

Abstract
Desmoid tumors (DTs) are rare soft tissue mesenchymal neoplasms that may be associated with impairments, disfigurement, morbidity, and (rarely) mortality. DT disease course can be unpredictable. Most DTs are sporadic, harboring somatic mutations in the gene that encodes for β‐catenin, whereas DTs occurring in patients with familial adenomatous polyposis have germline mutations in the APC gene, which encodes for a protein regulator of β‐catenin. Pathology review by an expert soft tissue pathologist is critical in making a diagnosis. Magnetic resonance imaging is preferred for most anatomic locations. Surgery, once the standard of care for initial treatment of DT, is associated with a significant risk of recurrence as well as avoidable morbidity because spontaneous regressions are known to occur without treatment. Consequently, active surveillance in conjunction with pain management is now recommended for most patients. Systemic medical treatment of DT has evolved beyond the use of hormone therapy, which is no longer routinely recommended. Current options for medical management include tyrosine kinase inhibitors as well as more conventional cytotoxic chemotherapy (e.g., anthracycline‐based or methotrexate‐based regimens). A newer class of agents, γ‐secretase inhibitors, appears promising, including in patients who fail other therapies, but confirmation in Phase 3 trials is needed. In summary, DTs present challenges to physicians in diagnosis and prognosis, as well as in determining treatment initiation, type, duration, and sequence. Accordingly, evaluation by a multidisciplinary team with expertise in DT and patient‐tailored management are essential. As management strategies continue to evolve, further studies will help clarify these issues and optimize outcomes for patients.
Keywords: active surveillance; antineoplastic agents; desmoid tumor; fibromatosis, aggressive; radiotherapy; tyrosine kinase inhibitors; γ‐secretase inhibitors
Short abstract
Desmoid tumors can be associated with significant morbidity and are challenging to diagnose and treat, generally requiring evaluation by a multidisciplinary team. Active surveillance has supplanted surgery as the primary approach to management in most patients with desmoid tumors, whereas newer treatments, such as tyrosine kinase inhibitors and γ‐secretase inhibitors, have shown promise in clinical trials.
INTRODUCTION
Desmoid tumor (DT), also known as aggressive fibromatosis, deep fibromatosis, and desmoid‐type fibromatosis, is a clonal fibroblastic proliferation arising in deep soft tissue and is characterized by infiltrative growth and a tendency toward recurrence but an inability to metastasize. 1 DTs can occur anywhere on the body but most commonly occur in the extremities in the case of sporadic DT and intra‐abdominally in patients with familial adenomatous polyposis (FAP). 2 , 3 , 4 Although not malignant, DTs are often locally aggressive and invasive and cause significant impairments, disfigurement, morbidity, and (rarely) mortality. 5 They may infiltrate adjacent organs, compress blood vessels and nerves, erode bones, invade muscle, and cause bowel obstructions. 2
DTs are rare, constituting <3% of soft tissue neoplasms, 6 with an estimated annual incidence of three to five cases per million worldwide. 3 , 7 , 8 , 9 , 10 , 11 Approximately 1000–1500 new cases are diagnosed in the United States each year. 12 Most DTs occur sporadically (non‐FAP), although DT is 1000‐fold more common in patients with FAP than in the general population. 2 , 13 DTs may be multifocal, typically in the same body part. 3 , 7 DTs occur predominantly in women (approximately 70% of cases), and the risk of DT development or progression appears to increase during and after pregnancy. 3 The most common age group for DT occurrence is 30–40 years, and trauma and prior surgery are known risk factors. 4
DTs are almost universally associated with alterations in the Wnt/β‐catenin pathway. 7 , 14 In 85%–90% of sporadic cases, DTs harbor somatic mutations in CTNNB1, the gene that encodes for β‐catenin, leading to its accumulation. 3 The point mutations are predominantly T41A (55%), S45F (35%), and S45P (10%). 5 , 15 , 16 , 17 , 18 , 19 In patients with FAP, DTs harbor germline mutations in the APC gene, which encodes for a protein regulating β‐catenin levels. 20 These two mutation types affect the same pathway yet are mutually exclusive and thus have diagnostic value. 7
The prognosis for patients with DT is notoriously variable. Tumors can be associated with an unpredictable disease course, including spontaneous regressions in 20%–30% of patients who are followed for 2–3 years. 21 , 22 Frequently, an initial growth phase is followed by stabilization. 23 , 24 , 25 Factors significantly associated with shorter progression‐free survival (PFS) include age (younger than 37 years), tumor size (>7 cm), and tumor location (extra‐abdominal). 2 , 26 , 27 , 28
DIAGNOSIS
Symptoms
Because of its rarity, DT may be misdiagnosed in as many as 30%–40% of cases, 3 , 29 resulting in inappropriate or delayed care. In one study, the time from patient‐reported symptom onset to DT diagnosis exceeded one year for 54% of patients. 30 Correctly diagnosing DT is key to optimizing management but, in practice, can prove challenging. Initial evaluation by a multidisciplinary team with expertise in the management of DT, including medical oncologists, radiation oncologists, radiologists, pathologists, surgeons, and geneticists, is recommended. 7 Clinical presentation varies and depends on tumor location. 3 Patients with DT often have a palpable mass at presentation. 31 Those with DT in the extremities may have pain and a limited range of motion that causes ambulatory difficulties. 2 Symptoms in patients with intra‐abdominal desmoids include weight loss, cachexia, and malaise. Both sporadic and FAP‐associated DT can compromise patient quality of life (QoL), adversely affecting physical, social, cognitive, and emotional domains. 32 , 33 , 34 , 35 , 36
Imaging
Magnetic resonance imaging (MRI) is the preferred method for imaging most DTs, with superior soft tissue imaging compared with computed tomography (CT) (Fig. 1A). 4 , 37 Signal intensity for DTs reflects the proportions of tumor components present (collagen fibers, spindle cells, extracellular matrix) (Fig. 1B). Tumors generally show moderate‐to‐marked enhancement with gadolinium. Low‐intensity, nonenhancing, linear bands called band sign are common and correspond with dense collagenous stroma seen by histology. However, this is not specific for DT. Imaging alone cannot distinguish DTs from other soft tissue tumors.
FIGURE 1.
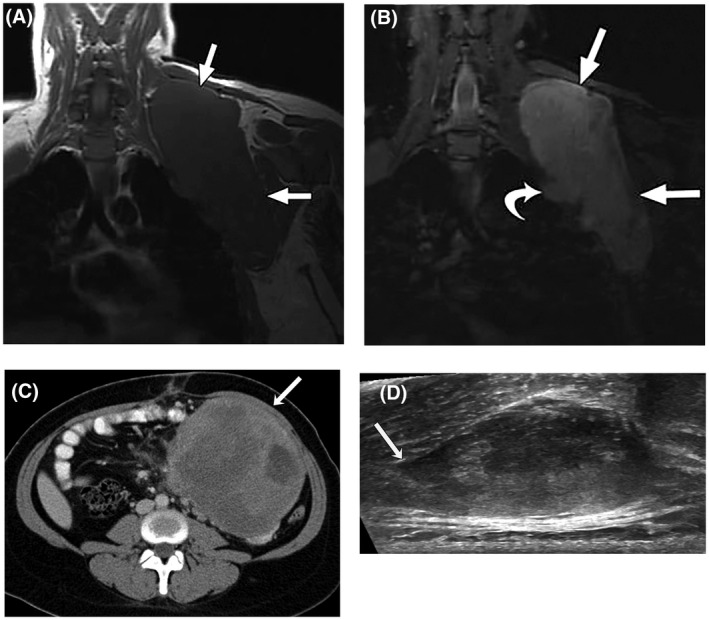
(A) T1‐weighted and (B) T2‐weighted, fat‐suppressed magnetic resonance images in the coronal plain of a 28‐year‐old woman with a large desmoid tumor (DT) in the shoulder region (straight arrows). The curved arrow indicates a nodular protrusion that raises concern for pleural invasion. (C) Axial, contrast‐enhanced computed tomography image from a 27‐year‐old woman with a nonresectable, solitary intra‐abdominal DT not associated with familial adenomatous polyposis. An arrow indicates a large, well defined mass adherent to the small bowel and mesenteric vessels. (D) Transverse ultrasound of a sporadic right paraspinal musculature extra‐abdominal DT in a 26‐year‐old woman. Linear fascial extension (tail sign) is indicated by the arrow. A and B reprinted from: Shinagare AB, Ramaiya NH, Jagannathan JP, et al. A to Z of desmoid tumors. AJR Am J Roentgenol. 2011;197(6):W1008–W1014, 4 with permission from the American Roentgen Ray Society. Copyright©2011, American Roentgen Ray Society. C and D reprinted from: Braschi‐Amirfarzan M, Keraliya AR, Krajewski KM, et al. Role of imaging in management of desmoid‐type fibromatosis: a primer for radiologists. Radiographics. 2016;36(3):767–782, 37 with permission from The Radiological Society of North America. Copyright©2016, The Radiological Society of North America.
CT scans can reveal a soft tissue mass, which typically is sharply marginated in abdominal wall tumors or has poorly defined, infiltrative margins in extra‐abdominal or mesenteric tumors (Fig. 1C), 4 , 37 and are useful for diagnosis and follow‐up of intra‐abdominal DTs and associated complications, such as small bowel obstruction. The extent of attenuation and enhancement varies, with most DTs demonstrating mild or moderate enhancement using an iodine‐based contrast agent. Ultrasound can be useful for the initial evaluation of tumors in extremities or in the abdominal wall (for pregnant patients) and for guiding biopsies. The sonographic appearance is variable, but a thin, linear extension along fascial planes called tail sign is sometimes seen (Fig. 1D). 32 Plain radiography and positron emission tomography CT have very limited roles in DT diagnosis. The latter may be helpful in patients with FAP to distinguish recurrent cancers (moderate uptake) from DT (mild uptake). 37
Pathologic features
DTs appear firm and white or gray, resembling scar tissue (Fig. 2). 32 , 37 , 38 An analysis of a biopsy specimen by an expert soft tissue pathologist is needed to distinguish DT from other neoplasms, such as lymphoma or sarcoma. 3 , 4 , 7 Histologic features include low‐to‐moderate cellularity, long fascicles of uniform cells, dense collagenous stroma, and a lack of malignant features. 39 DT immunohistochemistry is characterized by nuclear β‐catenin positivity along with positivity for smooth muscle actin, vimentin, cyclooxygenase‐2 (COX‐2), and frequently β‐estrogen receptors, and by negativity for desmin, S100, CD34, and KIT. 32 , 40
FIGURE 2.
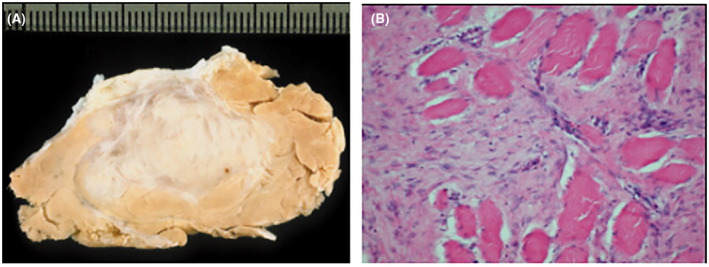
(A) Macroscopic view of the cut surface of an extra‐abdominal desmoid tumor. (B) Abdominal desmoid tumor showing typical infiltrative growth pattern of skeletal muscle (hematoxylin and eosin staining, original magnification ×200). A and B reprinted from: Leithner A, Gapp M, Radl R, et al. Immunohistochemical analysis of desmoid tumours. J Clin Pathol. 2005;58(11)1152–1156, 38 with permission from BMJ Publishing Group. Permission conveyed through Copyright Clearance Center, Inc.
Mutations in CTNNB1 or APC are the hallmarks of DT. Because mutations in these two genes are mutually exclusive, the finding of CTNNB1 mutation rules out FAP, and APC mutation rules out sporadic DT. Therefore, mutational analysis of β‐catenin has been proposed as a specific DT diagnostic tool, with a finding of wild‐type CTNNB1 suggestive of FAP. Next‐generation sequencing is preferable to Sanger sequencing, and all of codons 32 through 49 should be sequenced. 3 In practice, however, access or financial considerations often limit its use.
MANAGEMENT STRATEGIES
Active surveillance
Cumulative evidence of long‐term stabilization or spontaneous regression in many patients with sporadic DT has resulted in a paradigm shift from immediate surgical resection toward more conservative measures, particularly active surveillance (watchful waiting). 41 , 42 A large, prospective, observational study (ClinicalTrials.gov identifier NCT02547831) of patients with sporadic DT who were managed with active surveillance (MRI or CT every 3–6 months) recently reported a treatment‐free survival rate of 65.9% at 3 years, with 55% of patients experiencing spontaneous regression either initially or after progression. 41 A systematic review found that local control rates for surgery, surgery plus radiotherapy (RT), RT alone, and active surveillance were 75%, 78%, 85%, and 78%, respectively, for primary disease 43 ; however, selection and reporting bias as well as heterogeneity of patient and tumor characteristics require careful interpretation of these data. Of interest, among patients with recurrent disease, active surveillance was associated with significantly better local control than surgery (p = .001). 43
Recent guidelines, including those of the Desmoid Tumor Working Group, recommend active surveillance as the preferred front‐line approach to managing most patients with DT. 3 , 7 , 42 , 44 Of note, because pain caused by DT affects patient QoL, active surveillance requires an effective pain management strategy 42 ; however, further research is urgently needed to elucidate optimal approaches. 7 Patients should be monitored by clinical symptoms and MRI (or CT if MRI is not possible) at 3‐month to 6‐month intervals for at least 2–3 years and every 6–12 months thereafter, with shorter intervals if tumors are located at critical sites such as head and neck or mesentery. 7 , 42 , 44 Some degree of clinical or radiologic progression may be tolerated. 42 An algorithm illustrating this initial approach is shown in Figure 3. 7
FIGURE 3.
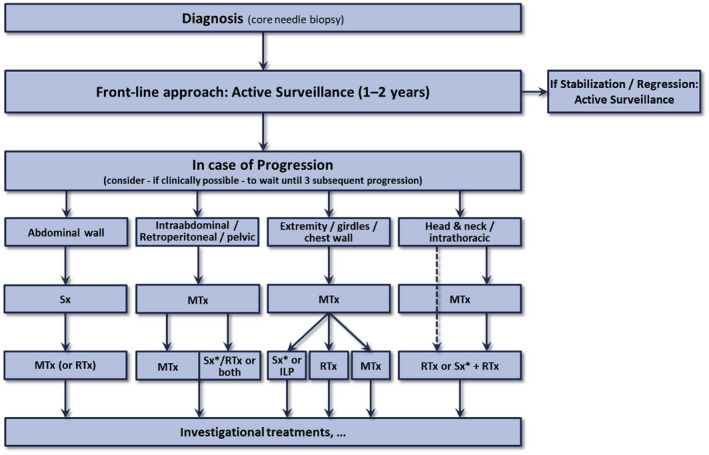
Schema for the management of patients with desmoid tumor recommended by the Desmoid Tumor Working Group. ILP, isolated limb perfusion; MTx, medical treatment; RTx, radiotherapy; Sx, surgery; Sx*, surgery is an option if morbidity is limited. Reprinted from: Desmoid Tumor Working Group. The management of desmoid tumours: a joint global consensus‐based guideline approach for adult and paediatric patients. Eur J Cancer. 2020;127:96–107, 7 with permission from Elsevier Science & Technology Journals. Permission conveyed through Copyright Clearance Center, Inc.
LOCAL CONTROL STRATEGIES
Surgery and radiotherapy
Until the early 2000s, the treatment for DT was similar to that for soft tissue sarcoma, with surgery considered the cornerstone of treatment. 3 , 45 , 46 However, postsurgical local recurrence rates at 5–10 years were reported to be in the range from 30% to 77%. 27 , 47 , 48 Furthermore, whether negative margins correlated with a decreased likelihood of recurrence was controversial. Microscopically margin‐negative (R0) resections were not achieved in most surgeries, and there was no consensus on whether a positive margin resection correlated with the risk of recurrence. 45 Postsurgical relapse rates were higher for extra‐abdominal DTs than for abdominal DTs and among juvenile patients versus adult patients, 46 and nomograms that incorporated tumor size to predict postsurgical recurrence were subsequently developed. 27 , 49
A recent Danish study reported that rates of surgery as initial DT treatment fell from 75% between 2009 and 2014 to 32% between 2015 and 2018. 50 Several factors have combined to unseat surgery as the de facto first‐line standard treatment for most patients with DT. Spontaneous DT regressions have been noted in 20%–55% of patients who underwent active surveillance. 21 , 41 Considering the relatively high postsurgical local recurrence rates, the trauma and functional impairments associated with surgery, and the introduction of newer treatment options, surgery is no longer considered the primary preferred therapy for patients with DT at diagnosis. 3 , 7 Surgical resection, however, remains an option in patients with symptomatic, disabling, or progressive DT when expected postsurgical morbidity is low and patients are carefully counselled. 51
A large meta‐analysis revealed that, although RT after R0 resection did not significantly lower the risk of local recurrence, this risk was almost doubled (relative risk, 1.78; 95% CI, 1.40–2.26) in patients who underwent R1 resections but did not receive RT. 51 This supports the potential role of adjuvant RT when surgery results in an incomplete resection 3 but bears a low level of evidence, and the risk of radiation‐induced sarcomas in a generally younger patient population needs to be considered. 7 A European Organization for Research and Treatment of Cancer study examined moderate‐dose RT alone in patients with inoperable, progressive DT. 52 Patients with primary, recurrent, or incompletely resected DTs (61.3% in extremities) received 56 grays in 28 fractions. The local control rate was 81.5% (13.6% had a complete response [CR]), and late toxic effects, including skin toxicity (41%), lymphedema (23%), and pain (18%), were reported. RT as a single modality appeared to be at least as effective as incomplete resection surgery followed by adjuvant RT. Therefore, RT alone may provide adequate local control in most patients who have progressive disease, for whom surgery is not an option, and for disease not otherwise controlled with medical therapy. 7
Other local control methods
Other methods of local DT control have been explored. High‐intensity focused ultrasound (HIFU) is minimally invasive, using ultrasound beams precisely focused on target locations to produce thermal coagulation necrosis. 53 In 111 patients with DTs, ultrasound‐guided HIFU provided a 36% 3‐month tumor volume reduction rate with the most common adverse events (AEs) being pain (14%; all Grade 1 or 2) and bone reaction (10%; all Grade 1). 54 Percutaneous cryoablation uses argon gas through a sealed, segmentally insulated probe to cause rapid cooling. 53 This has been used in a limited number of patients with extra‐abdominal DTs and was associated with tumor volume reductions and symptom improvements with low rates of complications. 55 , 56 This technique, however, may be limited to small‐to‐moderate sized extra‐abdominal tumors. 3 Radiofrequency ablation uses local tissue heating through an electrode to cause thermal necrosis but requires CT guidance of the probe, which may not clearly distinguish muscle from tumor. 57 Selective delivery of cytotoxic chemotherapy to DTs through intra‐arterial doxorubicin drug‐eluting embolization and subsequent tumor volume reduction has been reported in four pediatric patients with recurrent or refractory DTs 58 and, more recently, in a series of 11 adult women with symptomatic, progressively enlarging, extra‐abdominal DTs in which 10 patients (91%) reported improvement or abatement of pain. 59 Hyperthermic isolated limb perfusion may be considered in cases of progressive, unresectable disease for which medical treatments have failed or are contraindicated. 60
SYSTEMIC CONTROL STRATEGIES
Antiestrogens and nonsteroidal anti‐inflammatory drugs
Just as active surveillance has superseded surgery as the primary approach to DT, medical management has evolved to provide newer, more evidence‐based therapeutic options, although, at this time, no medication has received regulatory approval for the treatment of DT.
Estrogen has long been suspected of modulating DT. 2 Evidence includes estrogen receptor expression in DTs and the heightened DT risk during and shortly after pregnancy and among women taking estrogen‐containing oral contraceptives. Women of childbearing age appear to have greater DT growth rates than men or postmenopausal women, and menopause as well as tamoxifen have previously been associated with DT regression. Evidence for antiestrogen therapeutic effectiveness in DT is limited, however, to case series and single‐arm trials. A systematic review identified an overall response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST) criteria of 48%–51% for antiestrogen therapy, 61 although the lack of an active surveillance comparator makes this finding difficult to interpret. Therefore, treatment guidelines no longer routinely recommend hormone therapies. 7 , 44
The rationale for using nonsteroidal anti‐inflammatory drugs (NSAIDs) in patients with DT began with the observation that COX‐2 is overexpressed in these tumors. 62 NSAIDS that inhibit both COX‐1 and COX‐2, such as sulindac and indomethacin and the selective COX‐2 inhibitor celecoxib, have been investigated, often in combination with hormone therapy. 63 A wide range of response rates have been reported, as has favorable tolerability. 64 , 65 To date, however, no randomized, prospective studies of NSAIDs in DT have been reported, and NSAIDs are not currently deemed to be disease‐modifying agents. Guidelines now recommend their use for pain control only. 44
Cytotoxic chemotherapy
Evidence for the effectiveness of cytotoxic chemotherapy in DT comes from retrospective and prospective, nonrandomized studies. 7 Typically, low‐dose methotrexate plus vinblastine or vinorelbine, or, alternatively, a conventional anthracycline‐containing regimen is associated with disease control rates (DCRs) of 64%–100%. 66 In one report, the use of chemotherapy regimens (most commonly methotrexate plus vinblastine) in 62 children and adults with recurrent or progressive DT resulted in a 1.6% CR rate, a 19.4% partial response (PR) rate, and a 59.6% stable disease (SD) rate, according to RECIST criteria, with 19.4% of patients progressing at a median of 71.3 months. 67 The ORR was higher for anthracycline‐based regimens than for nonanthracycline regimens (54% vs. 12%; p = .0011), and toxicity was primarily hematologic, with AEs more common with the former (31% vs. 10%; p = .06).
Several recent retrospective studies of oral single‐agent vinorelbine have reported moderate response and clinical benefit rates in patients with DT. 68 , 69 Among 90 adults who had DT treated with oral vinorelbine with or without antiestrogen therapy, the best responses were 29% PR, 57% SD, and 14% progressive disease. 69 Concomitant antiestrogen therapy was associated with a significantly longer time to treatment failure in women (p = .03). The time to treatment failure was significantly longer in patients who had S45P or S45F mutations relative to those who had T41A or wild‐type (median not reached vs. 24.0 months; p = .04). Among the patients who were evaluable for pain, 74% had symptomatic improvement after 3 months. The most common grade ≥ 2 AE was nausea (39%).
Tyrosine kinase inhibitors
Although the exact mechanism(s) by which tyrosine kinase inhibitors (TKIs) act in DT has not been fully elucidated, 70 overexpression of platelet‐derived growth factor receptor β (PDGFRβ), which is inhibited by the TKI imatinib, has been postulated to drive DT development and growth. 71 Initial case reports suggested that imatinib had efficacy in patients with DT. 72 Subsequently, multiple prospective trials evaluated the safety and efficacy of TKIs in patients with DT (Table 1). 21 , 73 , 74 , 75 , 76 , 77 , 78 , 79 Of note, unlike cytotoxic chemotherapy, in which treatment cycles are limited, TKIs are generally used continuously until intolerance develops or disease progresses.
TABLE 1.
Summary of key prospective studies of tyrosine kinase inhibitors in patients with desmoid tumors
| Study | Phase | Design | No. | Patients | ORR, % | DCR, % | Other | Safety |
|---|---|---|---|---|---|---|---|---|
| Imatinib | ||||||||
| CSTIB2225 (Heinrich 2006 74 ) | 2 | OL, single‐arm | 19 | Aged ≥17 years; any line; 63% ABD | 16 | 84 | 1‐year DCR, 37% | Dose reductions (from 400 mg BID) required for most patients due to Grade ≥ 3 toxicities |
| SARC (Chugh 2010 75 ) | 2 | OL, single‐arm | 51 | Aged ≥10 years; not amenable to surgery; any line; 16% ABD; 16% FAP | 6 | 84 | 1‐year PFS, 66% | Grade 3–4 AEs: neutropenia (10%), rash (10%), fatigue (8%); dose reductions in 39% |
| FNCLCC/FSG (Penel 2011 76 ) | 2 | OL, single‐arm | 40 | Aged ≥18 years; any line; progressive DT not amenable to RT or surgery; 45% ABD; 14% FAP | 11 | 91 | 1‐year PFS, 67%; 2‐year PFS, 55%; 2‐year OS, 95% | Grade 3 AEs: rash (10%), abdominal pain (10%), vomiting (8%); four discontinuations (10%) due to AEs |
| NCT01137916 (Kasper 2017 77 ) | 2 | OL, single‐arm | 38 | Aged ≥18 years; any line; progressive DT (last 6 months) not amenable to RT or surgery; 18% ABD; 3% FAP | 19 | NS | 6‐month PAR, 65%; mDOR, 413 days | Grade 4 AEs, 3%; Grade 3 AEs, 11%, including neutropenia, leucopenia, nausea/vomiting, gastritis, rash, and contracture |
| Sunitinib | ||||||||
| Jo et al. (Jo 2014 78 ) | 2 | OL, single‐arm | 19 | Aged ≥18 years; not amenable to curative surgery; 63% ABD; 53% FAP | 26 | 68 | mDOR, 8.2 months; 2‐year PFS, 75%; 2‐year OS, 94% | Grade 4 AEs: neutropenia (5%); most common Grade 3 AE: neutropenia (26%); most common any‐grade AE: thrombocytopenia (67%; all Grade 1–2) |
| Miano et al. (Miano 2019 79 ) | 2 | OL RCT | 22 (SU) | Progressive, symptomatic, or recurrent DT | 75 | 100 | 2‐year PFS, 81% | Most common AEs: Grade 1–2 hypothyroidism (73%), fatigue (67%), hypertension (55%), diarrhea (51%) |
| 10 (TM) | 0 | NS | 2‐year PFS, 36% | NS | ||||
| Sorafenib | ||||||||
| NCT02066181 (Gounder 2018 21 )a | 3 | DB RCT | 49 (SOR) | Aged ≥18 years; progression ≥10% in 6 months; inoperable or requiring extensive surgery, or symptomatic; any line | 33 | NS | 1‐year PFS, 89%; 2‐year PFS, 81% | Grade 3–4 AEs: Papulopustular rash (12%), hypertension (8%); most common AEs: fatigue (73%), hand‐foot syndrome (71%); withdrawals due to AEs, 20% |
| 36 (PBO) | 20 | NS | 1‐year PFS, 46%; 2‐year PFS, 36% | Grade 3–4 AEs: abdominal pain (11%), vomiting (6%); most common AEs: fatigue (64%), nausea (42%); withdrawals due to AEs, 0% | ||||
| Pazopanib | ||||||||
| NCT01876082 (DESMOPAZ; Toulmonde 2019 73 )b | 2 | OL RCT | 48 (PAZ) | Aged ≥18 years; progressive disease; any line; FAP, 16% | 37 | 96 | 1‐year PFS, 86%; 2‐year PFS, 67% | Grade 3–4 AEs: hypertension (21%), diarrhea (15%); most common AEs: fatigue (81%), diarrhea (80%); withdrawals due to AEs, 8% |
| 22 (MV) | 25 | 75 | 1‐year PFS, 79%; 2‐year PFS, 79% | Grade 3–4 AEs: neutropenia (46%), ALAT or ASAT increase (18%); most common AEs: nausea and vomiting (73%), fatigue (69%); withdrawals due to AEs, 23% |
Abbreviations: ABD, abdominal; AE, adverse event; ALAT, alanine aminotransferase; ASAT, aspartate aminotransferase; BID, twice daily; DB, double‐blind; DCR, disease control rate; FAP, familial adenomatous polyposis; FNCLCC/FSG, Fédération Nationale des Centres de Lutte Contre Le Cancer/French Sarcoma Group; mDOR, median duration of response; MV, methotrexate and vinblastine; NCT, ClinicalTrials.gov identification number; NR, not reached; NS, not specified; OL, open‐label; OS, overall survival; PAR, progression arrest rate; PAZ, pazopanib; PBO, placebo; PC, placebo‐controlled; PFS, progression‐free survival; RCT, randomized controlled trial; RT, radiotherapy; SARC, Sarcoma Alliance for Research through Collaboration; SOR, sorafenib; SU, sunitinib; TM, tamoxifen and meloxicam.
Randomized, double‐blind, placebo‐controlled trial.
Noncomparative randomized, open‐label trial.
A retrospective review of sorafenib found that its activity warranted prospective evaluation in DT. 80 In a Phase 3, randomized, placebo‐controlled study, sorafenib demonstrated superior median PFS (not estimable vs. 11.3 months; hazard ratio, 0.13; p < .001) compared with placebo (Fig. 4). 21 The ORR was 33% and 20% in the sorafenib and placebo arms, respectively, the latter through spontaneous regressions. Discontinuations caused by AEs were reported in 20% and 0% of patients in the sorafenib and placebo arms, respectively. The most frequent AEs among patients who received sorafenib were grade 1–2 rash (73%), fatigue (67%), hypertension (55%), and diarrhea (51%). The most common grade ≥ 3 AEs were papulopustular rash (12%) and hypertension (8%) for sorafenib and abdominal pain (11%) and vomiting (6%) for placebo.
FIGURE 4.
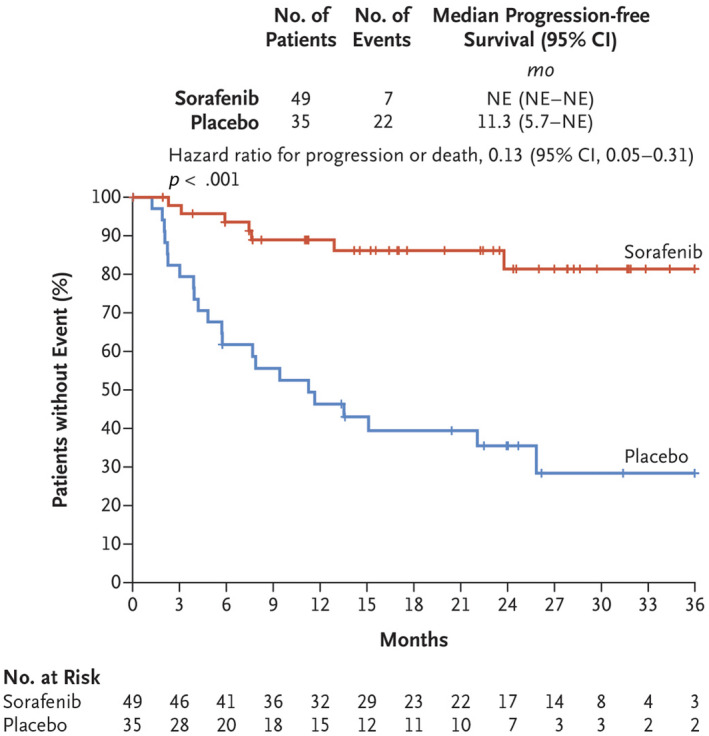
Kaplan–Meier plot of duration of progression‐free survival in patients with advanced and refractory desmoid tumors in the sorafenib and placebo arms of a clinical trial (ClinicalTrials.gov identifier NCT02066181). NE indicates not estimable. Reprinted from: Gounder MM, Mahoney MR, Van Tine BA, et al. Sorafenib for advanced and refractory desmoid tumors. N Engl J Med. 2018;379 (25):2417–2428, 21 with permission from Massachusetts Medical Society. Copyright©2018 Massachusetts Medical Society.
In the Phase 2 DESMOPAZ trial, pazopanib, a second‐generation multikinase inhibitor, was associated with higher objective response (37% vs. 25%) and 1‐year PFS (86% vs. 67%) rates than methotrexate plus vinblastine in adults with progressive DT, although no statistical comparisons between groups were performed. 73 Pain, as assessed using the Brief Pain Inventory, decreased by a clinically meaningful amount in the pazopanib arm only, and patient‐reported global health status was stable in the pazopanib arm but decreased in the methotrexate plus vinblastine arm. Fatigue and gastrointestinal AEs were the most common toxicities in both arms. The most common grade ≥ 3 AEs were hypertension (21%) and diarrhea (15%) for pazopanib and neutropenia (46%) and liver transaminitis (18%) for methotrexate plus vinblastine. Discontinuations caused by AEs were less frequent in the pazopanib arm (8% vs. 23%).
Although not approved by any regulatory agency, based on available evidence, TKIs have been recommended in guidelines as a systemic treatment option for patients with progressive DT. 3 , 7 However, caveats apply. Although convenient because of oral administration, TKIs could potentially result in permanent hypertension or thyroid dysfunction, which is of potential concern in younger patients. 81 The longer life expectancy of patients with DT stands in contrast to that of populations with metastatic cancer, for which these TKIs were initially developed. In addition, the tolerability of long‐term TKI use has not been fully assessed, nor have potential effects of TKIs on growth and fertility been explored, 70 although a diagnosis of DT, in itself, is not a contraindication to future pregnancy. 7 Finally, further studies are necessary to establish optimal dosing, duration, and sequencing of TKIs to better define their place in the treatment of DT. In the absence of comparative studies, the Desmoid Tumor Working Group recommends following a 5‐dimensional model that considers level of evidence, the ORR, the PFS rate, ease of administration, and expected toxicity associated with a particular agent, 7 generally moving from less toxic to more toxic treatments unless more aggressive treatment is indicated because of disease severity.
γ‐Secretase inhibitors
Notch signaling and dysregulation of cross‐talk between the Notch and Wnt/β‐catenin signaling pathways are implicated in tumorigenesis, progression, and treatment resistance 82 , 83 , 84 , 85 in multiple tumor types, including DT. 86 Inhibitors of γ‐secretase block Notch receptor proteolysis and subsequent translocation of the Notch intracellular domain to the nucleus. A selective γ‐secretase inhibitor (GSI), nirogacestat (PF‐03084014), inhibited cell growth and caused cell cycle arrest, providing in vitro validation for the potential use of GSIs in DT. 82
A first‐in‐patient study of oral nirogacestat in patients with advanced solid tumors resistant to therapy or for which no therapy was available reported that five of seven patients with DT (71.4%) achieved a PR, and the other two achieved SD, resulting in a DCR of 100%. 87 The most common AEs with nirogacestat were diarrhea (55% any grade; 9% grade 3), nausea (38% any grade; 2% grade ≥ 3), fatigue (30% any grade; 0% grade ≥ 3), and hypophosphatemia (27% any grade; 23% grade ≥ 3). Of seven evaluable patients with DT, there were no discontinuations because of AEs. All 5 patients who had a PR maintained their response for at least 48 months. 88 Furthermore, the mean duration of clinical benefit (≥63.8 months) was significantly longer than that observed with all prior interventions, including surgery (12.8 months; p < .001). Interestingly, the mean time to treatment response was 11.9 months by RECIST criteria but only 1.6 months by T2‐weighted MRI. 88
An open‐label Phase 2 study of nirogacestat in 17 heavily pretreated adults with recurrent, progressive DT reported a 29% ORR (all PRs) and a 100% DCR. 89 Symptom burden, according to the MD Anderson Symptom Inventory, was significantly and clinically meaningfully reduced in patients who achieved a PR. Clinical benefit was independent of CTNNB1 or APC mutational status, and four of five responders had DTs refractory to imatinib or sorafenib. The most common AEs were diarrhea (76%) and skin disorders (71%); the only grade ≥ 3 AE was hypophosphatemia (47%), which was reversible with supplementation.
Early studies have reported tumor regression with two other GSIs: AL101 90 and AL102. 91 Given the promising results obtained to date, several clinical trials of GSIs in DTs are underway (Table 2). 89 A Phase 2 trial (ClinicalTrials.gov identifier NCT04195399) is evaluating nirogacestat in patients aged 1–18 years with DT not amenable to surgery. A Phase 3 randomized, double‐blind, placebo‐controlled trial (ClinicalTrials.gov identifier NCT03785964; DeFi) of nirogacestat has completed accrual in adults with progressing DT. The primary end point is PFS, and secondary end points are ORR, tolerability, and patient‐reported outcomes. The RINGSIDE trial (ClinicalTrials.gov identifier NCT04871282) is a pivotal Phase 2/3 randomized, double‐blind, placebo‐controlled trial of AL102 in adults with progressing DT. The primary end point is PFS, and secondary end points include ORR and patient‐reported outcomes.
TABLE 2.
Clinical trials of investigational agents for patients with desmoid tumor (searched April 27, 2022)
| Trial identifier | Agent | Status | Phase | No. of patients | Key inclusion criteria | Primary endpoint | Estimated completion |
|---|---|---|---|---|---|---|---|
| NCT04871282 (RINGSIDE) | AL102 | Recruiting | 2/3 | 192 | Aged ≥18 years, TN or R/R DT | PFS | February 2025 |
| NCT01981551 (Kummar 2017 89 ) | Nirogacestat | Active, not recruiting | 2 | 17 | Aged ≥ 18 years, DT progressing after one or more prior systemic therapy and not amenable to surgery | ORR | September 2022 |
| NCT04195399 | Nirogacestat | Recruiting | 2 | 35 | Aged 1–18 years, progressing DT not amenable to surgery, one or more prior systemic therapy | PFS | December 2024 |
| NCT03785964 (DeFi) | Nirogacestat | Active, not recruiting | 3 | 142 | Aged ≥18 years, progressing TN and not amenable to surgery, or R/R DT | PFS | March 2023 |
| NCT03459469 | Tegavivint | Active, not recruiting | 1 | 24 | Aged ≥18 years, TN unresectable DT or progressing or symptomatic R/R DT | Safety, tolerability | November 2021 |
| NCT03802084 | Vactosertib + imatinib | Recruiting | 1/2 | 24 | Aged ≥19 years, DT not amenable to surgery or RT | Adverse events | December 2021 |
| NCT02834013 | Nivolumab + ipilimumab | Recruiting | 2 | 818 | Aged ≥18 years, histologically confirmed rare cancer, including DT | ORR | October 2023 |
| NCT01265030 | Sirolimus | Completed | 1/2 | 9 | Aged ≤29 years, TN or R/R DT planning to undergo surgery | mTOR pathway activation | June 2021 |
Abbreviations: DT, desmoid tumor; mTOR, mammalian target of rapamycin; NCT, ClinicalTrials.gov identification number; ORR, overall response rate; PFS, progression‐free survival; R/R, relapsed or refractory; TN, treatment‐naive.
Other investigational agents
Vactosertib, a TGFβR1 inhibitor, is being investigated in combination with imatinib in patients with advanced DTs in a Phase 1/2 trial (ClinicalTrials.gov identifier NCT03802084). Tegavivint, an inhibitor of transducing β‐like protein 1 (TBL1), a novel target in the Wnt/β‐catenin pathway, 92 was investigated for safety in the first‐in‐human trial (ClinicalTrials.gov identifier NCT0349469) and is being investigated in patients aged 1–30 years with recurrent or refractory solid tumors, including DT, in a Phase 1/2 trial (ClinicalTrials.gov identifier NCT04851119). Immunotherapy with the monoclonal antibodies nivolumab and ipilimumab is being investigated in a Phase 1/2 trial in adults with rare tumors, including DT (ClinicalTrials.gov identifier NCT02834013). Sirolimus, a drug that inhibits the mammalian target of rapamycin (mTOR) cell proliferation/survival pathway, was investigated in a pilot study to determine whether it decreases mTOR activation in children and young adults with surgically resectable DT (ClinicalTrials.gov identifier NCT01265030) (Table 2).
CONCLUSIONS
DT is often locally aggressive and invasive and, despite the lack of metastatic potential, is a source of chronic pain, disability, and disfigurement, with adverse effects on QoL. DT presents many clinical challenges to the treating physician. Given the rarity of the disease, the diagnosis of DT often requires consultation with an expert soft tissue pathologist because initial misdiagnoses can occur. Given the unpredictable disease course with the potential for spontaneous regressions, an active surveillance approach is currently the preferred management for patients who have DTs in noncritical locations. When treatment is needed, providers must be able to navigate an expanding range of locoregional and systemic options, and that requires the collaborative effort of a multidisciplinary team with expertise in the management of DT, including medical oncologists, radiation oncologists, radiologists, pathologists, surgical oncologists, orthopedic oncologists, geneticists, and supportive care. The difficulties are compounded because biomarkers predicting response to treatment have not been identified, and little evidence comparing the effectiveness of various DT treatments is currently available.
Although the DT treatment paradigm continues to evolve, several directions are clear. For most patients, surgery is no longer the preferred primary therapy and has been displaced by active surveillance. Except for DTs at critical sites, at least 1–2 years of active surveillance is now recommended, and some amount of progression may even be tolerated. Local management may be achieved nonsurgically in some patients by techniques like RT and HIFU. Regarding systemic options, hormone therapy is no longer recommended, and NSAIDs are largely used for pain control. Preferred options now include TKIs and chemotherapy. The optimal duration of treatment is based on cumulative dose limits and disease status for chemotherapy but is less well established for TKIs, although tolerability and disease status are typical factors.
Preliminary data suggest that novel GSIs may be active in DT. Upcoming Phase 3 data will provide additional information on GSI efficacy in treatment‐naive and refractory DT populations. Other novel therapeutic approaches are being explored. Evolutionary progress is driven by this continuing unmet need, and patient advocacy groups including, among others, the Desmoid Tumor Research Foundation (and its sister organizations 93 ), Rein in Sarcoma, Sarcoma Foundation of America, and Sarcoma Alliance, will continue to play an important role in advancing care for this rare disease.
CONFLICTS OF INTEREST
Richard F. Riedel reports institutional clinical research support from Aadi, AROG, Ayala, BioAtla, Daiichi‐Sankyo, Deciphera, GlaxoSmithKline, Ignyta, Inhibrx, Immune Design, Karyopharm, Lilly, NanoCarrier, Novartis, Oncternal, Philogen, Plexxikon, Rain Therapeutics, Roche, SpringWorks Therapeutics, Threshold, Tracon, and Trillium; personal/advisory fees from Aadi, Bayer, Blueprint, Daiichi‐Sankyo, Deciphera, EISAI, EMD Serono, Janssen, Lilly, Ignyta, NanoCarrier, and SpringWorks Therapeutics; and ownership in Limbguard, LLC (spouse), all outside the submitted work. Mark Agulnik reports research funding from Exelixis and personal/advisory fees from Aadi, Bayer, Adaptimmune, Regeneron, AstraZeneca, Bristol‐Myers Squibb, and Deciphera, all outside the submitted work.
ACKNOWLEDGMENTS
Assistance with article preparation was provided by Prescott Medical Communications Group (Chicago, Illinois), with financial support from SpringWorks Therapeutics (Stamford, Connecticut).
REFERENCES
- 1. World Health Organization Classification of Tumours Editorial Board . WHO Classification of Tumours: Soft Tissue and Bone Tumours. Soft Tissue and Bone Tumours. Volume 3. IARC Press; 2020. [Google Scholar]
- 2. Constantinidou A, Scurr M, Judson I, Litchman C. Clinical presentation of desmoid tumor. In: Litchman C, ed. Desmoid Tumor. Springer Science; 2012:5‐16. [Google Scholar]
- 3. Kasper B, Baumgarten C, Garcia J, et al. An update on the management of sporadic desmoid‐type fibromatosis: a European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol. 2017;28(10):2399‐2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shinagare AB, Ramaiya NH, Jagannathan JP, et al. A to Z of desmoid tumors. AJR Am J Roentgenol. 2011;197(6):W1008‐W1014. [DOI] [PubMed] [Google Scholar]
- 5. Penel N, Chibon F, Salas S. Adult desmoid tumors: biology, management and ongoing trials. Curr Opin Oncol. 2017;29(4):268‐274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Orphanet. Desmoid tumor. Accessed December 17, 2021. Orphanet; 2021. Available at: https://www.orpha.net/consor/cgi‐bin/Disease_Search.php?lng=EN&data_id=8665&Disease_Disease_Search_diseaseGroup=desmoid‐tumor&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Desmoid‐tumor&title=Desmoid%20tumor&search=Disease_Search_Simple
- 7. Desmoid Tumor Working Group . The management of desmoid tumours: a joint global consensus‐based guideline approach for adult and paediatric patients. Eur J Cancer. 2020;127:96‐107. [DOI] [PubMed] [Google Scholar]
- 8. van Broekhoven DL, Grunhagen DJ, den Bakker MA, van Dalen T, Verhoef C. Time trends in the incidence and treatment of extra‐abdominal and abdominal aggressive fibromatosis: a population‐based study. Ann Surg Oncol. 2015;22(9):2817‐2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kasper B, Strobel P, Hohenberger P. Desmoid tumors: clinical features and treatment options for advanced disease. Oncologist. 2011;16(5):682‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. National Library of Medicine . MedlinePlus: Desmoid tumor. National Library of Medicine, National Institutes of Health, US Department of Health and Human Services; 2022. Accessed February 17, 2022. https://medlineplus.gov/genetics/condition/desmoid‐tumor/
- 11. Orphanet. Prevalence of rare diseases: Bibliographic data. Orphanet Report Series, Rare Diseases Collection. Orphanet; January 2019, No. 1. Accessed February 26, 2022. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf
- 12. National Library of Medicine . MedlinePlus: Desmoid tumor. National Library of Medicine, National Institutes of Health, US Department of Health and Human Services; 2021. Accessed December 17, 2021. https://medlineplus.gov/genetics/condition/desmoid‐tumor/#frequency
- 13. Magid D, Fishman EK, Jones B, Hoover HC, Feinstein R, Siegelman SS. Desmoid tumors in Gardner syndrome: use of computed tomography. AJR Am J Roentgenol. 1984;142(6):1141‐1145. [DOI] [PubMed] [Google Scholar]
- 14. Crago AM, Chmielecki J, Rosenberg M, et al. Near universal detection of alterations in CTNNB1 and Wnt pathway regulators in desmoid‐type fibromatosis by whole‐exome sequencing and genomic analysis. Genes Chromosomes Cancer. 2015;54(10):606‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Colombo C, Miceli R, Lazar AJ, et al. CTNNB1 45F mutation is a molecular prognosticator of increased postoperative primary desmoid tumor recurrence: an independent, multicenter validation study. Cancer. 2013;119(20):3696‐3702. [DOI] [PubMed] [Google Scholar]
- 16. Lazar AJ, Tuvin D, Hajibashi S, et al. Specific mutations in the beta‐catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173(5):1518‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mullen JT, DeLaney TF, Rosenberg AE, et al. beta‐Catenin mutation status and outcomes in sporadic desmoid tumors. Oncologist. 2013;18(9):1043‐1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Timbergen MJM, Smits R, Grunhagen DJ, Verhoef C, Sleijfer S, Wiemer EAC. Activated signaling pathways and targeted therapies in desmoid‐type fibromatosis: a literature review. Front Oncol. 2019;9:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salas S, Chibon F, Noguchi T, et al. Molecular characterization by array comparative genomic hybridization and DNA sequencing of 194 desmoid tumors. Genes Chromosomes Cancer. 2010;49(6):560‐568. [DOI] [PubMed] [Google Scholar]
- 20. Miyoshi Y, Ando H, Nagase H, et al. Germ‐line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci U S A. 1992;89(10):4452‐4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gounder MM, Mahoney MR, Van Tine BA, et al. Sorafenib for advanced and refractory desmoid tumors. N Engl J Med. 2018;379(25):2417‐2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bonvalot S, Ternes N, Fiore M, et al. Spontaneous regression of primary abdominal wall desmoid tumors: more common than previously thought. Ann Surg Oncol. 2013;20(13):4096‐4102. [DOI] [PubMed] [Google Scholar]
- 23. Gronchi A, Raut CP. Optimal approach to sporadic desmoid tumors: from radical surgery to observation. Time for a consensus? Ann Surg Oncol. 2012;19(13):3995‐3997. [DOI] [PubMed] [Google Scholar]
- 24. Kim Y, Rosario MS, Cho HS, Han I. Factors associated with disease stabilization of desmoid‐type fibromatosis. Clin Orthop Surg. 2020;12(1):113‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stoeckle E, Coindre JM, Longy M, et al. A critical analysis of treatment strategies in desmoid tumours: a review of a series of 106 cases. Eur J Surg Oncol. 2009;35(2):129‐134. [DOI] [PubMed] [Google Scholar]
- 26. Salas S, Dufresne A, Bui B, et al. Prognostic factors influencing progression‐free survival determined from a series of sporadic desmoid tumors: a wait‐and‐see policy according to tumor presentation. J Clin Oncol. 2011;29(26):3553‐3558. [DOI] [PubMed] [Google Scholar]
- 27. Crago AM, Denton B, Salas S, et al. A prognostic nomogram for prediction of recurrence in desmoid fibromatosis. Ann Surg. 2013;258(2):347‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bishop AJ, Zarzour MA, Ratan R, et al. Long‐term outcomes for patients with desmoid fibromatosis treated with radiation therapy: a 10‐year update and re‐evaluation of the role of radiation therapy for younger patients. Int J Radiat Oncol Biol Phys. 2019;103(5):1167‐1174. [DOI] [PubMed] [Google Scholar]
- 29. Lucas A, Braggio D, Hernandez L, Mercier K. A retrospective collection of diagnostic data from the Desmoid Tumor Research Foundation natural history study [abstract]. J Clin Oncol. 2021;39(15 suppl):e23549. [Google Scholar]
- 30. Mercier KA, Hernandez L, Boulanger V, Seebald A, Rossov S, Milligan K. Quality of life and tumor location in patients with desmoid tumors: data from the Desmoid Tumor Research Foundation natural history study [abstract]. J Clin Oncol. 2019;37(15 suppl):e18291. [Google Scholar]
- 31. Zenzri Y, Yahyaoui Y, Charfi L, et al. The management of desmoid tumors: a retrospective study of 30 cases. Int J Surg Oncol. 2020;2020:9197216‐9197217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garcia‐Ortega DY, Martin‐Tellez KS, Cuellar‐Hubbe M, et al. Desmoid‐type fibromatosis. Cancers (Basel). 2020;12(7):1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bonvalot S, Desai A, Coppola S, et al. The treatment of desmoid tumors: a stepwise clinical approach. Ann Oncol. 2012;23(suppl 10):x158‐x166. [DOI] [PubMed] [Google Scholar]
- 34. Cuomo P, Scoccianti G, Schivo A, et al. Extra‐abdominal desmoid tumor fibromatosis: a multicenter EMSOS study. BMC Cancer. 2021;21(1):437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gounder MM, Maddux L, Paty J, Atkinson TM. Prospective development of a patient‐reported outcomes instrument for desmoid tumors or aggressive fibromatosis. Cancer. 2020;126(3):531‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rigaux P, Lefebvre‐Kuntz D, Penel N. SOS Desmoide. Pain burden in desmoid tumor patients: a survey of the French Advocacy Group SOS Desmoid. Bull Cancer. 2015;102(3):213‐216. [DOI] [PubMed] [Google Scholar]
- 37. Braschi‐Amirfarzan M, Keraliya AR, Krajewski KM, et al. Role of imaging in management of desmoid‐type fibromatosis: a primer for radiologists. Radiographics. 2016;36(3):767‐782. [DOI] [PubMed] [Google Scholar]
- 38. Leithner A, Gapp M, Radl R, et al. Immunohistochemical analysis of desmoid tumours. J Clin Pathol. 2005;58(11):1152‐1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zreik RT, Fritchie KJ. Morphologic spectrum of desmoid‐type fibromatosis. Am J Clin Pathol. 2016;145(3):332‐340. [DOI] [PubMed] [Google Scholar]
- 40. Mocellin S. Desmoid‐type fibromatosis. Soft Tissue Tumors. Springer; 2021:231‐237. [Google Scholar]
- 41. Colombo C, Vullo SL, Fiore M, et al. Active surveillance in primary desmoid tumor (DT): a prospective observational study [abstract]. J Clin Oncol. 2021;39(15 suppl):11570. [Google Scholar]
- 42. Gronchi A, Jones RL. Treatment of desmoid tumors in 2019. JAMA Oncol. 2019;5(4):567‐568. [DOI] [PubMed] [Google Scholar]
- 43. Seinen JM, Niebling MG, Bastiaannet E, Pras B, Hoekstra HJ. Four different treatment strategies in aggressive fibromatosis: a systematic review. Clin Transl Radiat Oncol. 2018;12:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. National Comprehensive Cancer Network (NCCN) . NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines). Soft Tissue Sarcoma. Version 1.2021. Accessed December 22, 2021. https://www.nccn.org/guidelines/guidelines‐detail?category=1&id=1464
- 45. Fiore M, MacNeill A, Gronchi A, Colombo C. Desmoid‐type fibromatosis: evolving treatment standards. Surg Oncol Clin N Am. 2016;25(4):803‐826. [DOI] [PubMed] [Google Scholar]
- 46. Reitamo JJ. The desmoid tumor. IV. Choice of treatment, results, and complications. Arch Surg. 1983;118(11):1318‐1322. [DOI] [PubMed] [Google Scholar]
- 47. Ballo MT, Zagars GK, Pollack A, Pisters PW, Pollack RA. Desmoid tumor: prognostic factors and outcome after surgery, radiation therapy, or combined surgery and radiation therapy. J Clin Oncol. 1999;17(1):158‐167. [DOI] [PubMed] [Google Scholar]
- 48. Easter DW, Halasz NA. Recent trends in the management of desmoid tumors. Summary of 19 cases and review of the literature. Ann Surg. 1989;210(6):765‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu H, Huang K, Li T, et al. Development, validation, and visualization of a web‐based nomogram for predicting the recurrence‐free survival rate of patients with desmoid tumors. Front Oncol. 2021;11:634648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anneberg M, Svane HML, Fryzek J, et al. The epidemiology of desmoid tumors in Denmark. Cancer Epidemiol. 2022;77:102114. [DOI] [PubMed] [Google Scholar]
- 51. Janssen ML, van Broekhoven DL, Cates JM, et al. Meta‐analysis of the influence of surgical margin and adjuvant radiotherapy on local recurrence after resection of sporadic desmoid‐type fibromatosis. Br J Surg. 2017;104(4):347‐357. [DOI] [PubMed] [Google Scholar]
- 52. Keus RB, Nout RA, Blay JY, et al. Results of a phase II pilot study of moderate dose radiotherapy for inoperable desmoid‐type fibromatosis—an EORTC STBSG and ROG study (EORTC 62991‐22998). Ann Oncol. 2013;24(10):2672‐2676. [DOI] [PubMed] [Google Scholar]
- 53. Zhang Z, Shi J, Yang T, Liu T, Zhang K. Management of aggressive fibromatosis. Oncol Lett. 2021;21(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang R, Chen JY, Zhang L, et al. The safety and ablation efficacy of ultrasound‐guided high‐intensity focused ultrasound ablation for desmoid tumors. Int J Hyperthermia. 2021;38(2):89‐95. [DOI] [PubMed] [Google Scholar]
- 55. Redifer Tremblay K, Lea WB, Neilson JC, King DM, Tutton SM. Percutaneous cryoablation for the treatment of extra‐abdominal desmoid tumors. J Surg Oncol. 2019;120(3):366‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schmitz JJ, Schmit GD, Atwell TD, et al. Percutaneous cryoablation of extraabdominal desmoid tumors: a 10‐year experience. AJR Am J Roentgenol. 2016;207(1):190‐195. [DOI] [PubMed] [Google Scholar]
- 57. Ilaslan H, Schils J, Joyce M, Marks K, Sundaram M. Radiofrequency ablation: another treatment option for local control of desmoid tumors. Skel Radiol. 2010;39(2):169‐173. [DOI] [PubMed] [Google Scholar]
- 58. Elnekave E, Atar E, Amar S, et al. Doxorubicin‐eluting intra‐arterial therapy for pediatric extra‐abdominal desmoid fibromatoses: a promising approach for a perplexing disease. J Vasc Intervent Radiol. 2018;29(10):1376‐1382. [DOI] [PubMed] [Google Scholar]
- 59. Kim D, Keohan ML, Gounder MM, Crago AM, Erinjeri JP. Transarterial chemoembolization with doxorubicin eluting beads for extra‐abdominal desmoid tumors: initial experience. Cardiovasc Intervent Radiol Published online April 19, 2022. 10.1007/s00270-022-03149-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bonvalot S, Rimareix F, Causeret S, et al. Hyperthermic isolated limb perfusion in locally advanced soft tissue sarcoma and progressive desmoid‐type fibromatosis with TNF 1 mg and melphalan (T1‐M HILP) is safe and efficient. Ann Surg Oncol. 2009;16(12):3350‐3357. [DOI] [PubMed] [Google Scholar]
- 61. Bocale D, Rotelli MT, Cavallini A, Altomare DF. Anti‐oestrogen therapy in the treatment of desmoid tumours: a systematic review. Colorectal Dis. 2011;13(12):e388‐e395. [DOI] [PubMed] [Google Scholar]
- 62. Mignemi NA, Itani DM, Fasig JH, et al. Signal transduction pathway analysis in desmoid‐type fibromatosis: transforming growth factor‐beta, COX2 and sex steroid receptors. Cancer Sci. 2012;103(12):2173‐2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Eastley NC, Hennig IM, Esler CP, Ashford RU. Nationwide trends in the current management of desmoid (aggressive) fibromatosis. Clin Oncol (R Coll Radiol). 2015;27(6):362‐368. [DOI] [PubMed] [Google Scholar]
- 64. Quast DR, Schneider R, Burdzik E, Hoppe S, Moslein G. Long‐term outcome of sporadic and FAP‐associated desmoid tumors treated with high‐dose selective estrogen receptor modulators and sulindac: a single‐center long‐term observational study in 134 patients. Fam Cancer. 2016;15(1):31‐40. [DOI] [PubMed] [Google Scholar]
- 65. Skapek SX, Anderson JR, Hill DA, et al. Safety and efficacy of high‐dose tamoxifen and sulindac for desmoid tumor in children: results of a Children's Oncology Group (COG) phase II study. Pediatr Blood Cancer. 2013;60(7):1108‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tsukamoto S, Takahama T, Mavrogenis AF, Tanaka Y, Tanaka Y, Errani C. Clinical outcomes of medical treatments for progressive desmoid tumors following active surveillance: a systematic review. Musculoskelet Surg. Published online February12, 2022. 10.1007/s12306-022-00738-x [DOI] [PubMed] [Google Scholar]
- 67. Garbay D, Le Cesne A, Penel N, et al. Chemotherapy in patients with desmoid tumors: a study from the French Sarcoma Group (FSG). Ann Oncol. 2012;23(1):182‐186. [DOI] [PubMed] [Google Scholar]
- 68. Gennatas S, Chamberlain F, Smrke A, et al. A timely oral option: single‐agent vinorelbine in desmoid tumors. Oncologist. 2020;25(12):e2013‐e2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mir O, Honore C, Chamseddine AN, et al. Long‐term outcomes of oral vinorelbine in advanced, progressive desmoid fibromatosis and influence of CTNNB1 mutational status. Clin Cancer Res. 2020;26(23):6277‐6283. [DOI] [PubMed] [Google Scholar]
- 70. Sparber‐Sauer M, Orbach D, Navid F, et al. Rationale for the use of tyrosine kinase inhibitors in the treatment of paediatric desmoid‐type fibromatosis. Br J Cancer. 2021;124(10):1637‐1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Napolitano A, Mazzocca A, Spalato Ceruso M, et al. Recent advances in desmoid tumor therapy. Cancers (Basel). 2020;12(8):2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mace J, Sybil Biermann J, Sondak V, et al. Response of extraabdominal desmoid tumors to therapy with imatinib mesylate. Cancer. 2002;95(11):2373‐2379. [DOI] [PubMed] [Google Scholar]
- 73. Toulmonde M, Pulido M, Ray‐Coquard I, et al. Pazopanib or methotrexate‐vinblastine combination chemotherapy in adult patients with progressive desmoid tumours (DESMOPAZ): a non‐comparative, randomised, open‐label, multicentre, phase 2 study. Lancet Oncol. 2019;20(9):1263‐1272. [DOI] [PubMed] [Google Scholar]
- 74. Heinrich MC, McArthur GA, Demetri GD, et al. Clinical and molecular studies of the effect of imatinib on advanced aggressive fibromatosis (desmoid tumor). J Clin Oncol. 2006;24(7):1195‐1203. [DOI] [PubMed] [Google Scholar]
- 75. Chugh R, Wathen JK, Patel SR, et al. Efficacy of imatinib in aggressive fibromatosis: results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res. 2010;16(19):4884‐4891. [DOI] [PubMed] [Google Scholar]
- 76. Penel N, Le Cesne A, Bui BN, et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): an FNCLCC/French Sarcoma Group phase II trial with a long‐term follow‐up. Ann Oncol. 2011;22(2):452‐457. [DOI] [PubMed] [Google Scholar]
- 77. Kasper B, Gruenwald V, Reichardt P, et al. Imatinib induces sustained progression arrest in RECIST progressive desmoid tumours: final results of a phase II study of the German Interdisciplinary Sarcoma Group (GISG). Eur J Cancer. 2017;76:60‐67. [DOI] [PubMed] [Google Scholar]
- 78. Jo JC, Hong YS, Kim KP, et al. A prospective multicenter phase II study of sunitinib in patients with advanced aggressive fibromatosis. Invest New Drugs. 2014;32(2):369‐376. [DOI] [PubMed] [Google Scholar]
- 79. Miano S, Francini G, Civitelli S, Petrioli R, Francini E. Clinical outcomes of sunitinib (Su) for patients (pts) with desmoid tumors (DT) [abstract]. J Clin Oncol. 2019;37(15 suppl):11052. [Google Scholar]
- 80. Gounder MM, Lefkowitz RA, Keohan ML, et al. Activity of sorafenib against desmoid tumor/deep fibromatosis. Clin Cancer Res. 2011;17(12):4082‐4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kasper B, Raut CP, Gronchi A. Desmoid tumors: to treat or not to treat, that is the question. Cancer. 2020;126(24):5213‐5221. [DOI] [PubMed] [Google Scholar]
- 82. McCaw TR, Inga E, Chen H, et al. Gamma secretase inhibitors in cancer: a current perspective on clinical performance. Oncologist. 2021;26(4):e608‐e621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Arcaroli JJ, Quackenbush KS, Purkey A, et al. Tumours with elevated levels of the Notch and Wnt pathways exhibit efficacy to PF‐03084014, a gamma‐secretase inhibitor, in a preclinical colorectal explant model. Br J Cancer. 2013;109(3):667‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rodilla V, Villanueva A, Obrador‐Hevia A, et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc Natl Acad Sci U S A. 2009;106(15):6315‐6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ronchini C, Capobianco AJ. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): implication for cell cycle disruption in transformation by Notch(ic). Mol Cell Biol. 2001;21(17):5925‐5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Shang H, Braggio D, Lee YJ, et al. Targeting the Notch pathway: a potential therapeutic approach for desmoid tumors. Cancer. 2015;121(22):4088‐4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Messersmith WA, Shapiro GI, Cleary JM, et al. A phase I, dose‐finding study in patients with advanced solid malignancies of the oral gamma‐secretase inhibitor PF‐03084014. Clin Cancer Res. 2015;21(1):60‐67. [DOI] [PubMed] [Google Scholar]
- 88. Villalobos VM, Hall F, Jimeno A, et al. Long‐term follow‐up of desmoid fibromatosis treated with PF‐03084014, an oral gamma secretase inhibitor. Ann Surg Oncol. 2018;25(3):768‐775. [DOI] [PubMed] [Google Scholar]
- 89. Kummar S, O'Sullivan Coyne G, Do KT, et al. Clinical activity of the gamma‐secretase inhibitor PF‐03084014 in adults with desmoid tumors (aggressive fibromatosis). J Clin Oncol. 2017;35(14):1561‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. El‐Khoueiry AB, Desai J, Iyer SP, et al. A phase I study of AL101, a pan‐NOTCH inhibitor, in patients (pts) with locally advanced or metastatic solid tumors [abstract]. J Clin Oncol. 2018;36(15 suppl):2515. [Google Scholar]
- 91. Chan D, Kaplan J, Gordon G, Desai J. Activity of the gamma secretase inhibitor AL101 in desmoid tumors: a case report of 2 adult cases. Curr Oncol. 2021;28(5):3659‐3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nomura M, Rainusso N, Lee YC, et al. Tegavivint and the beta‐catenin/ALDH axis in chemotherapy‐resistant and metastatic osteosarcoma. J Natl Cancer Inst. 2019;111(11):1216‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. The Desmoid Tumor Research Foundation . Other Desmoid Tumor Patient Advocacy Groups. Accessed February 26, 2022. https://dtrf.org/sisterorgs/