was applied to mixture samples before imputing cell fractions and 1000 permutations were set for significance. Moreover, the SpatialDecon algorithm [8] was additionally used for Nanostring data (safeTME profile matrix was applied). The differences in CIBERSORTx cell fractions among clusters were investigated using the Wilcoxon‐Mann‐Whitney test. Similarly, the differences in SpatialDecon cell fractions among ROI subgroups were investigated using the Kruskal–Wallis test.
To compare the GEx expression across different DLBCL genetic subtype groups relative to Schmitz et al. [6] and Chapuy at al. datasets [5], we have calculated a GEx hallmark gene score as the difference between the total GEx‐UP gene expression and the total GEx‐DOWN gene expression. The bootstrap t‐test [33] has been used for the pairwise comparisons among genetic subtype groups.
All statistical analyses have been performed using R statistical software (v4.0.2, http://www.R‐project.org).
Author contributions
V.L.I. and G.M.: Conceptualization, data curation, formal analysis, investigation, visualization, writing–original draft, writing–review and editing. V.C.: Resources, formal analysis, data curation, investigation, visualization. M.C.V., G.B., S.M.: Resources, data curation, software, formal analysis, and methodology. B.B. and A.M.: Resources, data curation. P.B. and L.C.: Resources, formal analysis, visualization. G.L.: Resources, formal analysis, data curation, visualization. A.D.N: Resources, investigation, methodology, formal analysis. F.F.: Resources, formal analysis, methodology, investigation, visualization. F.P.: Conceptualization, formal analysis, supervision, investigation, visualization, writing–original draft, writing–review and editing. C.T: Conceptualization, data curation, formal analysis, supervision, validation, investigation, visualization, writing–original draft, writing–review and editing. All authors contributed to the manuscript and approved the submitted version.
Conflict of interest
The authors declare no commercial or financial conflict of interest.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/eji.202149746
Abbreviations
- COO
cell of origin
- DLBCL
diffuse large B‐cell lymphoma
- DZ
dark zone
- FISH
fluorescence in situ hybridization
- GEx
germinotropic expansion
- IGH
Immunoglobulin heavy chain
- IGK
Immunnoglobulin light chain
- LZ
light zone
- PCA
Principal component analysis
- Peri
perifollicular region
- QC
quality control
- ROIs
regions of interest
- VAF
variant allele frequency
Supporting information
Supporting Information
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Acknowledgements
The Authors wish to acknowledge Prof. Maurilio Ponzoni for helpful discussion. This study has been supported by the Italian Foundation for Cancer Research (AIRC) through the IG‐2018 22145 Investigator Grant to C.T.; 5×1000 22759 Grant to C.T.; and by the Italian Ministry of Education, University and Research (MIUR) Grant 2017K7FSYB to C.T.
Open Access Funding provided by Universita degli Studi di Palermo within the CRUI‐CARE Agreement.
Contributor Information
Fabio Pagni, Email: fabio.pagni@unimib.it.
Claudio Tripodo, Email: claudio.tripodo@unipa.it.
Data availability statement
The data that support the findings of this study are available in the Supporting information of this article. Normalized gene expression data generated in the Digital Spatial Profiling experiment are available in Supporting information Table S3. Targeted next‐generation sequencing data are available in the Sequence Read Archive (ID: PRJNA818013).
References
- 1. De Silva, N. S. and Klein, U. , Dynamics of B cells in germinal centres. Nat. Rev. Immunol., 2015. 15: 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allen, C. D. , Okada, T. and Cyster, J. G. , Germinal‐center organization and cellular dynamics. Immunity 2007. 27: 190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yam‐Puc, J. C. , Zhang, L. , Maqueda‐Alfaro, R. A. , Garcia‐Ibanez, L. , Zhang, Y. , Davies, J. , Senis, Y. A. , et al. Enhanced BCR signaling inflicts early plasmablast and germinal center B cell death. iScience 2021. 24: 102038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alizadeh, A. A. , Eisen, M. B. , Davis, R. E. , Ma, C. , Lossos, I. S. , Rosenwald, A. , Boldrick, J. C. ,, et al. Distinct types of diffuse large B‐cell lymphoma identified by gene expression profiling. Nature 2000. 403: 503–511. [DOI] [PubMed] [Google Scholar]
- 5. Chapuy, B. , Stewart, C. , Dunford, A. J. , Kim, J. , Kamburov, A. , Redd, R. A. , Lawrence, M. S. , et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018. 24: 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schmitz, R. , Wright, G. W. , Huang, D. W. , Johnson, C. A. , Phelan, J. D. , Wang, J. Q. , Roulland, S. , et al. Genetics and pathogenesis of diffuse large B‐cell lymphoma. N. Engl. J. Med. 2018. 378: 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oshima, K. , Khiabanian, H. , da Silva‐Almeida, A. C. , Tzoneva, G. , Abate, F. , Ambesi‐Impiombato, A. , Sanchez‐Martin, M. , et al. Mutational landscape, clonal evolution patterns, and role of RAS mutations in relapsed acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2016. 113: 11306–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Danaher, P. , Kim, Y. , Nelson, B. , Griswold, M. , Yang, Z. , Piazza, E. and Beechem, J. M. , Advances in mixed cell deconvolution enable quantification of cell types in spatially‐resolved gene expression data. bioRxiv 2020. 10.1101/2020.08.04.235168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tripodo, C. , Zanardi, F. , Iannelli, F. , Mazzara, S. , Vegliante, M. , Morello, G. , Di Napoli, A. , et al. A spatially resolved dark‐ versus light‐zone microenvironment signature subdivides germinal center‐related aggressive B cell lymphomas. iScience 2020. 23: 101562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varano, G. , Raffel, S. , Sormani, M. , Zanardi, F. , Lonardi, S. , Zasada, C. , Perucho, L. , et al. The B‐cell receptor controls fitness of MYC‐driven lymphoma cells via GSK3β inhibition. Nature 2017. 546: 302–306. [DOI] [PubMed] [Google Scholar]
- 11. Tarte, K. , Zhan, F. , De Vos, J. , Klein, B. and Shaughnessy, J. Jr. , Gene expression profiling of plasma cells and plasmablasts: toward a better understanding of the late stages of B‐cell differentiation. Blood 2003. 102: 592–600. [DOI] [PubMed] [Google Scholar]
- 12. De Silva, N. S. , Simonetti, G. , Heise, N. and Klein, U. , The diverse roles of IRF4 in late germinal center B‐cell differentiation. Immunol. Rev. 2012. 247: 73–92. [DOI] [PubMed] [Google Scholar]
- 13. Boy, S. , van Heerden, M. , Pool, R. , Willem, P. and Slavik, T. , Plasmablastic lymphoma versus diffuse large B cell lymphoma with plasmablastic differentiation: proposal for a novel diagnostic scoring system. J. Hematopathol. 2015. 8: 3–11. [Google Scholar]
- 14. Ortega‐Molina, A. , Boss, I. W. , Canela, A. , Pan, H. , Jiang, Y. , Zhao, C. , Jiang, M. , et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 2015. 21: 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O'Connor, B. P. , Raman, V. S. , Erickson, L. D. , Cook, W. J. , Weaver, L. K. , Ahonen, C. , Lin, L. ‐ L. , et al. BCMA is essential for the survival of long‐lived bone marrow plasma cells. J. Exp. Med. 2004. 199: 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jourdan, M. , Caraux, A. , Caron, G. , Robert, N. , Fiol, G. , Rème, T. , Bolloré, K. , et al. Characterization of a transitional preplasmablast population in the process of human B cell to plasma cell differentiation. J. Immunol. 2011. 187: 3931–3941. [DOI] [PubMed] [Google Scholar]
- 17. Yoon, S. O. , Zhang, X. , Lee, I. Y. , Spencer, N. , Vo, P. and Choi, Y. S. , CD9 is a novel marker for plasma cell precursors in human germinal centers. Biochem. Biophys. Res. Commun. 2013. 431: 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Won, W. J. and Kearney, J. F. , CD9 is a unique marker for marginal zone B cells, B1 cells, and plasma cells in mice. J. Immunol. 2002. 168: 5605–5611. [DOI] [PubMed] [Google Scholar]
- 19. Xia, Y. , Xu‐Monette, Z. Y. , Tzankov, A. , Li, X. , Manyam, G. C. , Murty, V. , Bhagat, G. , et al. Loss of PRDM1/BLIMP‐1 function contributes to poor prognosis of activated B‐cell‐like diffuse large B‐cell lymphoma. Leukemia 2017. 31: 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hans, C. P. , Weisenburger, D. D. , Greiner, T. C. , Gascoyne, R. D. , Delabie, J. , Ott, G. , Müller‐Hermelink, H. K. , et al. Confirmation of the molecular classification of diffuse large B‐cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004. 103: 275–282. [DOI] [PubMed] [Google Scholar]
- 21. Swerdlow, S. H. , Campo, E. , Harris, N. L. , Jaffe, E. S. , Pileri, S. A. , Stein, H. , Thiele, J. , et al. WHO classification of tumours of haematopoietic and lymphoid tissues, 4th ed., IARC, 2 Lyon, France, 2017. [Google Scholar]
- 22. Hunter, Z. R. , Xu, L. , Yang, G. , Tsakmaklis, N. , Vos, J. M. , Liu, X. , Chen, J. , et al. Transcriptome sequencing reveals a profile that corresponds to genomic variants in Waldenström macroglobulinemia. Blood 2016. 128: 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maezawa, Y. , Nakajima, H. , Suzuki, K. , Tamachi, T. , Ikeda, K. , Inoue, J. , Saito, Y. , et al. Involvement of TNF receptor‐associated factor 6 in IL‐25 receptor signaling. J. Immunol. 2006. 176: 1013–1018. [DOI] [PubMed] [Google Scholar]
- 24. Ahonen, C. , Manning, E. , Erickson, L. D. , O'Connor, B. , Lind, E. F. , Pullen, S. S. , Kehry, M. R. , et al. The CD40‐TRAF6 axis controls affinity maturation and the generation of long‐lived plasma cells. Nat. Immunol. 2002. 3: 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Green, J. A. and Cyster, J. G. , S1PR2 links germinal center confinement and growth regulation. Immunol. Rev. 2012. 247: 36–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Myers, R. C. , King, R. G. , Carter, R. H. and Justement, L. B. , Lymphotoxin α1β2 expression on B cells is required for follicular dendritic cell activation during the germinal center response. Eur. J. Immunol. 2013. 43: 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gars, E. , Butzmann, A. , Ohgami, R. , Balakrishna, J. P. and O'Malley, D. P. , The life and death of the germinal center. Ann. Diagn. Pathol. 2020. 44: 151421. [DOI] [PubMed] [Google Scholar]
- 28. Wright, G. W. , Huang, D. W. , Phelan, J. D. , Coulibaly, Z. A. , Roulland, S. , Young, R. M. , Wang, J. Q. ,, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell 2020. 37: 551–568.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kotlov, N. , Bagaev, A. , Revuelta, M. V. , Phillip, J. M. , Cacciapuoti, M. T. , Antysheva, Z. , Svekolkin, V. , et al. Clinical and biological subtypes of B‐cell lymphoma revealed by microenvironmental signatures. Cancer Discov. 2021. 11: 1468–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xie, Z. , Bailey, A. , Kuleshov, M. V. , Clarke, D J B. , Evangelista, J. E. , Jenkins, S. L. , Lachmann, A. , et al. Gene set knowledge discovery with Enrichr. Current Protocols 2021. 1: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ritchie, M. E. , Phipson, B. , Wu, D. , Hu, Y. , Law, C. W. , Shi, W. , Smyth, G. K. , et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic. Acids. Res. 2015. 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rusk, N. , Expanded CIBERSORTx. Nat. Methods 2019. 16: 577. [DOI] [PubMed] [Google Scholar]
- 33. Efron, B. , Tibshirani, R. and Tibshirani, R. J. , An introduction to the bootstrap. Chapman & Hall/CRC monographs on statistics and applied probability, Chapman & Hall/CRC, Philadelphia, PA, 1994. 10.1007/978-1-4899-4541-9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Table S1
Table S2
Table S3
Table S4
Table S5
Table S6
Table S7
Table S8
Table S9
Data Availability Statement
The data that support the findings of this study are available in the Supporting information of this article. Normalized gene expression data generated in the Digital Spatial Profiling experiment are available in Supporting information Table S3. Targeted next‐generation sequencing data are available in the Sequence Read Archive (ID: PRJNA818013).