

Abstract
Introduction
Allergen exposure worsens viral‐triggered asthma exacerbations and could predispose the host to secondary bacterial infections. We have previously demonstrated that exposure to house dust mite (HDM) reduced TLR‐3‐induced IFN‐β in human bronchial epithelial cells (HBECs) from healthy donors. We hypothesize that HDM sensitization in different ways may be involved in both viral and bacterial resistance of HBECs in asthma. In this study, the role of HDM sensitization and effects of HDM exposure on viral stimulus‐challenged HBECs from asthmatic donors have been explored with regard to expression and release of molecules involved in anti‐viral and anti‐bacterial responses, respectively.
Methods
HBECs from HDM‐sensitized (HDM+) and unsensitized (HDM‐) patients with asthma were used. HBECs were exposed to HDM or heat inactivated (hi)‐HDM (20 μg/ml) for 24 h prior to stimulation with the viral infection mimic, Poly(I:C), for 3 or 24 h. Samples were analyzed with ELISA and RT‐qPCR for β‐defensin‐2, IFN‐β, TSLP, and neutrophil‐recruiting mediators: IL‐8 and TNF‐⍺. NFκB signaling proteins p105, p65, and IκB‐⍺ were analyzed by Western blot.
Results
Poly(I:C)‐induced IFN‐β expression was reduced in HBECs from HDM + compared to HDM‐ patients (p = 0.05). In vitro exposure of HBECs to HDM furthermore reduced anti‐microbial responses to Poly(I:C) including β‐defensin‐2, IL‐8, and TNF‐⍺, along with reduced NFκB activity. This was observed in HBECs from asthma patients sensitized to HDM, as well as in non‐sensitized patients. By contrast, Poly (I:C)‐induced release of TSLP, a driver of T2 inflammation, was not reduced with exposure to HDM.
Conclusion
Using HBECs challenged with viral infection mimic, Poly(I:C), we demonstrated that allergic sensitization to HDM was associated with impaired anti‐viral immunity and that HDM exposure reduced anti‐viral and anti‐bacterial defense molecules, but not TSLP, across non‐allergic as well as allergic asthma. These data suggest a role of HDM in the pathogenesis of asthma exacerbations evoked by viral infections including sequential viral‐bacterial and viral‐viral infections.
Keywords: allergy, asthma, bronchial epithelium, exacerbation
In this study, we investigate the role of HDM sensitization and effects of HDM exposure on viral mimic‐challenged HBECs from asthmatic donors. In vitro exposure of viral mimic‐challenged HBECs to HDM reduces IFN‐β and anti‐microbial agents including β‐def‐2, IL‐8, and TNF‐α, but not TSLP, across non‐allergic as well as allergic asthma. Data suggest that both HDM sensitization and acute exposure to HDM contribute to reducing infection‐evoked immune protection in asthma and thus increase the risk of symptomatic infections and exacerbations.Abbreviations: β‐def‐2, β‐defensin‐2; HBECs, human bronchial epithelial cells; HDM, house dust mite; IFN‐β, interferon‐β; MDA‐5, melanoma differentiation‐associated protein; NF‐κB, nuclear factor‐kappa B; Poly (I:C), polyinosinic‐polycytidylic acid; RIG‐I, retinoic acid‐inducible gene I; TLSP, thymic stromal lymphopoietin
Abbreviations
- COPD
Chronic obstructive pulmonary disease
- FENO
Fractional exhaled nitric oxide
- FEV
Forced expiratory volume in first second
- FVC
Forced vital capacity
- HBEC
Human bronchial epithelial cells
- HDM
House dust mite
- HI
Heat inactivated
- IC
Inhaled corticosteroids
- IFN
Interferon
- IL‐8
Interleukin‐8
- IRF
Interferon regulatory factor
- IκB
Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor
- MDA5
Melanoma differentiation‐associated protein
- NF‐κB
Nuclear factor‐kappa‐light‐chain‐enhancer of activated B cells
- PRR
Pattern recognition receptors
- RIG‐I
Retinoic acid‐inducible gene I
- RV
Rhinovirus
- TLR
Toll‐like receptor
- TNF
Tumor necrosis factor
- TSLP
Thymic stromal lymphopoietin
1. INTRODUCTION
Asthma is a chronic airway disease affecting about 8% of the European adult population, and with an increasing prevalence among young adults. 1 Asthma is a heterogeneous disease, and allergy with sensitization to aeroallergens such as house dust mite (HDM) is a significant contributing factor in a majority of patients. HDM is highly immunogenic due to its interaction with the innate immune system. For example, as HDM and its fecal pellets hold compounds including lipopolysaccharide and β‐glucans that can bind to pattern recognition receptors (PRRs). 2 Further, HDM contains several proteases, which can affect directly epithelial membrane integrity or activate protease‐activated receptors. 3 Due to the heterogeneous immune properties of HDM aeroallergens, effects may be seen in non‐allergic as well as allergic asthma. 4
Although most patients with asthma suffer from a mild to moderate disease, 5%–10% develop a severe disease with frequent exacerbations despite high doses of inhaled corticosteroids. Rhinovirus (RV) infection is estimated to trigger up to 80% of asthma exacerbations. 5 RV primarily targets human bronchial epithelial cells (HBECs), which are considered central responders in airways innate immunity. 6 During replication, RV produces intermediate, double‐stranded‐(ds)‐RNA molecules responsible for initiating the biological effects of the infection. 7 dsRNA is detected by PRRs, including toll‐like receptor‐3 (TLR‐3), MDA5, and RIG‐I, which activate downstream transcription factors, including interferon regulatory factor (IRF)‐3 and nuclear factor‐kappa B (NF‐κB). 8 This leads to the production of anti‐viral interferon‐β (IFN‐β) and other important proteins involved in healthy epithelium host defense. However, in patients with asthma, RV infection, or dsRNA exposure, causes overexpression and overproduction of TSLP. 9 , 10 Based on the data from a clinical trial using intervention monoclonal antibodies targeting TSLP, this cytokine seems critically involved in asthma exacerbations. 11 , 12
In response to RV infection, an anti‐microbial innate immune response is triggered in the bronchial mucosa with increased production of IFNs including IFN‐β. 13 , 14 Several studies have demonstrated that viral stimulus‐induced anti‐viral IFN‐β is reduced in HBECs from patients with asthma compared with healthy individuals. 9 , 15 RV infection also induces bronchial epithelial release of several anti‐microbial proteins from HBECs that potentially limit further adjoining infections. 5 Among pro‐inflammatory, epithelium‐derived cytokines, IL‐8, and TNF‐α have a crucial role in the activation of neutrophils. 16 Other important epithelial mediators include β‐defensin‐2, an anti‐microbial peptide produced in response to RV infection, and previously shown to be associated with chronic obstructive pulmonary disease (COPD) exacerbations. 17 Additionally, a decreased release of β‐defensin‐2 in atopic dermatitis (a type‐2 mediated disease) was shown to be associated with increased risk of infections. 18 Despite the capacity of inducing anti‐microbial molecules, studies have highlighted that RV infection in asthma may actually increase the risk of getting a secondary bacterial airway infection. 19 , 20 , 21 , 22 The mechanisms underlying this association are currently unknown, but exposure to allergens that may affect the bronchial epithelial anti‐microbial response to viral infection may be one link. 27 , 28 This consideration is also underscored by clinical observations. 23
As suggested by microbiome studies, the lungs of patients with asthma have an increased prevalence of colonizing pathogenic bacteria compared with healthy subjects. 24 It has further been demonstrated that patients with allergic asthma have higher prescription rates of antibiotics than patients with non‐allergic asthma, which is also linked with the severity of the disease. 23 Supporting an important role of allergy, children sensitized to HDM have a defective antibody response toward bacteria. 25 Hales et al. (2009) also demonstrated an association between bacterial antibodies in HDM asthma and asthma exacerbations, apparently independent of prior viral infection. 26 In a previous work, we have demonstrated that HDM stands out among other allergens (Alternaria alternata, Artemisia vulgaris, and Betula pendula) 27 in that 24 h exposure to HDM of the bronchial epithelial cell‐line BEAS‐2B caused reduced IFN‐β response to viral stimulation. 28
We do not know how the effect of HDM and HDM sensitization is affecting the immune reactivity of HBECs from patients with asthma after viral infection. Hence, the aim of the study was to investigate HBECs from a clinically defined cohort of patients with asthma, the effects of HDM on anti‐viral, anti‐bacterial, and TSLP response after dsRNA challenges. We hypothesized that patients sensitized to HDM may have an impaired interferon response. Further, we hypothesized that HDM‐sensitized patients may have a decreased viral and bacterial resistance, without reducing epithelial indices of poor tolerance to viral infection, exemplified here by epithelial expression of TSLP.
2. MATERIALS AND METHODS
2.1. Study population
Twenty patients with asthma were randomly selected from the patients included in the UPSTREAM study, 29 and were included in the present study (Table 1). The asthma diagnosis was confirmed according to GINA criteria. 30 All patients had a positive mannitol challenge test at inclusion, and had moderate to severe asthma according to GINA severity (2018). Before undergoing bronchoscopy patients were assessed with measures of lung function, fractional exhaled nitric oxide (FeNO) and blood and induced sputum cell differential count (Table 1). A skin prick test to 10 aeroallergens (birch [Betula species], grass [Phleum pratense] mugwort, horse, dog, cat [Felis domesticus], house dust mite [Der p 1 and Der f 2], and fungi [Alternaria and Cladosporium species]; (ALK‐Abello, Hørsholm, Denmark) was performed (Table 1). Allergic sensitization to HDM (HDM+) was defined as a wheal of at least 3mm to either Der p1 and/or Der f2 (dia1+dia2/2) independent of whether patients reported to be symptomatic with exposure or not. Atopy was defined as a positive skin prick test to the common allergens exposed before (Table 1). All subjects provided informed consent prior to inclusion. The approval was obtained from the local ethical review board (H‐16002008) in Copenhagen. 29
TABLE 1.
Clinical and characteristics of study population
| All | HDM + | HDM ‐ | p‐value | |
|---|---|---|---|---|
| N | 20 | 7 | 13 | – |
| Age years (mean (range)) | 40.9 (20–73) | 32.4 (20–60) | 43.0 (23–73) | n.s. |
| Sex (M/F) | 8/12 | 2/5 | 5/8 | n.s. |
| Allergic sensitization | ||||
| Atopy (Yes/No) | 12/8 | 7/0 | 5/8 | 0.0074 |
| House dust mite (Yes/No) | 7/13 | 7/0 | 0/13 | <0.0001 |
| A. alternata (Yes/No) | 0/20 | 0/7 | 0/13 | n.s. |
| C. herbarum (Yes/No) | 0/20 | 0/7 | 0/13 | n.s. |
| A. vulgaris (Yes/No) | 4/16 | 2/5 | 2/11 | n.s. |
| B. pendula (Yes/No) | 6/14 | 3/4 | 3/10 | n.s. |
| P. pretense (Yes/No) | 8/12 | 5/2 | 3/10 | 0.0353 |
| Cat (Yes/No) | 7/13 | 5/2 | 2/11 | 0.0122 |
| Dog (Yes/No) | 7/13 | 3/4 | 4/9 | n.s. |
| Horse (Yes/No) | 2/18 | 1/6 | 1/12 | n.s. |
| FEV1 (% predicted) | 90.0 (82.5–101.5) | 86.0 (80.0–92.0) | 94.0 (83.0–104.0) | n.s. |
| FVC (% predicted) | 100.5 (93.0–116.5) | 101.0 (88.0–108.0) | 100.0 (94.5–118.5) | n.s. |
| FEV1/FVC (% predicted) | 73.5 (70.2–78.2) | 74.0 (68.0–76.0) | 73.0 (70.5–79.5) | n.s. |
| PD15 (mg) | 130.4 (37.4–206.3) | 37.5 (27.9–125.7) | 146.4 (104.9–214.8) | 0.0456 |
| ACQ6 | 2.1 (1.7–2.7) | 2.7 (1.7–3.5) | 2.0 (1.6–2.6) | n.s. |
| ICS dose (μg/day) | 1200 (800–1600) | 1200 (800–1600) | 1000 (800–1600) | n.s. |
| FeNO (ppb) | 24.5 (13.4–46.4) | 32.8 (17.2–62.1) | 22.7 (10.6–35.7) | n.s. |
| IgE (IU/ml) | 47.0 (23.0–197.0) | 110.0 (35.0–271.0) | 40.0 (15.5–189.5) | n.s. |
| Blood Eosinophils (x109/L; abs) | 0.30 (0.14–0.44) | 0.32 (0.14–0.48) | 0.26 (0.13–0.38) | n.s. |
| Blood Neutrophils (x109/L; abs) | 4.45 (2.65–5.65) | 5.20 (4.50–6.1) | 3.90 (2.40–4.75) | n.s. |
| Sputum Eosinophils (%) | 1.5 (0.37–16.5) | 19.5 (4.3–21.6) | 1.2 (0.16–7.2) | 0.0339 |
| Sputum Neutrophils (%) | 46.5 (21.7–75.7) | 63.5 (33.4–80.9) | 45.9 (20.0–75.9) | n.s. |
Data are presented as median value (Interquartile range), unless otherwise expressed.
Mann–Whitney U‐test for continuous variables and chi‐squared test for categorical variables.
Abbreviations: ACQ6, Asthma Control Questionnaire; FeNO, fractional exhaled nitric oxide; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; HDM, house dust mite; ICS, inhaled corticosteroids; n.s, not significant; PD15, Dose of mannitol required to reduce the FEV1 by 15% of the baseline value.
2.2. Human bronchial epithelial cell cultures
HBECs from the selected patients with asthma were obtained during the bronchoscopy by bronchial brushing (bronchoscope: Olympus BF‐1TQ180/BF‐1TH190, Olympus, Hamburg, Germany) with standard sterile‐sheared nylon cytology brushes, as previously described. 31 HBECs were cultured in bronchial epithelial growth medium (BEGM, Lonza) supplemented with SingleQuots (Lonza) but gentamycin was replaced with 0.1% Primocin (InvivoGen). Cells were cultured in standard conditions (5% CO2 and 37° C). At passage 2, cells were seeded into collagen coated 12‐well plates in BEGM medium and were grown to 70%–80% confluence. Then, HBECs were challenged with HDM (20 ug/mL; Greer) extract or heat inactivated‐HDM (hi‐HDM; 30 minutes at 65°C) in BEGM medium for 24 h, as previously described. 28 Cells were then stimulated with polyinosinic:polycytidylic acid (Poly(I:C); dsRNA)) for 3 or 24 h (Figure S1). The growth medium was then replaced with bronchial epithelial basal medium supplemented with 1% insulin‐transferrin‐selenium (Gibco) and 1.2% bovine serum albumin (Millepore).
2.3. In vitro TLR‐3, MDA5, and RIG‐I stimulation
In this study, we used polyinosinic:polycytidylic acid (Poly(I:C); dsRNA)) as a viral mimic. During replication, RV produces intermediate, dsRNA molecules that bind to TLR‐3 within the endosomes or MDA5/RIG‐I in the cytoplasm. RV‐induced production of interferons and pro‐inflammatory cytokines are mainly dependent on the dsRNA rather than the viral proteins, indicating that UV‐inactivated RV does not induce any response in epithelial cells. 10 By using TLR‐3 agonist Poly(I:C) high molecular weight (InvivoGen) (10 μg/mL) or the MDA5/RIG‐I agonist Poly(I:C)‐lyovec (InvivoGen) (0.25 μg/ml), we mimic RV infection with a lower variability in the cytokine response compared with a RV infection to obtain more standardized experiments.
2.4. RNA extraction and mRNA analysis
Total RNA was extracted from HBECs by using an RNA extraction kit (Nucleospin® RNA II, Macherey‐Nagel) following commercial protocol. Briefly, 1 μg of RNA was reverse transcribed to cDNA (Precision Nanoscript 2 Reverse Transcription Kit, PrimerDesign), and real‐time quantitative PCR was performed on a Mx3005P qPCR system (Stratagene) using standard cycling parameters. Primers were obtained from PrimerDesign and Qiagen (Table S1). Samples were analyzed by the ΔCt method and normalized to UBC/GAPDH expression.
2.5. Immunoblotting
The protein expression of NFκ‐B p105, NFκB p65, IκB‐α, and GAPDH was quantified by Western blot. Cells were lysed in a lysis buffer containing 1% TritonX‐100, 10 mM Tris‐HCl, 50 mM NaCl, 5 mM EDTA, 30 mM sodium pyrophosphate, 50 mM NaF, 0.1 mM Na3VO4, and protease and phosphatase inhibitors (Sigma‐Aldrich). Protein concentrations were determined by BCA protein assay (Pierce Thermo Scientific) for each sample and equal amounts of protein were loaded on a 5%–10% TGX stain‐free gel (Bio‐Rad Laboratories AB), before blotting on a Trans‐Blot Turbo Transfer System (Bio‐Rad Laboratories AB). This was followed by blocking of the membrane in 5% BSA in Tris‐buffered (5% Tween‐20) and overnight incubation at 4°C with primary antibodies from Cell Signaling Technology (anti‐ NFκ‐B p105 Rabbit mAb, NFκ‐B p65 Rabbit mAb, IκB‐α Rabbit mAb, anti‐GAPDH Rabbit mAb). IκB‐α were stripped for GAPDH with Western blot stripping buffer (Thermo). Then, the membrane was washed and incubated for 1 h with secondary antibodies (anti‐Rabbit IgG HRP‐linked Ab; Cell Signaling Technology). Detection was performed by chemiluminescence using Clarity MAX ECL substrate (Bio‐Rad Laboratories AB), and immunoblots were visualized by LI‐COR Odyssey Fc Imager (LI‐COR Biosciences).
2.6. ELISA
IL‐8, TNF‐α, and TSLP were measured in cell supernatants using DuoSet ELISA from R&D systems. β‐defensin‐2 was measured using ELISA‐kit from Phoenix Pharmaceuticals Inc. All protocols were performed according to manufactures instructions and compared with a standard curve.
2.7. Statistics
Data are presented as median (interquartile range). Mann–Whitney U‐test was used for comparison between two groups. For comparison between more than two groups, Kruskal–Wallis with Dunn´s multiple comparisons test was performed. p‐values of <0.05 were considered statistically significant. All statistical analyses were performed using GraphPad Prism version 9.0 software (GraphPad Software).
3. RESULTS
3.1. HDM sensitization and exposure impairs Poly(I:C)‐induced bronchial epithelial IFN‐β expression in patients with asthma
We have previously shown that HDM exposure affects TLR3‐mediated induction of ΙFNs after Poly (I:C) stimulation HBEC from healthy individuals and in a mouse model of asthma exacerbation. 28 Expanding these previous results, we have now included primary HBECs from asthmatic patients with (HDM +) or without (HDM –) sensitization to HDM (Table 1) to evaluate the combined effect of HDM sensitization and the viral mimic Poly(I:C) on IFN‐β expression. Demographics and clinical characteristics of the study population are presented in (Table 1).
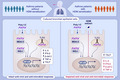
Stimulation of HBECs with Poly(I:C) alone increased IFN‐β expression after 24 h stimulation whereas this was not the case for HDM or hi‐HDM alone (Figure 1A). Further, pre‐treatment with HDM resulted in a down‐modulation of IFN‐β gene expression in response to 24 h Poly(I:C)‐stimulation in both HDM + and HDM‐ patients (Figure 1B). Strikingly, HDM + patients exhibited a diminished IFN‐β expression in response to Poly(I:C) compared with HDM – patients at 24 h (p = 0.011; Figure 1B). The same result was obtained when comparing HDM + patients with non‐atopic patients (negative sensitization to all allergens measured) (Figure S2A). However, no effect on Poly (I:C)‐induced IFN‐β expression was seen between atopic vs non‐atopic patients, suggesting this effect was specific for HDM sensitization (Figure S2B).
FIGURE 1.
Bronchial epithelial cells from patients with asthma were exposed to HDM or heat inactivated (hi)‐HDM (20μg/ml) for 24 h, then stimulated with the viral mimic Poly(I:C) (A). Cells were then divided into HDM negative (HDM‐; n = 2–13) and HDM positive (HDM+; n = 6–7) (B). Gene expression of IFN‐β 24 h (A‐B) after Poly(I:C). Samples were analyzed by the ΔCt method and normalized to UBC/GAPDH expression. Data are presented as median (interquartile range)
3.2. Poly (I:C)‐induced production of IL‐8, TNF‐⍺, and β‐defensin‐2 is impaired by HDM exposure in HBECs, independently of HDM sensitization
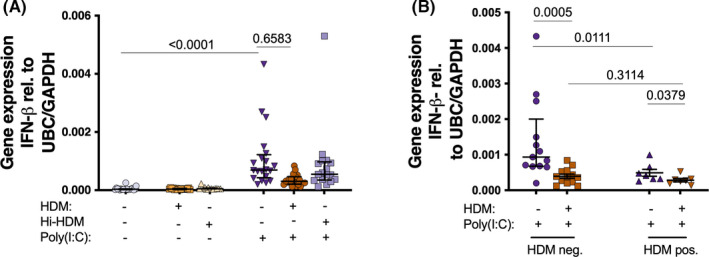
Next, we studied the role of HDM on the HBECs production of several cytokines and chemokines involved in anti‐bacterial immunity after viral stimulation. IL‐8 and TNF‐α, crucial molecules in the neutrophilic response against virus and bacteria, as well as the anti‐microbial peptide β‐defensin‐2, were found increased in HBECs after 24 h Poly(I:C) stimulation both at gene (Figure 2A–C) and protein level (Figure 2D–F). HBECs anti‐bacterial response to Poly(I:C) was clearly impaired after 24 h pre‐treatment with HDM, both at gene (Figure 2A–C) and protein levels (Figure 2D–F). A similar induction of IL‐8, TNF‐α, and β‐defensin‐2 was found after 3 h Poly(I:C) stimulation, but HDM pre‐treatment had no effect at this early time point (Figure S3A–C). Of note, pre‐treatment with hi‐HDM did not affect the Poly(I:C) induction of IL‐8, TNF‐α, and β‐defensin‐2 at 24 h (Figure 2A–F), suggesting that HDM‐mediated effects are dependent on HDM‐associated heat‐sensitive components. Interestingly, the decreasing effect of HDM pre‐treatment on HBECs anti‐bacterial response to 24 h Poly(I:C) was similar in both HDM + and HDM – patients (Figure 2G–I). Moreover, the 24 h Poly(I:C)‐ induction of IL‐8, TNF‐α, and β‐defensin‐2 in HBECs not exposed to HDM in vitro was not influenced by HDM sensitization, or atopy (Figure 2G–I, S2C‐H). We only observed a higher gene expression of IL‐8 in HDM – compared with HDM + patients after 3 h of Poly(I:C) stimulation (Figure S3D–F).
FIGURE 2.
Bronchial epithelial cells from patients with asthma were exposed to HDM or heat inactivated (hi)‐HDM (20 μg/ml) for 24 h, then stimulated with the viral mimic (Poly(I:C)) (10 μg/ml) for 24 h. 24 h gene expression of IL‐8 (A), TNF‐α (B), and β‐defensin‐2 (C) after Poly(I:C) (n = 20 except hi‐HDM n = 16). Protein levels of IL‐8 (D), TNF‐α (E), and β‐defensin‐2 (F) 24 h after Poly(I:C) (n = 7 except hi‐HDM n = 3). Cells were also divided into HDM negative (HDM‐; n = 13) and HDM positive (HDM+; n = 7) cells (G‐I) with 24 h gene expression of IL‐8 (G), TNF‐α (H), and β‐defensin‐2 (I). Samples were analyzed by the ΔCt method and normalized to UBC/GAPDH expression. Data are presented as median (interquartile range)
3.3. Poly(I:C)‐induced production of TSLP in HBECs from patients with asthma is not modified by HDM exposures or HDM sensitization
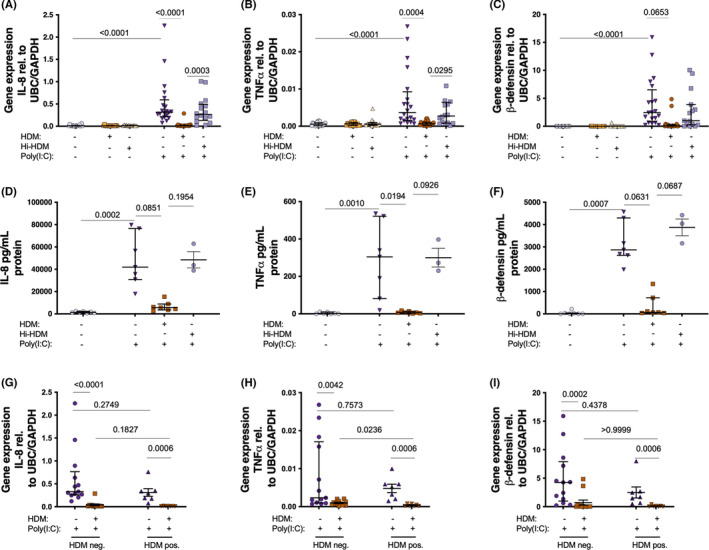
TSLP is an alarmin suggested to be involved in asthma exacerbations. 9 , 12 Hence, we wanted to study the effect of HDM on viral stimulus‐induced TSLP expression. Both 3 h gene expression (there was no Poly(I:C)‐induced TSLP at 24 hours) and 24 h protein expression of TSLP was increased after Poly (I:C) stimulation (Figure 3A,B). However, 24 h pre‐treatment with HDM did not affect Poly(I:C)‐induced TSLP expression neither in the overall study population (Figure 3A,B), nor after dividing the patients in HDM + and HDM – groups (Figure 3C). There was no difference in Poly(I:C)‐induced TSLP expression in HBECs between HDM + vs HDM‐ patients (Figure 3C).
FIGURE 3.
Bronchial epithelial cells from patients with asthma were exposed to HDM or heat inactivated (hi)‐HDM (20 μg/ml) for 24 h, then stimulated with the viral mimic Poly(I:C) (10 μg/ml) for 3 h and 24 h. Cells were then divided according to HDM negative (HDM‐; n = 12) and HDM positive (HDM+; n = 7) patients. 3h gene expression of TSLP (A, C), 24 h, protein expression of TSLP (B). n = 20 except hi‐HDM n = 16 in (A, B). Samples were analyzed by the ΔCt method and normalized to UBC/GAPDH expression. Data are presented as median (interquartile range)
3.4. HDM‐mediated down‐modulation of anti‐viral and anti‐bacterial defense is not dependent on PRR expression
Rhinovirus infection, as well as the viral replication mimic Poly (I:C) (dsRNA) are TLR‐3 agonists, but they also activate the cytosolic dsRNA sensors MDA5 and RIG‐I. A direct modification of these PRRs expression could be one potential explanation of the impaired anti‐bacterial and anti‐viral response in HDM‐challenged HBECs. Therefore, we measured the expression of TLR‐3, MDA5, and RIG‐I after 24 h Poly (I:C) stimulation in HBECs pre‐treated with or without HDM or hi‐HDM.
As expected, 24 h Poly(I:C) stimulation of HBECs increased gene expression of TLR‐3, MDA5, and RIG‐I (Figure 4A–C). However, no effect of HDM or hi‐HDM pre‐treatment on the magnitude of induction for the three PRRs was observed (Figure 4A–C).
FIGURE 4.
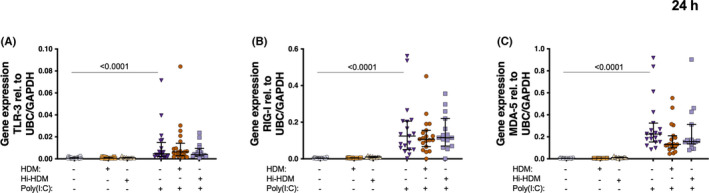
Bronchial epithelial cells from patients with asthma were exposed to HDM or heat inactivated (hi)‐HDM (20 μg/ml) for 24 h, then stimulated with the viral mimic (Poly(I:C)) (10 μg/ml) for 24 h. Gene expression of (A) TLR‐3, (B) MDA5 (C) RIG‐I at 24 h after Poly(I:C). n = 20 except hi‐HDM n = 16. Samples were analyzed by the ΔCt method and normalized to UBC/GAPDH expression. Data are presented as median (interquartile range)
3.5. HDM‐mediated down‐modulation of anti‐viral and anti‐bacterial defense occurs in a TLR‐3 dependent manner
To further study the involvement of MDA5 and RIG‐I in the HDM‐dependent down‐modulation of IFN‐β and pro‐inflammatory mediator expression after viral stimulation, we use Poly(I:C)‐lyovec (MDA5/RIG‐I agonist) as an agonist for these receptors. 32
The MDA5/RIG‐I agonist Poly(I:C)‐lyovec produced similar effects to Poly(I:C) on the expression of IL‐8, TNF‐α, and IFN‐β in HBECs (Figure 5A–C). However, the expression of both pro‐inflammatory and anti‐viral mediators was not changed with HDM pre‐treatment, suggesting that the HDM‐mediated decrease of these anti‐viral and neutrophilic molecules is TLR‐3 dependent (Figure 5A–C).
FIGURE 5.
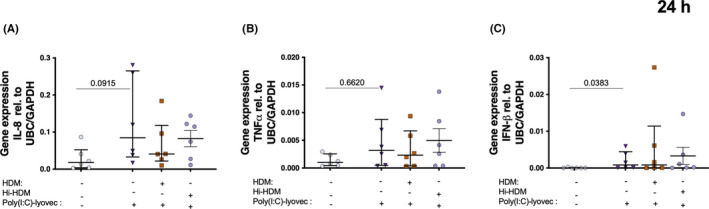
Bronchial epithelial cells from patients with asthma were exposed to HDM or heat inactivated (hi)‐HDM (20μg/ml) for 24 h, then stimulated with the viral mimic dsRNA‐Lyovec for 24 h. Gene expression of (A) IL‐8 and (B) TNF‐⍺ n = 6. Samples were analyzed by the ΔCt method and normalized to UBC/GAPDH expression. Data are presented as median (interquartile range)
3.6. Heat‐sensitive components in HDM impair Poly(I:C) activation of NFκB
Since NFκB is one of the main transcription factors involved in the regulation of anti‐bacterial and anti‐viral responses of HBEC’s downstream of TLR‐3 signaling, we wanted to investigate the interaction between Poly(I:C) and HDM in the context of NF‐κB activation. Poly(I:C) stimulation alone, both increased phosphorylation of p105 and p65 NFκB family members and trended to decrease IκB‐α (negative regulator) expression (Figure 6A–D). Pre‐treatment with HDM, on the other hand, decreased Poly(I:C)‐induced phosphorylation of both p105 and p65. However, Hi‐HDM did not affect Poly(I:C)‐induced phosphorylation of p105 and p65 (Figure 6A–D).
FIGURE 6.
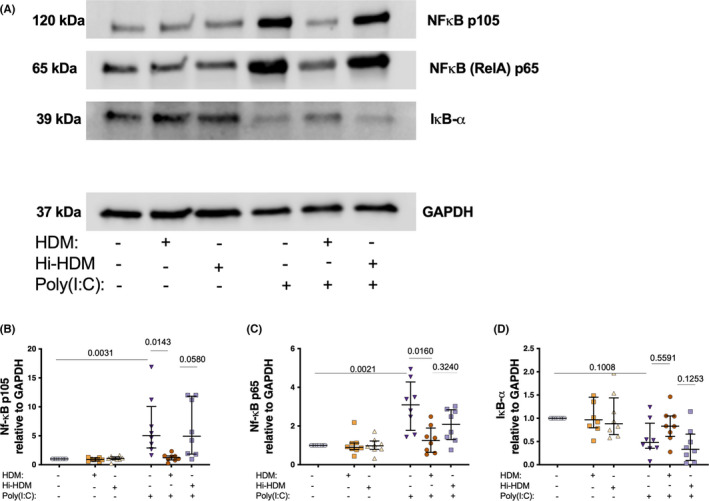
Bronchial epithelial cells from patients with asthma were exposed to HDM or heat inactivated (hi)‐HDM (20 μg/ml) for 24 h, then stimulated with the viral mimic dsRNA (Poly(I:C) 10 μg/mL) for 24h (A) Immunoblot of NfκB p105, NFκB p65, IκB‐α, and GAPDH from primary cell lysate after 24 h. Quantification of NfκB p105 (B), NFκB p65 (C), and IκB‐α (D). n = 6. Data are presented as median (interquartile range)
4. DISCUSSION
Using human bronchial epithelial cells from patients with asthma, this study demonstrated that in vitro exposure to HDM reduced Poly(I:C)‐induced expression independently of HDM sensitization of airway anti‐viral and anti‐bacterial mediators IFN‐β, TNF‐ α, IL‐8, and β‐defensin‐2. As further suggested by the present observations, NFκB may be causally involved in this reductive effect. We further demonstrated that HDM sensitization alone was associated with reduced responses to Poly(I:C), especially regarding HBECs expression of anti‐viral IFN‐β. In contrast, Poly(I:C)‐induced release of an upstream T2 pathogenic cytokine, TSLP, from HBECs was not altered by the HDM exposure. The present data on innate immune responses of bronchial epithelium in asthma are of interest with regard to risk factors contributing to secondary airway infections following an initial rhinovirus infection.
We have previously demonstrated that HDM impairs viral stimulus‐induced production of IFN‐β in a bronchial epithelial cell‐line (BEAS‐2B) and HBEC from healthy individuals. 28 In the present study, we demonstrated that in vitro exposure to HDM of HBECs from patients with asthma resulted in a dampening of Poly(I:C)‐induced IFN‐β expression in both HDM + and HDM– patients. However, HDM + patients displayed a reduced Poly(I:C)‐induced epithelial IFN‐β expression compared with HDM– patients. Such a phenotype‐dependent IFN‐β response may prompt further research to potentially explain contradictory results regarding the deficient epithelial IFN‐β expression after viral stimulation in different cohorts of patients with asthma. 33 In line with our results, previous studies have shown that T2 cytokines added to HBECs cause a deficient IFN‐β response to viral stimulation. 34 Indeed, the latter type of interactions has underpinned the idea that T2 inflammation facilitates asthma exacerbations through reducing resistance to viral infections. 35 However, host's‐tolerance to T2 cytokines (e.g., IL‐4, IL‐13, and TSLP), produced during viral infections, may be even more important in asthma exacerbations 9 , 36 ; for example, the pathogenic effects of TSLP, which is overproduced during viral infections, 9 may suffice to contribute to asthma exacerbations. 6 , 11 In this study, we confirm viral stimulus‐induced epithelial expression of TSLP. 9 We further demonstrate that exposure to HDM inhibited Poly(I:C)‐induced increased expression/release of several cytokines and that this occurred without reducing TSLP release, which is known to be regulated by several different transcription factors. 37
Type I IFNs, prominently represented by IFN‐β, are a cytokine family considered essential for early innate anti‐viral defense of the respiratory tract. 33 However, IFN‐β also exhibits direct anti‐bacterial functions, 38 which is in line with a focus of this study. We have thus analyzed gene expression and release of additional molecules that are induced by Poly(I:C) challenge of HBECs, which are likely involved in airway anti‐bacterial activity. Among them, β‐defensin‐2, which has attracted interest as an anti‐microbial peptide with bactericidal properties. 17 Differing from an anti‐microbial peptide such as cathelicidin, which has appeared in challenged asthmatic airways as a result of plasma exudation mechanisms, 39 defensins are likely produced by bronchial epithelium in asthma. 40 Baines et al reported that sputum defensin levels in asthma were independent of inflammatory phenotype. 40 In line with this, we evidenced equal baseline and equal Poly(I:C)‐induced β‐defensin‐2 expression in HBECs from HDM +and HDM – patients. Also, the present 24 h exposure to HDM inhibited the Poly(I:C)‐induced defensin equally in HBECs from each phenotype where the gene expression and release of defensin were practically abolished, suggesting that participation of defensin in anti‐bacterial defense can be severely compromised in asthmatic individuals following exposure to HDM. In the same line, previous studies have shown that T2 cytokines have a suppressive effect on NFκB mediated expression of β‐defensins‐2. 18
β‐defensin‐2 also works as an alarmin, promoting T cell and dendritic cell‐mediated inflammation. 41 Similarly, other molecules analyzed in this study, IL‐8 and TNF‐⍺ are pro‐inflammatory mediators that can be both beneficial (anti‐microbial defense) or pathogenic. They are major recruiters of neutrophils to airway mucosal tissue and surface 42 , 43 supporting the role of these cells in the defense against invading microorganisms, but also facilitating overreactions. 44 Although neutrophilic inflammation is suggested as a potential pathogenic factor, especially in severe asthma, 45 this role still awaits confirmation by targeted drug treatment effects. Indeed, a balance toward a role in defense is suggested by interventions with anti‐TNF‐⍺ biologics that reduce neutrophils but have not produced the expected anti‐inflammatory remedial effect in asthma, but rather increased susceptibility to infection. 46 Here we demonstrate that a mere 24 h exposure of HBECs to HDM, independent of HDM sensitization, causes inhibition of viral stimulus‐induced release of both IL‐8 and TNF‐⍺, strikingly different from the unchanged release of TSLP. Although other molecules may be involved in attracting neutrophils to the airways, it is suggested that depression of viral induced TNF‐⍺ and IL‐8, caused by exposure of airway lining epithelium to HDM, contributes to the increased risk of secondary bacterial infection upon a primary viral infection.
Expectedly, Poly(I:C) increased epithelial expression of major PRRs, including TLR‐3, MDA5, and RIG‐I. Supporting a key role of TLR‐3 activation on IL‐8 and TNF‐α regulation. A lower response on these mediators´ expression was observed after specific stimulation of the cytosolic receptors MDA5 and RIG‐I with Poly(I:C)‐lyovec. In addition, these effects were not affected by HDM exposure, suggesting that HDM‐induced impairment of anti‐viral and anti‐bacteria mediator expression is in a TLR‐3 dependent manner. Although HDM challenge did not affect Poly(I:C)‐induced gene levels of TLR‐3 (or MDA5 and RIG‐I), it inhibited the activation of the downstream transcription factor NFκB. NFκB‐signaling plays a key role in the anti‐bacterial response. 47 Mice with impaired NFκB‐signaling have high susceptibility to bacterial lung infections due to decreased cytokine expression and defective neutrophil recruitment. 48 , 49 Several studies have indicated that NFκB inhibition increases the risk of pneumonia. 50 , 51
The present study has limitations. Our data concern a limited number of patients with HDM‐sensitized asthma. Although the same results were found when compared HDM+with non‐atopic asthma, and no effect was evidenced between atopic and non‐atopic patients, we cannot exclude the possibility that other specific allergen sensitizations have an effect on the results. Further studies are thus warranted to confirm our findings and address their relevance to atopic asthma in general. The usage of Poly(I:C) as a viral mimic has been accepted to reflect physiological relevant pro‐inflammatory and anti‐viral mediators during rhinovirus infections. 9 Thus, our observations suggest the importance of investigating the effect of rhinovirus infection in patients with allergic asthma. Further, new studies are warranted to address the mechanism by which HDM blocks specific cytokines induced by Poly(I:C) while not affecting others.
In this study, we demonstrated that HDM exposure of HBECs from patients with asthma results in the reduction of IFN‐β and several anti‐microbial agents (β‐defensin‐2, TNF‐⍺, and IL‐8) in response to viral infection, but not TSLP. Moreover, anti‐viral IFN‐β expression in response to Poly(I:C) was also reduced in patients with HDM sensitization compared with those not sensitized to HDM. If the anti‐microbial effects, known to be induced by a primary viral infection, are subnormal, protection against ongoing infection as well as additional respiratory infections could be compromised. Hence, we suggest that both HDM sensitization and acute exposure to HDM contribute to reducing infection‐evoked immune protection in asthma and thus increase the risk of symptomatic infections and exacerbations.
CONFLICT OF INTEREST
C.P had grant and personal fees from Astra Zeneca, GSK, Novartis, TEVA, Sanofi, Chiesi and ALK‐ABELLO outside the present work. J.J.N.F had grant from GSK, ISCIII and SEPAR outside the present work. All the other authors had nothing to disclose.
AUTHOR CONTRIBUTIONS
A.S., M.H., and C.P. contributed with clinical samples. S.C., S.R., and S.T. contributed to the collection of data. S.C., J.J.N.F., L.U., and C.A. contributed to the analysis of data. S.C., A.S., S.R., M.M., H.A., J.J.N.F., M.H., C.P., and L.U. contributed to the interpretation of data and S.C., J.J.N.F., and L.U. drafted the manuscript. All authors gave input and approved the submitted version. L.U. conceived and supervised the study and secured funding.
Supporting information
Supplementary Material
ACKNOWLEDGEMENT
The authors acknowledge support from Vetenskapsrådet (Swedish medical research council) grant numbers: 2020‐00922_VR and 2017‐00806_VR as well as Swedish Heart and lung foundation. grant number: 20180207_HLF.
Cerps S, Sverrild A, Ramu S, et al. House dust mite sensitization and exposure affects bronchial epithelial anti‐microbial response to viral stimuli in patients with asthma. Allergy. 2022;77:2498–2508. doi: 10.1111/all.15243
Funding information
This research has been funded by Vetenskapsrådet (Swedish medical research council) grant numbers: 2020‐00922_VR and 2017‐00806_VR as well as Swedish Heart and lung foundation. grant number: 20180207_HLF
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Borna E, Nwaru BI, Bjerg A, et al. Changes in the prevalence of asthma and respiratory symptoms in western Sweden between 2008 and 2016. Allergy. 2019;74(9):1703‐1715. [DOI] [PubMed] [Google Scholar]
- 2. Fahlbusch B, Koch A, Douwes J, et al. The effect of storage on allergen and microbial agent levels in frozen house dust. Allergy. 2003;58(2):150‐153. [DOI] [PubMed] [Google Scholar]
- 3. Chapman MD, Wunschmann S, Pomes A. Proteases as Th2 adjuvants. Curr Allergy Asthma Rep. 2007;7(5):363‐367. [DOI] [PubMed] [Google Scholar]
- 4. Langley SJ, Goldthorpe S, Craven M, Woodcock A, Custovic A. Relationship between exposure to domestic allergens and bronchial hyperresponsiveness in non‐sensitised, atopic asthmatic subjects. Thorax. 2005;60(1):17‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Edwards MR, Strong K, Cameron A, Walton RP, Jackson DJ, Johnston SL. Viral infections in allergy and immunology: how allergic inflammation influences viral infections and illness. J Allergy Clin Immunol. 2017;140(4):909‐920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uller L, Persson C. Viral induced overproduction of epithelial TSLP: role in exacerbations of asthma and COPD? J Allergy Clin Immunol. 2018;142(2):712. [DOI] [PubMed] [Google Scholar]
- 7. Wang Q, Nagarkar DR, Bowman ER, et al. Role of double‐stranded RNA pattern recognition receptors in rhinovirus‐induced airway epithelial cell responses. J Immunol. 2009;183(11):6989‐6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Triantafilou K, Vakakis E, Richer EA, Evans GL, Villiers JP, Triantafilou M. Human rhinovirus recognition in non‐immune cells is mediated by Toll‐like receptors and MDA‐5, which trigger a synergetic pro‐inflammatory immune response. Virulence. 2011;2(1):22‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uller L, Leino M, Bedke N, et al. Double‐stranded RNA induces disproportionate expression of thymic stromal lymphopoietin versus interferon‐beta in bronchial epithelial cells from donors with asthma. Thorax. 2010;65(7):626‐632. [DOI] [PubMed] [Google Scholar]
- 10. Calven J, Yudina Y, Hallgren O, et al. Viral stimuli trigger exaggerated thymic stromal lymphopoietin expression by chronic obstructive pulmonary disease epithelium: role of endosomal TLR3 and cytosolic RIG‐I‐like helicases. J Innate Immun. 2012;4(1):86‐99. [DOI] [PubMed] [Google Scholar]
- 11. Corren J, Karpefors M, Hellqvist A, Parnes JR, Colice G. Tezepelumab reduces exacerbations across all seasons in patients with severe, uncontrolled asthma: a post hoc analysis of the PATHWAY phase 2b study. J Asthma Allergy. 2021;14:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Menzies‐Gow A, Corren J, Bourdin A, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. 2021;384(19):1800‐1809. [DOI] [PubMed] [Google Scholar]
- 13. Persson C. Early humoral defence: contributing to confining COVID‐19 to conducting airways? Scand J Immunol. 2021;93(6):e13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwasaki A, Foxman EF, Molony RD. Early local immune defences in the respiratory tract. Nat Rev Immunol. 2017;17(1):7‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wark PA, Johnston SL, Bucchieri F, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201(6):937‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Devalia JL, Davies RJ. Airway epithelial cells and mediators of inflammation. Respir Med. 1993;87(6):405‐408. [DOI] [PubMed] [Google Scholar]
- 17. Arnason JW, Murphy JC, Kooi C, et al. Human beta‐defensin‐2 production upon viral and bacterial co‐infection is attenuated in COPD. PLoS One. 2017;12(5):e0175963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chieosilapatham P, Ogawa H, Niyonsaba F. Current insights into the role of human beta‐defensins in atopic dermatitis. Clin Exp Immunol. 2017;190(2):155‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iikura M, Hojo M, Koketsu R, et al. The importance of bacterial and viral infections associated with adult asthma exacerbations in clinical practice. PLoS One. 2015;10(4):e0123584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hanada S, Pirzadeh M, Carver KY, Deng JC. Respiratory viral infection‐induced microbiome alterations and secondary bacterial pneumonia. Front Immunol. 2018;9:2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kloepfer KM, Lee WM, Pappas TE, et al. Detection of pathogenic bacteria during rhinovirus infection is associated with increased respiratory symptoms and asthma exacerbations. J Allergy Clin Immunol. 2014;133(5):1301‐1307, 7 e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bashir H, Grindle K, Vrtis R, et al. Association of rhinovirus species with common cold and asthma symptoms and bacterial pathogens. J Allergy Clin Immunol. 2018;141(2):822‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woehlk C, von Bulow A, Kriegbaum M, Backer V, Porsbjerg C. Allergic asthma is associated with increased risk of infections requiring antibiotics. Ann Allergy Asthma Immunol. 2018;120(2):169‐76 e1. [DOI] [PubMed] [Google Scholar]
- 24. Hilty M, Burke C, Pedro H, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5(1):e8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hales BJ, Chai LY, Elliot CE, et al. Antibacterial antibody responses associated with the development of asthma in house dust mite‐sensitised and non‐sensitised children. Thorax. 2012;67(4):321‐327. [DOI] [PubMed] [Google Scholar]
- 26. Hales BJ, Pearce LJ, Kusel MM, Holt PG, Sly PD, Thomas WR. Differences in the antibody response to a mucosal bacterial antigen between allergic and non‐allergic subjects. Thorax. 2008;63(3):221‐227. [DOI] [PubMed] [Google Scholar]
- 27. Ramu S, Menzel M, Bjermer L, Andersson C, Akbarshahi H, Uller L. Allergens produce serine proteases‐dependent distinct release of metabolite DAMPs in human bronchial epithelial cells. Clin Exp Allergy. 2018;48(2):156‐166. [DOI] [PubMed] [Google Scholar]
- 28. Akbarshahi H, Menzel M, Ramu S, Mahmutovic Persson I, Bjermer L, Uller L. House dust mite impairs antiviral response in asthma exacerbation models through its effects on TLR3. Allergy. 2018;73(5):1053‐1063. [DOI] [PubMed] [Google Scholar]
- 29. Sverrild A, Hansen S, Hvidtfeldt M, et al. The effect of tezepelumab on airway hyperresponsiveness to mannitol in asthma (UPSTREAM). Eur Respir J. 2021;59(1):2101296. [DOI] [PubMed] [Google Scholar]
- 30. Bateman ED, Hurd SS, Barnes PJ, et al. "Global strategy for asthma management and prevention: GINA executive summary". Eur Respir J. 2008;31:143‐178. Eur Respir J. 2018;51(2). [DOI] [PubMed] [Google Scholar]
- 31. Menzel M, Akbarshahi H, Tufvesson E, Persson C, Bjermer L, Uller L. Azithromycin augments rhinovirus‐induced IFNbeta via cytosolic MDA5 in experimental models of asthma exacerbation. Oncotarget. 2017;8(19):31601‐31611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kvarnhammar AM, Petterson T, Cardell LO. NOD‐like receptors and RIG‐I‐like receptors in human eosinophils: activation by NOD1 and NOD2 agonists. Immunology. 2011;134(3):314‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sadler AJ, Williams BR. Interferon‐inducible antiviral effectors. Nat Rev Immunol. 2008;8(7):559‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Contoli M, Ito K, Padovani A, et al. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy. 2015;70(8):910‐920. [DOI] [PubMed] [Google Scholar]
- 35. Dunican EM, Fahy JV. The role of type 2 inflammation in the pathogenesis of asthma exacerbations. Ann Am Thorac Soc. 2015;12(Suppl. 2):S144‐S149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Herbert C, Do K, Chiu V, et al. Allergic environment enhances airway epithelial pro‐inflammatory responses to rhinovirus infection. Clin Sci (Lond). 2017;131(6):499‐509. [DOI] [PubMed] [Google Scholar]
- 37. Ganti KP, Mukherji A, Surjit M, Li M, Chambon P. Similarities and differences in the transcriptional control of expression of the mouse TSLP gene in skin epidermis and intestinal epithelium. Proc Natl Acad Sci U S A. 2017;114(6):E951‐E960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaplan A, Lee MW, Wolf AJ, et al. Direct antimicrobial activity of IFN‐beta. J Immunol. 2017;198(10):4036‐4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu MC, Xiao HQ, Brown AJ, Ritter CS, Schroeder J. Association of vitamin D and antimicrobial peptide production during late‐phase allergic responses in the lung. Clin Exp Allergy. 2012;42(3):383‐391. [DOI] [PubMed] [Google Scholar]
- 40. Baines KJ, Wright TK, Simpson JL, et al. Airway beta‐defensin‐1 protein is elevated in COPD and severe asthma. Mediators Inflamm. 2015;2015:407271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tewary P, de la Rosa G, Sharma N, et al. beta‐Defensin 2 and 3 promote the uptake of self or CpG DNA, enhance IFN‐alpha production by human plasmacytoid dendritic cells, and promote inflammation. J Immunol. 2013;191(2):865‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smart SJ, Casale TB. TNF‐alpha‐induced transendothelial neutrophil migration is IL‐8 dependent. Am J Physiol. 1994;266(3 Pt 1):L238‐L245. [DOI] [PubMed] [Google Scholar]
- 43. Kuhns DB, Young HA, Gallin EK, Gallin JI. Ca2+‐dependent production and release of IL‐8 in human neutrophils. J Immunol. 1998;161(8):4332‐4339. [PubMed] [Google Scholar]
- 44. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. 2012;30:459‐489. [DOI] [PubMed] [Google Scholar]
- 45. Trejo Bittar HE, Yousem SA, Wenzel SE. Pathobiology of severe asthma. Annu Rev Pathol. 2015;10:511‐545. [DOI] [PubMed] [Google Scholar]
- 46. Wenzel SE, Barnes PJ, Bleecker ER, et al. A randomized, double‐blind, placebo‐controlled study of tumor necrosis factor‐alpha blockade in severe persistent asthma. Am J Respir Crit Care Med. 2009;179(7):549‐558. [DOI] [PubMed] [Google Scholar]
- 47. Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med. 2008;358(7):716‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Alcamo E, Mizgerd JP, Horwitz BH, et al. Targeted mutation of TNF receptor I rescues the RelA‐deficient mouse and reveals a critical role for NF‐kappa B in leukocyte recruitment. J Immunol. 2001;167(3):1592‐1600. [DOI] [PubMed] [Google Scholar]
- 49. Quinton LJ, Jones MR, Simms BT, et al. Functions and regulation of NF‐kappaB RelA during pneumococcal pneumonia. J Immunol. 2007;178(3):1896‐1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sadikot RT, Zeng H, Joo M, et al. Targeted immunomodulation of the NF‐kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J Immunol. 2006;176(8):4923‐4930. [DOI] [PubMed] [Google Scholar]
- 51. Sadikot RT, Han W, Everhart MB, et al. Selective I kappa B kinase expression in airway epithelium generates neutrophilic lung inflammation. J Immunol. 2003;170(2):1091‐1098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.