

Abstract
Aims
Chronic heart failure (CHF) is a systemic syndrome with a poor prognosis and a need for novel therapies. We investigated whether whole blood transcriptomic profiling can provide new mechanistic insights into cardiovascular (CV) mortality in CHF.
Methods and results
Transcriptome profiles were generated at baseline from 944 CHF patients from the BIOSTAT‐CHF study, of whom 626 survived and 318 died from a CV cause during a follow‐up of 21 months. Multivariable analysis, including adjustment for cell count, identified 1153 genes (6.5%) that were differentially expressed between those that survived or died and strongly related to a validated clinical risk score for adverse prognosis. The differentially expressed genes mainly belonged to five non‐redundant pathways: adaptive immune response, proteasome‐mediated ubiquitin‐dependent protein catabolic process, T‐cell co‐stimulation, positive regulation of T‐cell proliferation, and erythrocyte development. These five pathways were selectively related (RV coefficients >0.20) with seven circulating protein biomarkers of CV mortality (fibroblast growth factor 23, soluble ST2, adrenomedullin, hepcidin, pentraxin‐3, WAP 4‐disulfide core domain 2, and interleukin‐6) revealing an intricate relationship between immune and iron homeostasis. The pattern of survival‐associated gene expression matched with 29 perturbagen‐induced transcriptome signatures in the iLINCS drug‐repurposing database, identifying drugs, approved for other clinical indications, that were able to reverse in vitro the molecular changes associated with adverse prognosis in CHF.
Conclusion
Systematic modelling of the whole blood protein‐coding transcriptome defined molecular pathways that provide a link between clinical risk factors and adverse CV prognosis in CHF, identifying both established and new potential therapeutic targets.
Keywords: Chronic heart failure, RNA, T‐cells, Interleukins, Fibroblast growth factor 23, Iron, Drug‐repurposing
Introduction
The prognosis of patients with chronic heart failure (CHF) remains poor, despite substantial improvements in CHF diagnosis and treatment. Several prognostic risk scores have been developed for CHF, 1 , 2 yet the precise mechanistic links between such clinical risk scores and survival remain unclear. Furthermore, beyond natriuretic peptides, 3 several circulating protein biomarkers that have been shown to be independently associated with prognosis in CHF 4 , 5 , 6 remain to have the underpinning molecular nature of those relationships elucidated.
Whole blood transcriptomic profiling is a practical and powerful methodology to study genome‐wide biology in large numbers of patients. Despite CHF being a systemic syndrome, no study has investigated whether whole blood transcriptomic profiles can inform about the molecular processes underpinning the observed relationships between clinical or protein biomarkers and cardiovascular (CV) mortality in these patients. If the whole blood transcriptome can be shown to reflect important drivers of mortality, then modelling whole blood global gene expression may help identify clinically relevant pathways to target therapeutics development. 7
In this study, we report the first large‐scale whole blood transcriptomic profiles from patients with CHF. Our main aims were to (i) contrast such profiles between patients who survived or died, (ii) examine whether this provides new insights into molecular mechanisms through which clinical and protein biomarkers associated with adverse outcome might affect prognosis, and (iii) explore the utility of blood‐based transcriptomic signatures as a novel tool to aid in vitro drug‐repurposing analysis in CHF.
Methods
Participants
This was a nested case–control study undertaken within the BIOSTAT‐CHF study. 8 Briefly, BIOSTAT‐CHF enrolled an index cohort of 2516 patients from 69 hospital centres in 11 European countries between 2010 and 2014. Patients were aged >18 years with symptoms of new‐onset or worsening CHF, confirmed either by a left ventricular ejection fraction (LVEF) of ≤40% or B‐type natriuretic peptide (BNP) level >400 pg/ml and/or N‐terminal pro‐B‐type natriuretic peptide (NT‐proBNP) plasma levels >2000 pg/ml, treated with either oral or intravenous furosemide ≥40 mg/day or equivalent at the time of inclusion. After enrolment and obtaining a blood sample, all participants had their heart failure medication optimized to guideline‐recommended or maximally tolerated doses. The median follow‐up of the cohort was 21 months, with an interquartile range of 15 months. The study complied with the 2008 Declaration of Helsinki and was approved by the relevant ethics committee in each centre. All participants gave written informed consent to participate.
For the current transcriptome analysis, participants were categorized into two groups: survivors (those participants who survived during the follow‐up period without hospitalization), and non‐survivors (those participants who died from a CV cause). Cause of death (CV or non‐CV) was adjudicated by a senior clinician (A.A.V.) based on reports of mortality events using previously reported criteria. 9 There were 441 CV deaths (out of a total of 657 deaths), and 1437 participants survived without hospitalization. After the exclusion of participants with no usable RNA samples, 332 who died and 1061 who survived, were available to study. The two groups, those that survived or died, were matched for age and sex with an approximate ratio of 1:2, to match the number of arrays available (online supplementary Figure S1 ).
Generation of transcriptomic profiles
Whole blood was collected into PAXgene tubes (Qiagen) and RNA was isolated using TRizol® (Life Technologies), dissolved in 20 µl RNAse‐free water and profiled using the Affymetrix Human Transcriptomic Array 2.0 (HTA, Thermo Fisher Scientific) which contains ∼7 million probes to provide a comprehensive view of the transcriptome. Samples were processed following the manufacturer's protocol (Sutton Bonington Array facility, University of Nottingham). Briefly, 250 ng of RNA was processed to single‐stranded sense fragmented DNA using the WT PLUS Reagent Kit. Five µg of fragmented, end‐labelled sense stranded target cDNA was hybridized to each array, and scanned in batches of 24, using a 30007G scanner. The running order used a random list generator to ensure even distribution of cases and controls during profiling. Full details of the processing of the arrays and quality checks of the gene expression data are provided in online supplementary Appendix S1 . A total of 944 samples were successfully profiled, of whom 626 (66%) were survivors and 318 (34%) died of a CV cause.
Statistical and bioinformatics analysis
RNA expression values were derived for 17 748 protein‐coding genes (ENSG), then normalized and logarithm (base 2) transformed. We applied a multivariable linear model analysis with group (survival and non‐survival), age, sex and estimates of major white cell populations as covariates using the limma package in R, to identify differentially expressed ENSGs. Estimated white cell proportions were derived by applying ABIS, 10 with absolute values being scaled to 100 prior to further analysis (online supplementary Appendix S1 ). P‐values were adjusted using the Benjamini–Hochberg false discovery rate (FDR) method; an FDR value of <5% was used. Gene ontology (GO) profiles (Biological Processes) were assessed using Metascape, 11 with the detectable transcriptome used as the GO background to calculate enriched pathway statistics. 12 The main drivers to GO enrichment were identified by removing associated processes, using the topGO hierarchical analysis package in R.
To assess whether there was a relationship between the observed transcriptomic differences between survivors and non‐survivors, and clinical and biochemical risk factors associated with CHF prognosis, we used a reproducible prognostic risk score for mortality developed in BIOSTAT‐CHF (BIOSTAT‐CHF risk score, BRS). 2 The BRS model considered 42 clinical and biochemical variables, identifying five variables (more advanced age, higher blood urea nitrogen and NT‐proBNP, lower haemoglobin, and failure to prescribe a beta‐blocker) as the strongest independent predictor of mortality with a combined C‐statistic of 0.73. 2 We undertook linear correlation analysis of gene expression with the BRS and compared the overlap of this set of genes with those differentially expressed between survivors and non‐survivors. The effect of adjusting for the BRS on the latter set of genes was also investigated.
To gain insight into potential protein drivers of the transcriptional changes, we correlated the transcriptomic changes with plasma levels of >150 circulating proteins available in the BIOSTAT‐CHF cohort, some of which have been reported to be associated with prognosis in the cohort. 5 , 6 The majority of proteins were measured using two commercial platforms (Olink Proseek Multiplex and Alere Luminex Panel), with a minority derived from routinely available hospital clinical assays or bespoke assays. Specifically, fibroblast growth factor 23 (FGF23) data were generated using the c‐terminal ELISA (Immutopics, Inc., San Clemente, CA, USA) while hepcidin (HEPC) was measured using the competitive ELISA developed by Kroot et al. 13 To examine the relationship between each of the non‐redundant GO categories, and the protein biomarkers, we estimated the RV coefficient, a multivariate generalization of the squared Pearson correlation coefficient. Pearson correlation coefficients, adjusting for multiple testing (Bonferroni correction), were also calculated for the significant associated proteins and individual genes within the top‐ranked GO categories. For proteins most associated (RV coefficient >0.20) with at least one significant GO, we examined the relationship with CV mortality, using a Cox proportional hazards model.
Finally, to explore the potential of our approach to identify new drug treatments, we utilized the CMap‐L1000v1 7 in vitro drug signature database (https://clue.io/) as previously described. 14 Briefly, to represent the prognosis‐related transcriptomics signature, we used the 60 most upregulated and 60 most down‐regulated genes that were also present in the CMap‐L1000v1 database. Choice of 60/60 genes is empirical and informed by our recent work on insulin resistance drug repurposing 14 that yielded a drug list composed of ∼50% true‐positive drug classes and/or drug targets. The matching score was calculated by aggregating the transcriptional pattern across nine independent cell lines for >2500 compounds, many of which include Food and Drug Administration approved drugs. 14 For each matching drug, the known protein targets were identified using PubChem and the small molecule suite. 15 A network of significant pathway interactions was derived using metascape.org, using the CHF survival associated genes and the top ranked known protein target of each drug as input values, and the detectable transcriptome as background.
Results
Subjects
The two groups (survivors and non‐survivors) were, by design, closely matched for age and sex (Table 1 and online supplementary Figure S1 ). Patients were predominantly Caucasian; there was no difference in hypertension but there was a higher prevalence of both type 2 diabetes mellitus (T2DM) and CHF of ischaemic aetiology, in the non‐survivor group. Baseline use of CHF medication was similar in both groups, except for a greater use of beta‐blockers in the survivor group. The echo‐estimated LVEF was similar between groups (≈31%) as was heart rate. However, patients that died had worse symptoms, lower systolic and diastolic blood pressure and higher plasma concentrations of natriuretic peptides, and lower estimated glomerular filtration rate (eGFR), haemoglobin and body mass index (BMI), at baseline.
Table 1.
Demographic, clinical and laboratory variables for study groups
| Variable | Survivor (n = 626) | Non‐survivor (n = 318) | p‐value |
|---|---|---|---|
| Demographics | |||
| Male sex | 75.7 (474) | 74.5 (237) | 0.748 |
| Age (years) | 71 (10.7) | 71.4 (11) | 0.584 |
| BMI (kg/m2) | 27.7 (6.2) | 26.6 (7.3) | 0.206 |
| Clinical profile | |||
| NYHA class | <0.001 | ||
| I | 2.9 (18) | 0.9 (3) | |
| II | 44.2 (277) | 22.6 (72) | |
| III | 41.2 (258) | 58.5 (186) | |
| IV | 8.6 (54) | 14.2 (45) | |
| LVEF (%) | 31.6 (9.9) | 31.3 (12.4) | 0.747 |
| Heart rate (bpm) | 79.2 (20.2) | 80.4 (19.4) | 0.386 |
| Systolic blood pressure (mmHg) | 128.0 (21.7) | 121.1 (22.3) | <0.001 |
| Diastolic blood pressure (mmHg) | 76.6 (13.7) | 71.7 (12.3) | <0.001 |
| HF history | |||
| Ischaemic aetiology | 61.2 (340) | 70.3 (206) | 0.01 |
| HF hospitalization in previous year | 22.7 (142) | 42.8 (136) | <0.001 |
| Medical history | |||
| Hypertension | 65.2 (408) | 63.2 (201) | 0.599 |
| Diabetes mellitus | 28.3 (177) | 39.0 (124) | 0.001 |
| Medication at baseline | |||
| ACE inhibitors or ARB | 74.8 (468) | 69.2 (220) | 0.081 |
| Beta‐blockers | 84 (526) | 77 (245) | 0.011 |
| Mineralocorticoid receptor antagonist | 51.3 (321) | 50.9 (162) | 0.977 |
| Laboratory measurements | |||
| Haemoglobin (g/dl) | 13.4 (1.8) | 12.6 (1.8) | <0.001 |
| Erythrocytes (million cells/µl) | 4.5 (0.6) | 4.4 (0.9) | 0.31 |
| Leucocytes (×109/L) | 7.8 [6.6, 9.3] | 7.8 [6.4, 9.6] | 0.908 |
| eGFR (ml/min/1.73 m2; CKD‐EPI) | 62.0 [46.9, 76.7] | 52.3 [34.8, 69.4] | <0.001 |
| BNP (pg/ml) | 174.7 [78.3, 371.1] | 338.1 [176.2, 652] | <0.001 |
| NT‐proBNP (ng/L) | 2005 [949, 4642] | 4121 [2332, 9275] | <0.001 |
Values are expressed as % (n), mean (standard deviation), or median [interquartile range].
ACE, angiotensin‐converting enzyme; ARB, angiotensin II receptor blocker; BMI, body mass index; BNP, B‐type natriuretic peptide; CKD‐EPI, Chronic Kidney Disease Epidemiology Collaboration equation; eGFR, estimated glomerular filtration rate; HF, heart failure; LVEF, left ventricular ejection fraction; NT‐proBNP, N‐terminal pro‐B‐type natriuretic peptide; NYHA, New York Heart Association.
Whole blood transcriptomic differences between survivors and non‐survivors
Laboratory‐measured total leucocyte count did not differ between those that survived or died (Table 1 ). However, counts modelled for individual blood cell types using specific gene expression markers (see Methods) identified significant reduction in T‐cells (−17%, p < 1 × 10−10) and B‐cells (−11%, p < 1 × 10−3) in the non‐survivor group, and an increase in neutrophils (+5%, p < 1 × 10−5) and basophils (+14%, p < 1 × 10−3; online supplementary Figure S2A ). After including the estimated cell subtypes as covariates in the analysis together with age and sex, we found that 1153 genes (6.5%) were differentially expressed (FDR <5% and >5% difference, 557 up‐regulated and 596 down‐regulated) between survivors and non‐survivors (Figure 1A and online supplementary Table S1 ). Up‐ and down‐regulated genes often belonged to the same biological pathways (online supplementary Figure S2B ) and mapped to several interrelated biological pathways (Figure 1B and online supplementary Table S2 ). The processes most responsible for the GO results were identified by removing the redundant topological features of the GO structure (online supplementary Figure S3 ). This identified five main ‘driver’ GO categories, each with a robust fold enrichment (FE) and low FDR: adaptive immune response (FE = 2.3, FDR <0.1%), proteasome‐mediated ubiquitin‐dependent protein catabolic process (UPP; FE = 1.7, FDR <3%), T‐cell co‐stimulation (FE = 3.9, FDR <0.1%), positive regulation of T‐cell proliferation (FE = 3.0, FDR <0.1%), and erythrocyte development (FE = 4.3, FDR <0.1%).
Figure 1.
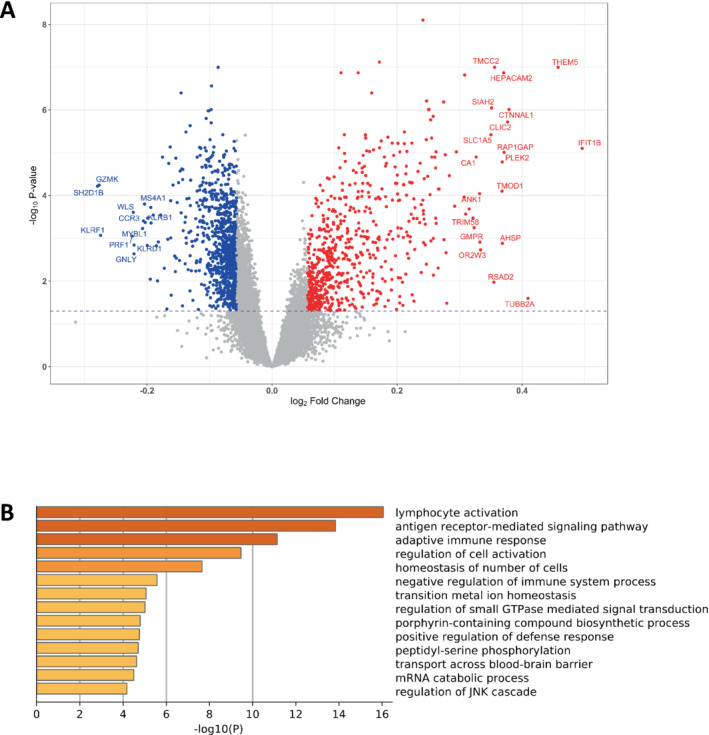
Differential gene expression and biological process gene ontology analysis. (A) A volcano plot of the differential gene expression pattern between groups, calculated using limma and the following model: ENSG ∼ Age + Sex + log(sum T‐cells) + log(sum B‐cells) + log(neutrophils) + log(basophils) + group. (B) Global gene ontology (biological processes) analysis carried out using Metascape and the list of 1153 differentially regulated genes. The 17 748 detected protein‐coding genes were used as the background and the pre‐analysis settings were: threshold >2.0 and non‐adjusted p‐values threshold p < 0.0001. The plot x‐axis is p‐values (log base 10) while the displayed categories had a false discovery rate of 1.4% or better (see online supplementary Table S2 ).
The differentially expressed genes in each of these pathways are shown in Figure 2 . Overall, the pattern of changes in gene expression indicates an impairment of the adaptive immune response, down‐regulation of T‐cell co‐stimulation and disruption of positive regulation of T‐cell proliferation in non‐survivors. In contrast, the UPP pathway and the erythrocyte development pathway were both activated in non‐survivors (see Discussion).
Figure 2.

Individual gene expression responses within the topology adjusted top‐ranked gene ontology categories. Differential gene expression is presented across the five topology adjusted gene ontology categories identified using weighted Fisher's exact test, fold enrichment calculations and BH correction of Fisher's test statistics using the R package topGO. FC, fold change.
BIOSTAT‐CHF prognostic risk score and transcriptomic changes
Linear regression analysis of gene expression versus the BRS 2 identified 1086 correlated genes, and these predominantly belonged to the same pathways as the genes differentially expressed between survivors and non‐survivors (online supplementary Figure S4 ). On an individual gene level basis, 390 genes were common to both lists. Inclusion of the BRS as a covariate in the analysis of the expression differences between survivors and non‐survivors removed all but one of the gene expression differences between groups. This suggests that the observed variation in the blood transcriptome is strongly related to clinical factors associated with prognosis.
Transcriptomic changes and circulating protein biomarkers
To examine whether circulating proteins represented potential drivers of the transcriptomic changes, and therefore potential intermediaries between the clinical prognostic variables and the observed gene expression differences, we modelled the relationships between >150 circulating proteins, and the five top‐ranked GO categories. While most demonstrated limited association (online supplementary Table S3 ), seven proteins demonstrated an RV coefficient of 0.2 or greater, with at least one of the five GOs (Table 2 ). These were, in order of association: FGF23, soluble ST2 receptor (sST2), adrenomedullin (ADM), HEPC, pentraxin‐3 (PTX3), WAP 4‐disulfide core domain 2 (WFDC2) and interleukin‐6 (IL‐6). Notably, six of these proteins positively covaried with each other, while HEPC demonstrated a negative correlation, especially with FGF23 (online supplementary Figure S5 ). These seven proteins also demonstrated a significant univariate association with mortality in the present nested case–control sample (Table 2 ), as previously reported for some of these proteins in the full BIOSTAT‐CHF cohort. 5 , 6 Notably, although T2DM was more common in non‐survivors at baseline, we did not observe any pathway‐level association with glycated haemoglobin (online supplementary Table S3 ).
Table 2.
Top ontologies and RV coefficient estimates with circulating protein biomarkers
| GO pathway (no. of genes) | FGF23 | sST2 | ADM | HEPC | PTX3 | WFDC2 | IL‐6 |
|---|---|---|---|---|---|---|---|
| Adaptive immune response (56) | 0.20 | 0.25 | 0.20 | 0.04 | 0.22 | 0.21 | 0.20 |
| T‐cell co‐stimulation (14) | 0.16 | 0.23 | 0.21 | 0.01 | 0.22 | 0.20 | 0.18 |
| Positive regulation of T‐cell proliferation (18) | 0.25 | 0.25 | 0.22 | 0.08 | 0.21 | 0.19 | 0.18 |
| Erythrocyte development (11) | 0.17 | 0.09 | 0.13 | 0.18 | 0.04 | 0.04 | 0.04 |
| Proteasome‐mediated ubiquitin‐dependent protein catabolic process (47) | 0.30 | 0.18 | 0.21 | 0.22 | 0.10 | 0.09 | 0.09 |
| Univariate association with survival (HR) | 1.64 | 1.70 | 1.92 | 0.84 | 1.57 | 1.88 | 1.43 |
| p < 1 × 10−34 | p < 1 × 10−20 | p < 1 × 10−22 | p < 1 × 10−4 | p < 1 × 10−9 | p < 1 × 10−28 | p < 1 × 10−12 |
They are listed in rank order from the RV analysis as follows: ADM, adrenomedullin; FGF23, fibroblast growth factor 23; HEPC, hepcidin; IL‐6, interleukin 6; sST2, soluble ST2 receptor (decoy receptor for IL‐33); PTX3, pentraxin‐3; WFDC2, WAP 4‐disulfide core domain 2. The table presents the RV coefficients – calculated between the expression of all genes identified within each of the top‐ranked gene ontology pathways and the levels of plasma protein biomarkers – for those proteins that demonstrated at least a 0.2 or > relationship with one of the five top‐ranked gene ontology pathways (all p < 0.001). The number of genes in each gene ontology pathway is listed in brackets. FE is the fold enrichment over the gene ontology database.
Co‐expression analysis
To further delineate the inter‐relationships between genes within each of the five GO categories and their relationship with the protein biomarkers, and how these might relate to prognosis, we created Pearson correlation heatmaps for each category, for survivors and non‐survivors separately (Figure 3 and online supplementary Figure S6 ). To facilitate identification of altered co‐expression relationships between survivors and non‐survivors we also created differential co‐expression plots (online supplementary Figures S7 and S8 ). The direction of gene co‐expression was largely conserved between survivors and non‐survivors, partly speaking to the robustness of the methodologies utilized. However, these analyses highlighted several individual examples of altered gene co‐expression, e.g. the iron transporter, transferrin receptor was upregulated in non‐survivors (Figure 2 ) and correlated to IL6ST (gp130) expression only in non‐survivors (Figure 3B and online supplementary Figure S8B ). Gene expression within the UPP pathway was subject to numerous shifts in the correlative structure (Figure 3 and online supplementary Figure S7B ) with genes like HECTD3 (up‐regulated in non‐survivors) becoming more positively co‐expressed with other members of the pathway (except for SMURF2 and RNF216). Likewise, the correlations between the seven protein biomarkers (FGF23, sST2, ADM, HEPC, PTX3, WFDC2 and IL‐6) tended to be stronger in non‐survivors, except for IL‐6 (Figure 3A vs. online supplementary Figure S3B ). The correlation between the seven individual proteins and individual genes varied substantially (online supplementary Figures S7 and S8 ) and this provided insight into some likely connections between the transcriptome profile and survival (see Discussion).
Figure 3.
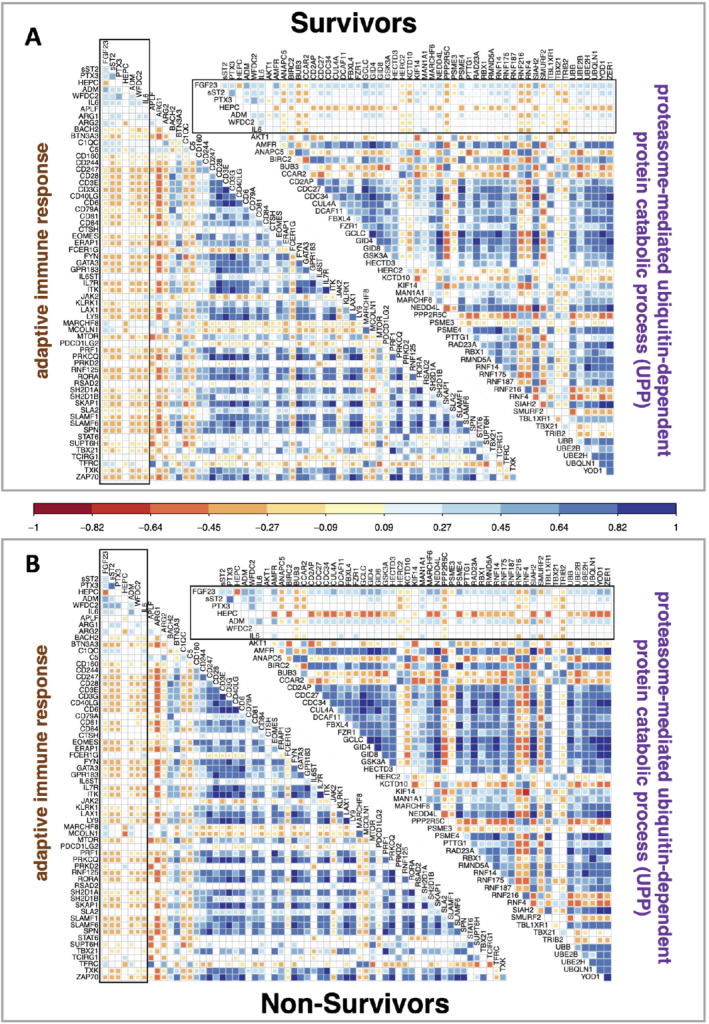
Top gene ontology pathway gene and protein biomarkers inter‐relationships. Gene expression was correlated with the top protein biomarkers using Pearson correlation coefficients for the two largest significant pathways ‘adaptative immune response’ and ‘proteasome‐mediated ubiquitin‐dependent protein catabolic process’ – the other three top ranked gene ontology pathways are presented in online supplementary Figure S6 . The protein values are enclosed by a black oblong box. Data are plotted separately (grey boxes) for survivors (n = 626) and non‐survivors (n = 318). Correlation values are represented by colour and are plotted for significant correlations (Bonferroni corrected threshold, p < 1.5 × 10−4).
Using mortality‐related transcript patterns to identify new drug targets
A search, using the combined 60 most up‐regulated and 60 most down‐regulated genes between survivors and non‐survivors that mapped to the iLINCS database, against >2500 in vitro drug signatures, found 45 matching compounds (online supplementary Table S4 ). This included 29 that reversed the mortality associated gene expression pattern, and these drugs had 47 known protein targets (<1 µM potency). Combined pathway analysis of the drug targets and the top regulated mortality associated genes established that the 47 proteins mapped to biological processes identified by the CHF mortality related transcriptome. This included the ‘immune response‐activating cell receptor signalling’ and ‘lymphocyte activation’ pathways (Figure 4 ).
Figure 4.
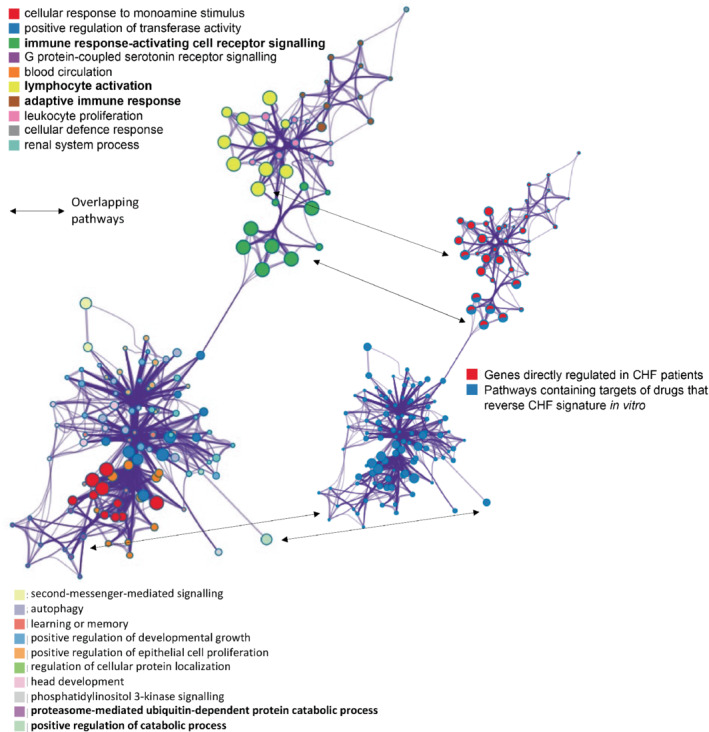
Pathway level overlaps between survival‐related genes and the protein targets of drugs that reverse the survival‐related gene expression signature in vitro. A network of significant pathways (p = 10−18–10−4) was derived using metascape.org. The input genes were the 120 chronic heart failure (CHF) survival‐associated genes used to identify drugs that regulate the CHF signature in vitro (https://clue.io/) and the 47 protein targets of the 29 drugs (identified using PubChem and the small‐molecule suite) that reverse the CHF signature in vitro. There are two identical plots. The large plot, on the left‐hand side, presents the significant pathways for this combined gene list. Edges represent connected gene ontology biological processes (>0.3), and nodes within each cluster are coloured/named by their most statistically enriched gene ontology term, scaled in size by the total number of terms represented. The smaller plot, on the right‐hand side, is same network structure but now colour coded by input list membership. This identifies if the drug targets appear within CHF survival‐associated pathways (pathways common to Figure 2 ) or whether the drug target falls within a pathway more indirectly connected with the patient transcriptomic signature. Each node is presented as a pie‐chart, with the ‘slices’ coloured and scaled to indicate which gene list the terms originate from, and what proportion the lists contribute to the ontology groupings.
Discussion
Whole blood transcriptomic profiling is a potentially powerful methodology to study genome‐wide biology directly in patients. Here, in the first prospective study of this type in heart failure, we provide novel insights into molecular processes associated with adverse CV prognosis. Specifically, we show that such profiles differ at baseline between those patients with CHF who died of a CV cause (over a median follow‐up of 21 months) compared to those that survived, with more than 1 in 20 of the detectable protein‐coding transcriptome showing a difference. Although the overall fold differences in gene expression between survivors and non‐survivors were small, these differences are likely to be driven by a subset of immune cells and therefore the fold change values would be ‘diluted’ by other cell types contributing to the global RNA profile. Thus, the magnitude of the measured differences should not be equated with their biological significance. Indeed, the differences could be mapped to specific biological pathways, several of which have been postulated to be disrupted in CHF. Importantly, we show that the differentially regulated transcriptome is strongly related to clinical and protein predictors of survival. While these findings are correlative, they highlight potential mechanisms and intermediaries through which the risks related to clinical factors are mediated. Finally, we provide initial evidence that the mortality‐associated RNA pathways in CHF can be reversed in vitro by drugs, some already approved for clinical use, identifying them – and their targets – as being potentially relevant to drug development for CHF.
Altered biological pathways and prognosis in heart failure
In cross‐sectional analysis, lymphopenia has been associated with more severe NYHA class and poorer outcomes in CHF. 16 , 17 Our study provides further detailed evidence that CHF patients with a poorer prognosis not only have fewer T‐ and B‐cell, but they also have an altered immune cell phenotype. Specifically, T‐cell receptor (TCR) signal‐transducing molecules such as CD3 epsilon and gamma, as well the co‐stimulatory receptor CD28, are down‐regulated in non‐survivors. Further, a number of genes encoding protein adaptors of TCR signals (such as Lck, Zap70, PKC‐θ, ITK and SKAP1), as well as genes that act to amplifying TCR signals such as CD6 18 were all down‐regulated (Figure 2 ). Many of these ‘adaptative immune response’ pathway members, and those involved in TCR signalling in particular, were negatively correlated (Figure 3 ) with both arginase gene transcripts (ARG1 and ARG2); which were 14%–16% more abundant in those that subsequently died (online supplementary Table S1 ). Arginase degrades arginine and decreased arginine, preceded by a measurable increase in arginase activity, is associated with poorer 6‐month outcomes in patients with ST‐elevation myocardial infarction. 19 Arginase gene expression, induced by lactate production consequent to hypoxia, can limit T‐cell proliferation. 20 A negative association between ARG2 and the pro‐survival IL‐7 receptor (IL7R/CD127) was observed in survivors (Figure 3A ) potentially linking arginine metabolism and IL7R signalling role in T‐cell survival and memory responses. 21 These observations provide support for altered metabolism contributing to the altered T‐cell molecular profile, which our transcriptome‐wide analysis indicates is a central feature of those patients that died.
Increased net catabolic activity has long been associated with poorer prognosis in heart failure 22 and our transcriptomics approach also identified that up‐regulation of the UPP pathway occurs in circulating blood cells (Figure 2 ). Siah E3 ubiquitin protein ligase 2 (SIAH2) was one of the most up‐regulated genes in the non‐survivor group (28%, p = 9 × 10−7) and it mediates degradation of heme oxygenase‐1, 23 a cardioprotective enzyme involved with immunomodulation. 24 HECTD3 – which has a role in both cardiac hypertrophy 25 and T helper‐17 development 26 – was expressed to a greater extent in those that died, showing a stronger (positive) co‐expression (online supplementary Figure S7B ) with other members of the activated UPP pathway (Figure 2 ). In contrast SMURF2 and RNF216 (two down‐regulated genes in those that died) were negatively associated with HECTD3 (Figure 3B ). This is interesting as SMURF2 is a E3 ubiquitin ligase targeting degradation of pro‐inflammatory TGFB/SMAD signalling, while RNF216 contributes to ubiquitin‐mediated protein degradation of TLR4. 27 Presumably targeted acceleration of protein degradation, through UPP, is required to facilitate proinflammatory processes and these more specific events may represent more tractable therapeutic targets.
It is plausible that one driver of the catabolic profile in those that died was disrupted iron homeostasis. As discussed below, altered iron regulatory protein, HEPC (which regulates ferroportin capacity 28 ) and induction of erythrocyte development revealed a number of key observations. For example, there was a strong positive association with the up‐regulated transferrin receptor (which imports iron into cells) and up‐regulated APFL (aprataxin PNK‐like factor) – a ubiquitous DNA damage response enzyme 29 potentially responding to inappropriate iron accumulation. 30 These observations indicate that molecular processes associated with cachexia in CHF, previously identified in cardiac and peripheral tissues, 22 are also evident in the whole blood transcriptome where a pervasive role for disrupted iron homeostasis appears across several of the altered pathways.
Do the mortality‐associated protein biomarkers drive the transcriptional responses to chronic heart failure?
As the seven protein biomarkers associated with the transcriptomic changes co‐varied with each other (online supplementary Figure S5 ), it is more challenging to identify which proteins are directly linked to the transcriptome differences, particularly as most have plausible mechanistic roles in CHF. For example, PTX3 can skew T‐cell differentiation towards the IL‐17 producing phenotype 31 while ADM has a positive impact on the endothelial integrity and anti‐inflammatory properties. 32 FGF23, a protein secreted that regulates calcium and phosphate homeostasis, 33 is produced by osteocytes and cardiac cells, and promotes hypertrophy and fibrosis in a calcineurin‐dependent manner. 34 WFDC2 (also known as HE4) suppresses osteopontin and promotes cell survival in an osteopontin‐ and interferon‐γ‐dependent manner. 35 WFDC2 is also linked to increased ARG1 activity and immune suppression. 36
Exploration of single transcript and protein biomarker relationships does, however, provide several clues as to the order of events, particularly for the altered T‐cell biology and iron homeostasis (Figure 3 and online supplementary Figure S6 ). For example, HEPC links to both altered UPP activity and altered T‐cell biology. HEPC demonstrated a strong association with the UPP and ‘erythrocyte development’ pathways (Figure 3 and online supplementary Figure S6 ). Notably, the association between HEPC protein and expression of genes from the UPP pathway was more striking in the non‐survivors (Figure 3 B, e.g. GCLC, PSME4, SIAH2, TBL1XR1, YOD1 and ZER1). In contrast, mucolipin (MCOLN1) was negatively associated with HEPC and one of the few up‐regulated genes in the ‘adaptive immune response’ pathway. MCOLN1 has an important role in calcium‐mediated cardiac remodelling 37 and is also implicated in iron homeostasis. 38 Together, these examples, and those listed above, imply that alterations in iron status may be a key integrator for the immune‐related changes in transcriptome and catabolic pathway activation. sST2 was also up‐regulated in those that died, consistent with its relationship with outcome following myocardial infarction, where it subdues IL‐33 signalling. 39 sST2 levels were positively correlated with the up‐regulated erythrocyte development pathway and of note, in the non‐survivor group, the increased ‘erythropoiesis development’ signature was accompanied by reduced haemoglobin (p < 1 × 10−6) and iron (Table 1 ). IL‐33 can be cardioprotective and is released upon tissue damage, stimulating T‐helper type 1 (Th1), Th2 and pro‐inflammatory Th17 cells, 40 such that loss of IL‐33 through increased sST2 may help explain the observed immune‐related transcriptome profile. Overall, our analysis indicates that each of these seven survival‐related proteins could drive the transcriptome response, coupling immunological and iron homeostasis together with clinical risk factors.
Using mortality‐related transcript patterns to identify new drug treatments
Not all circulating protein biomarkers or regulated genes make relevant therapeutic targets. For instance, despite being modulated in CHF, targeting tumour necrosis factor‐α or IL‐6 pathways has not led to improved outcomes in heart failure, or been found to reduce major CV events. 41 Notably, the present analysis did not rank either of these cytokines as being a dominant feature of the mortality‐related pathways. Instead, our analysis indicates that antagonizing FGF23 signalling and/or down‐regulating sST2 expression (or its binding to IL‐33) represent more enticing targets. Beyond these individual proteins, use of transcriptome signatures represents a promising alternative strategy to discover therapeutics and pathways that can modulate diseases. 7 , 14 , 42 In fact, many of the protein targets of the 29 drugs that reversed the mortality‐related transcriptome in vitro, are not regulated in patients, but rather represent members of the same pathways regulated in those that died (Figure 4 ). Notably, several positively acting drugs are known to target heart failure‐related molecular processes (online supplementary Table S4 ), and several have in vivo support for their potential utility in CHF, including a monoamine oxidase inhibitor which can be cardioprotective, 43 forskolin which elevates cAMP and cardiac function, 44 and the experimental epigenetic drug, chaetocin (a histone lysine methyltransferase inhibitor), which appears to be cardioprotective. 45 Our findings encourage a further appraisal of the potential benefit in CHF of some of these drugs and pathways.
Limitations
Our study has several limitations. Despite being the largest whole blood genome‐wide transcriptomics study in CHF conducted in a very well‐characterized cohort, further large scale independent transcriptomic studies are required to validate our observations. An important component of our strategy was to adjust for differences in cell counts between survivors and non‐survivors. We modelled changes in individual white cell subtypes using the transcriptomics data and studies using fresh blood samples from patients with CHF are needed to fully explore the changes we predicted. Most patients in BIOSTAT‐CHF had heart failure with reduced ejection fraction and whether our findings extend to those with preserved ejection fraction remains to be determined. Finally, our results are correlative and experimental studies are required to confirm the direct involvement of the various pathways we have identified in adverse prognosis in CHF.
Conclusion
In conclusion, our analysis provides evidence that whole blood transcriptomics can identify molecular pathways that associate with CV mortality in CHF. These reflect clinical factors that are associated with poor prognosis and may be driven by specific intermediary protein factors. We demonstrate the potential utility of such transcriptomic profiling for identifying novel therapeutic targets for CHF.
Supporting information
Appendix S1. Supporting Information
Supplementary Tables
Acknowledgement
We acknowledge the contribution of members of the BIOSTAT‐CHF consortium.
Funding
The BIOSTAT‐CHF project was funded by a grant from the European Commission (FP7‐242209‐BIOSTAT‐CHF). Development of the gene‐chip informatics methodologies were supported by the Medical Research Council UK (G1100015) to JAT. Dr Hamby is funded by the UK National Institute for Health Research and Drs Webb, Nelson, Timmons and Marelli‐Berg by the British Heart Foundation.
Conflict of interest: none declared.
References
- 1. Canepa M, Fonseca C, Chioncel O, Laroche C, Crespo‐Leiro M, Coats A, et al. Performance of prognostic risk scores in chronic heart failure patients enrolled in the European Society of Cardiology Heart Failure Long‐Term Registry. JACC Heart Fail. 2018;6:452–62. [DOI] [PubMed] [Google Scholar]
- 2. Voors AA, Ouwerkerk W, Zannad F, van Veldhuisen DJ, Samani NJ, Ponikowski P, et al. Development and validation of multivariable models to predict mortality and hospitalization in patients with heart failure. Eur J Heart Fail. 2017; 19:627–34. [DOI] [PubMed] [Google Scholar]
- 3. Brunner‐La Rocca HP, Sanders‐van Wijk S. Natriuretic peptides in chronic heart failure. Card Fail Rev. 2019;5:44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ibrahim NE, Januzzi JL. Established and emerging roles of biomarkers in heart failure. Circ Res. 2018;123:614–29. [DOI] [PubMed] [Google Scholar]
- 5. Ter Maaten JM, Voors A, Damman K, van der Meer P, Anker S, Cleland J, et al. Fibroblast growth factor 23 is related to profiles indicating volume overload, poor therapy optimization and prognosis in patients with new‐onset and worsening heart failure. Int J Cardiol. 2018;253:84–90. [DOI] [PubMed] [Google Scholar]
- 6. Markousis‐Mavrogenis G, Tromp J, Ouwerkerk W, Devalaraja M, Anker SD, Cleland JG, et al. The clinical significance of interleukin‐6 in heart failure: results from the BIOSTAT‐CHF study. Eur J Heart Fail. 2019;21:965–73. [DOI] [PubMed] [Google Scholar]
- 7. Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Voors AA, Anker SD, Cleland JG, Dickstein K, Filippatos G, van der Harst P, et al. A systems BIOlogy study to TAilored treatment in chronic heart failure: rationale, design, and baseline characteristics of BIOSTAT‐CHF. Eur J Heart Fail. 2016;18:716–26. [DOI] [PubMed] [Google Scholar]
- 9. Ferreira JP, Ouwerkerk W, Tromp J, Ng L, Dickstein K, Anker S, et al. Cardiovascular and non‐cardiovascular death distinction: the utility of troponin beyond N‐terminal pro‐B‐type natriuretic peptide. Findings from the BIOSTAT‐CHF study. Eur J Heart Fail. 2020;22:81–9. [DOI] [PubMed] [Google Scholar]
- 10. Monaco G, Lee B, Xu W, Mustafah S, Hwang YY, Carré C, et al. RNA‐Seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 2019;26:1627–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun. 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Timmons JA, Szkop KJ, Gallagher IJ. Multiple sources of bias confound functional enrichment analysis of global‐omics data. Genome Biol. 2015;16:15–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kroot JJC, Laarakkers CMM, Geurts‐Moespot AJ, Grebenchtchikov N, Pickkers P, Van Ede AE, et al. Immunochemical and mass‐spectrometry‐based serum hepcidin assays for iron metabolism disorders. Clin Chem. 2010;56:1570–9. [DOI] [PubMed] [Google Scholar]
- 14. Timmons JA, Anighoro A, Brogan RJ, Stahl J, Wahlestedt C, Farquhar DG, et al. A human‐based multi‐gene signature enables quantitative drug repurposing for metabolic disease. Elife. 2022;11: e68832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moret N, Clark NA, Hafner M, Wang Y, Lounkine E, Medvedovic M, et al. Cheminformatics tools for analyzing and designing optimized small‐molecule collections and libraries. Cell Chem Biol. 2019;26:765–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fukunaga T, Soejima H, Irie A, Sugamura K, Oe Y, Tanaka T, et al. Relation between CD4+ T‐cell activation and severity of chronic heart failure secondary to ischemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 2007;100:483–8. [DOI] [PubMed] [Google Scholar]
- 17. Yücel H, Ege MR, Zorlu A, Kaya H, Beton O, Güngör H, et al. Lymphocytopenia is associated with poor NYHA functional class in chronic heart failure patients with reduced ejection fraction. Turk Kardiyol Dern Ars. 2015;43:427–33. [DOI] [PubMed] [Google Scholar]
- 18. Hwang JR, Byeon Y, Kim D, Park SG. Recent insights of T cell receptor‐mediated signaling pathways for T cell activation and development. Exp Mol Med. 2020;52:750–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Molek P, Zmudzki P, Wlodarczyk A, Nessler J, Zalewski J. The shifted balance of arginine metabolites in acute myocardial infarction patients and its clinical relevance. Sci Rep. 2021;11:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, et al. Dichloroacetate improves immune dysfunction caused by tumor‐secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. 2013;133:1107–18. [DOI] [PubMed] [Google Scholar]
- 21. Belarif L, Mary C, Jacquemont L, Le Mai H, Danger R, Hervouet J, et al. IL‐7 receptor blockade blunts antigen‐specific memory T cell responses and chronic inflammation in primates. Nat Commun. 2018;9:4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rauchhaus M, Doehner W, Francis DP, Davos C, Kemp M, Liebenthal C, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102:3060–7. [DOI] [PubMed] [Google Scholar]
- 23. Chillappagari S, Belapurkar R, Möller A, Molenda N, Kracht M, Rohrbach S, et al. SIAH2‐mediated and organ‐specific restriction of HO‐1 expression by a dual mechanism. Sci Rep. 2020;10:2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tomczyk M, Kraszewska I, Dulak J, Jazwa‐Kusior A. Modulation of the monocyte/macrophage system in heart failure by targeting heme oxygenase‐1. Vascul Pharmacol. 2019;112:79–90. [DOI] [PubMed] [Google Scholar]
- 25. Rangrez AY, Borlepawar A, Schmiedel N, Deshpande A, Remes A, Kumari M, et al. The E3 ubiquitin ligase HectD3 attenuates cardiac hypertrophy and inflammation in mice. Commun Biol. 2020;3:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cho JJ, Xu Z, Parthasarathy U, Drashansky TT, Helm EY, Zuniga AN, et al. Hectd3 promotes pathogenic Th17 lineage through Stat3 activation and Malt1 signaling in neuroinflammation. Nat Commun. 2019;10:701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malonis RJ, Fu W, Jelcic MJ, Thompson M, Canter BS, Tsikitis M, et al. RNF11 sequestration of the E3 ligase SMURF2 on membranes antagonizes SMAD7 down‐regulation of transforming growth factor β signaling. J Biol Chem. 2017;292:7435–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823:1434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Corbeski I, Dolinar K, Wienk H, Boelens R, Van Ingen H. DNA repair factor APLF acts as a H2A‐H2B histone chaperone through binding its DNA interaction surface. Nucleic Acids Res. 2018;46:7138–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Camarena V, Sant DW, Huff TC, Mustafi S, Muir RK, Aron AT, et al. cAMP signaling regulates DNA hydroxymethylation by augmenting the intracellular labile ferrous iron pool. Elife. 2017;6:e29750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gupta G, Mou Z, Jia P, Sharma R, Zayats R, Viana SM, et al. The long pentraxin 3 (PTX3) suppresses immunity to cutaneous leishmaniasis by regulating CD4+ T helper cell response. Cell Rep. 2020;33:108513. [DOI] [PubMed] [Google Scholar]
- 32. Voors AA, Kremer D, Geven C, ter Maaten JM, Struck J, Bergmann A, et al. Adrenomedullin in heart failure: pathophysiology and therapeutic application. Eur J Heart Fail. 2019;21:163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ho BB, Bergwitz C. FGF23 signalling and physiology. J Mol Endocrinol. 2021;66:R23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Leifheit‐Nestler M, Haffner D. Paracrine effects of FGF23 on the heart. Front Endocrinol. 2018;9:278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. James NE, Cantillo E, Oliver MT, Rowswell‐Turner RB, Ribeiro JR, Kim KK, et al. HE4 suppresses the expression of osteopontin in mononuclear cells and compromises their cytotoxicity against ovarian cancer cells. Clin Exp Immunol. 2018;193:327–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rowswell‐Turner RB, Singh RK, Urh A, Yano N, Kim KK, Khazan N, et al. HE4 overexpression by ovarian cancer promotes a suppressive tumor immune microenvironment and enhanced tumor and macrophage PD‐L1 expression. J Immunol. 2021;206:2478–88. [DOI] [PubMed] [Google Scholar]
- 37. Falcón D, Galeano‐Otero I, Calderón‐Sánchez E, Del Toro R, Martín‐Bórnez M, Rosado JA, et al. TRP channels: current perspectives in the adverse cardiac remodelling. Front Physiol. 2019;10:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Grishchuk Y, Peña KA, Coblentz J, King VE, Humphrey DM, Wang SL, et al. Impaired myelination and reduced brain ferric iron in the mouse model of mucolipidosis IV. Dis Model Mech. 2015;8:1591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ciccone MM, Cortese F, Gesualdo M, Riccardi R, Di Nunzio D, Moncelli M, et al. A novel cardiac bio‐marker: ST2: a review. Molecules. 2013;18:15314–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Griesenauer B, Paczesny S. The ST2/IL‐33 axis in immune cells during inflammatory diseases. Front Immunol. 2017;8:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thanigaimani S, Phie J, Krishna S, Moxon J, Golledge J. Effect of disease modifying anti‐rheumatic drugs on major cardiovascular events: a meta‐analysis of randomized controlled trials. Sci Rep. 2021;11:6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wagner A, Cohen N, Kelder T, Amit U, Liebman E, Steinberg DM, et al. Drugs that reverse disease transcriptomic signatures are more effective in a mouse model of dyslipidemia. Mol Syst Biol. 2015;11:791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huuskonen C, Hämäläinen M, Paavonen T, Moilanen E, Mennander A. Monoamine oxidase a inhibition protects the myocardium after experimental acute volume overload. Anatol J Cardiol. 2019;21:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baumann G, Felix S, Sattelberger U, Klein G. Cardiovascular effects of forskolin (HL 362) in patients with idiopathic congestive cardiomyopathy – a comparative study with dobutamine and sodium nitroprusside. J Cardiovasc Pharmacol. 1990;16:93–100. [DOI] [PubMed] [Google Scholar]
- 45. Ono T, Kamimura N, Matsuhashi T, Nagai T, Nishiyama T, Endo J, et al. The histone 3 lysine 9 methyltransferase inhibitor chaetocin improves prognosis in a rat model of high salt diet‐induced heart failure. Sci Rep. 2017;7:39752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information
Supplementary Tables