

Abstract
POIKiloderma, tendon contractures, myopathy, pulmonary fibrosis is a congenital multisystem disorder due to FAM111B dominant variants. We present a literature review focusing on the frequency and the impact of hepatic involvement and a case report of a patient with severe end‐stage liver disease. Whole exome sequencing (WES) was conducted on the proband and his parents. A de novo FAM111B: c.1879A > G; (p.Arg627Gly) variant was identified. Hepatic involvement is present in 11 out of the 30 patients described in the literature, with different levels of dysfunction ranging from mild transaminitis to liver fibrosis found in three different cases by liver biopsies. Liver involvement seems to be a significant cause of morbidity. We propose to modify the previous acronym in POIK‐TMPL: including POIKiloderma, tendon contractures, myopathy, pulmonary fibrosis/pancreas insufficiency and cancer, liver involvement/lymphedema. Moreover, we suggest screening patients with FAM111B variants for liver involvement from the first month of life and continue with an appropriate follow‐up. Further studies are needed to better understand this frequent complication.
Keywords: cirrhosis, FAM111B, liver, POIKTMP, POIK‐TMPL, poikiloderma
Abbreviations
- CLD
chronic liver disease
- CT
computed tomography
- DLCO
diffusion of lung CO2
- EGD
esophagogastroduodenoscopy
- EMG
electromyography
- ESLD
end stage liver disease
- MRI
muscle magnetic resonance imaging
- POIKTMP
POIKiloderma, tendon contractures, myopathy, pulmonary fibrosis
- SSc
systemic sclerosis
- US
doppler ultrasound
1. BACKGROUND
Poikiloderma, tendon contractures, myopathy, pulmonary fibrosis (POIKTMP) (MIM#615704) is a congenital multisystemic disorder due to FAM111B (homo sapiens family with sequence similarity 111, member B) dominant variants, located on chromosome 11 (11q12.1), encoding a trypsin‐like cysteine/serine peptidase (Mercier et al., 2013). It is characterized by skin findings of poikiloderma (typically appearing in the first 6 months and mainly localized to the face and sun‐exposed areas), alopecia, hypohidrosis with heat intolerance, mild lymphedema of the extremities, muscle contractures (usually seen in childhood) and progressive muscle weakness of all four limbs. Some adults develop progressive interstitial pulmonary fibrosis with progressive dyspnea and restrictive lung function impairment. Other features are exocrine pancreatic insufficiency, liver impairment, hematologic abnormalities, relative short stature with growth retardation, and cataracts (Chen et al., 2019). Recently FAM111B has been reported as a susceptibility locus for prostate cancer (Akamatsu et al., 2012), whereas there is a high probability of pancreatic cancer predisposition in the POIKTMP condition: two patients died at the age of 64 (Goussot et al., 2017) and 32 years respectively (Mercier et al., 2019).
The exact prevalence of POIKTMP is unknown.
The diagnosis can be established by the concomitant presence of characteristic clinical features and histological findings signs. Muscle magnetic resonance imaging (MRI) and histological examination are helpful for diagnosis, as they show atrophy and extensive fatty infiltration of the muscle regardless of age. Microscopic analysis of skin biopsies reveals a scleroderma‐like aspect of the lesions with fibrosis and alterations of the elastic network. Genetic testing is the gold standard for the diagnosis of POIKTMP.
The phenotype of POIKTMP differs from the other well‐documented types of inherited poikiloderma: the Rothmund‐Thomson syndrome type 2 has overlapping clinical findings with POIKTMP, and it is caused by a mutation in RECQL4, with an autosomal recessive inheritance. Other syndromes characterized by poikiloderma are Werner syndrome, Clericuzio‐type poikiloderma with neutropenia, Kindler syndrome, Weary hereditary sclerosing poikiloderma, Baller‐Gerold syndrome and dyskeratosis congenita.
Late onset of poikiloderma characterizes the latter. Chromosome instability syndromes such as Bloom syndrome (sister chromatid exchange studies), Fanconi pancytopenia syndrome, ataxia‐telangiectasia and Nijmegen breakage syndrome (diepoxybutane, bleomycin, and mitomycin‐induced chromosome challenge), xeroderma pigmentosum (ultraviolet‐induced unscheduled DNA synthesis) and Charcot–Marie‐Tooth neuropathy should as well be considered in the differential diagnosis.
In 2006, a hereditary form of fibrosing poikiloderma (HFP) was observed for the first time associated with tendon contractures, myopathy, and pulmonary fibrosis in two generations of a South African family (Khumalo et al., 2006).
In 2013 Mercier et al. identified the causative gene FAM111B by whole‐exome sequencing (WES) in a French boy, born to unrelated parents, presented with poikiloderma at 1 month of age, eczematous lesions of the hands and feet, alopecia, hypohidrosis, lymphoedema of all limbs, progressive muscle weakness and tendon contractures of both feet. The authors cross‐referenced his heterozygous de novo variants to the list of heterozygous potentially damaging variants present in the affected members in the South African family: FAM111B appeared as the only candidate gene in common between the two families. Subsequently, POIKTMP was diagnosed in three affected persons of Algerian, Italian, and Moroccan origins. (Mercier et al., 2013)
To date 30 patients with FAM111B variants have been reported in the literature (in particular, 14 independent families and 12 sporadic patients). (Chen et al., 2019; Dokic et al., 2020; Goussot et al., 2017; Mercier et al., 2015; Roversi et al., 2021; Seo et al., 2016; Takeichi et al., 2017; Zhang et al., 2019); the patient we report was recently included in the paper of Roversi et al., describing the chromosomal instability in peripheral blood lymphocytes from two FAM111B‐mutated patients. (Roversi et al., 2021)
The principal features of patients described in the literature have been summarized in Table 1, end of search August 2021.
TABLE 1.
Main clinical features in patients with FAM111B variants reported in literature
| Mercier et al., 2015 | Seo et al., 2016 | Takeichi et al., 2017 | Goussot et al., 2017 | Zhang et al., 2019 | Chen et al., 2019 | Dokic et al., 2020 | Roversi et al., 2021 (present report) | Total N = 31 | ||
|---|---|---|---|---|---|---|---|---|---|---|
| N = 14 | N = 7 | N = 1 | N = 4 | N = 2 | N = 1 | N = 1 | N = 1 | |||
| Main clinical features | Poikiloderma | 14 | 7 | 1 | 4 | 2 | 1 | 1 | 1 | 31(100%) |
| Tendon contractures | 9 | 6 | 0 | 4 | 1 | 0 | 0 | 1 | 21 (68%) | |
| Myopathy | 8 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 10 (32%) | |
| Pulmonary fibrosis | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 10 (32%) | |
|
Exocrine pancreatic insufficiency |
4 | 5 | 0 | 3 | 0 | 0 | 0 | 0 | 12 (39%) | |
| Pancreas cancer | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 2 (6%) | |
| Liver involvement | 4 | 4 | 0 | 1 | 1 | 0 | 1 | 1 | 12 (39%) | |
| Lymphedema | 7 | 7 | 0 | 3 | 0 | 0 | 0 | 1 | 18 (58%) |
Note: Patients of different ages were grouped in a single table. Pulmonary fibrosis was assesed at radiological and functional test. Myopathy was assessed at MR or biopsy.
Table 2 reports main clinical, laboratory and imaging data of patients with FAM111B variants by age groups.
TABLE 2.
Major signs and symptoms of 31 individuals with FAM111B variants by age groups
| Age groups (years) | All patients | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0–6 | 6–12 | 12–30 | 30–50 | >50 | n = 31 | ||||||||||||||
| n = 7 | n = 5 | n = 11 | n = 4 | n = 4 | |||||||||||||||
| + | − | NA | + | − | NA | + | − | NA | + | − | NA | + | − | NA | + | − | NA | ||
| Phenotype | |||||||||||||||||||
| General | Consanguinity | 0 | 7 | 0 | 0 | 5 | 0 | 1 | 10 | 0 | 1 | 3 | 0 | 0 | 4 | 0 | 2 | 29 | 0 |
| Growth retardation | 6 | 8 | 17 | ||||||||||||||||
| Delayed puberty | 2 | 2 | 27 | ||||||||||||||||
| ↓ Heigh‐weigh CT/BMI | 7 | 2 | 22 | ||||||||||||||||
| Normal IQ | 14 | 0 | 17 | ||||||||||||||||
| Skin | Poikiloderma | 7 | 0 | 0 | 5 | 0 | 0 | 11 | 0 | 0 | 4 | 0 | 0 | 4 | 0 | 0 | 31 | 0 | 0 |
| Photosensitivity | 3 | 1 | 3 | 3 | 0 | 2 | 5 | 0 | 6 | 3 | 0 | 1 | 1 | 1 | 2 | 15 | 2 | 18 | |
| Truncal hypopigmentation | 2 | 2 | 3 | 2 | 2 | 1 | 6 | 3 | 2 | 1 | 3 | 0 | 3 | 1 | 0 | 14 | 11 | 6 | |
| Bullous lesions | 3 | 1 | 3 | 3 | 2 | 0 | 0 | 6 | 5 | 0 | 4 | 0 | 0 | 3 | 1 | 6 | 16 | 9 | |
| Eczema like | 7 | 0 | 0 | 3 | 2 | 0 | 4 | 5 | 2 | 2 | 2 | 0 | 1 | 3 | 0 | 17 | 12 | 2 | |
| Lymphoedema | 2 | 2 | 3 | 4 | 1 | 0 | 7 | 2 | 2 | 2 | 2 | 0 | 3 | 1 | 0 | 18 | 8 | 5 | |
| Sclerosis/scleroderma | 0 | 3 | 4 | 0 | 5 | 0 | 4 | 3 | 4 | 3 | 1 | 0 | 2 | 2 | 0 | 9 | 14 | 8 | |
| Cellulitis/erysipelas | 1 | 2 | 4 | 1 | 2 | 2 | 2 | 3 | 6 | 0 | 3 | 1 | 1 | 1 | 2 | 5 | 11 | 15 | |
| Hypoidrosis/heat intolerance | 6 | 1 | 0 | 4 | 1 | 0 | 8 | 0 | 3 | 3 | 0 | 1 | 4 | 0 | 0 | 25 | 2 | 4 | |
| Adnexa | Hypotrichosis/nonscarring alopecia | 6 | 0 | 1 | 5 | 0 | 0 | 7 | 1 | 3 | 4 | 0 | 0 | 3 | 0 | 1 | 25 | 1 | 5 |
| Nails dysplasia | 1 | 4 | 2 | 0 | 5 | 0 | 2 | 3 | 6 | 0 | 3 | 1 | 0 | 2 | 2 | 3 | 17 | 11 | |
| Muscles | Muscle weakness | 1 | 4 | 2 | 3 | 0 | 2 | 4 | 1 | 6 | 1 | 1 | 2 | 0 | 0 | 4 | 9 | 6 | 18 |
| Limbs | 1 | 4 | 2 | 4 | 0 | 1 | 2 | 1 | 8 | 1 | 1 | 2 | 0 | 0 | 4 | 8 | 6 | 17 | |
| Axial | 5 | 0 | 2 | 1 | 2 | 2 | 2 | 1 | 8 | 1 | 1 | 2 | 0 | 0 | 4 | 9 | 4 | 18 | |
| Amyotrophy | 5 | 0 | 2 | 2 | 1 | 2 | 2 | 2 | 7 | 1 | 1 | 2 | 0 | 0 | 4 | 10 | 4 | 17 | |
| Joints | Lower limbs contractures | 3 | 3 | 1 | 4 | 1 | 0 | 7 | 2 | 2 | 2 | 2 | 0 | 3 | 0 | 1 | 19 | 8 | 4 |
| Upper limbs contractures | 1 | 5 | 1 | 2 | 3 | 0 | 8 | 2 | 1 | 2 | 2 | 0 | 3 | 0 | 1 | 16 | 12 | 3 | |
| Skeletal | Scoliosis | 0 | 2 | 5 | 0 | 2 | 3 | 1 | 0 | 10 | 1 | 0 | 3 | 0 | 0 | 4 | 2 | 4 | 25 |
| Mouth | Dysphagia/velopharyngeal insufficiency | 0 | 3 | 4 | 1 | 2 | 2 | 1 | 1 | 9 | 1 | 1 | 2 | 0 | 0 | 4 | 3 | 7 | 21 |
| Liver | Hepatomegaly | 1 | 4 | 2 | 3 | 0 | 2 | 1 | 3 | 7 | 0 | 3 | 1 | 0 | 1 | 3 | 5 | 11 | 15 |
| Pancreas | Exocrine insufficiency | 1 | 2 | 4 | 3 | 2 | 0 | 1 | 0 | 10 | 0 | 1 | 3 | 0 | 0 | 4 | 5 | 5 | 21 |
| Steatorrhea/diarrhea | 2 | 1 | 4 | 2 | 3 | 0 | 2 | 0 | 9 | 0 | 2 | 2 | 1 | 0 | 3 | 7 | 6 | 18 | |
| Enzyme replacement | 1 | 1 | 5 | 1 | 1 | 3 | 0 | 0 | 11 | 0 | 0 | 4 | 0 | 0 | 4 | 2 | 2 | 27 | |
| Eyes | Cataract/amblyopia | One amblyopia | One cataract | ||||||||||||||||
| Laboratory | |||||||||||||||||||
| Blood tests | Eosinophilia | 1 | 0 | 6 | 2 | 2 | 1 | 0 | 3 | 8 | 0 | 1 | 3 | 0 | 0 | 4 | 3 | 6 | 22 |
| ↑ GOT‐AST/GPT‐ALT | 3 | 1 | 3 | 2 | 0 | 3 | 5 | 1 | 5 | 1 | 0 | 3 | 0 | 1 | 3 | 11 | 3 | 17 | |
| ↑ Gamma GT | 1 | 1 | 5 | 1 | 0 | 4 | 0 | 1 | 10 | 0 | 0 | 4 | 0 | 0 | 4 | 2 | 2 | 27 | |
| ↑ Alkaline phosphatase | 2 | 0 | 5 | 1 | 0 | 4 | 3 | 1 | 7 | 0 | 0 | 4 | 0 | 0 | 4 | 6 | 1 | 24 | |
| ↑ SCK | 1 | 2 | 4 | 1 | 1 | 3 | 2 | 1 | 8 | 1 | 1 | 2 | 0 | 0 | 4 | 5 | 5 | 21 | |
| Muscle exploration | Altered EMG | 0 | 0 | 7 | 2 | 0 | 3 | 1 | 1 | 9 | 0 | 0 | 4 | 0 | 0 | 4 | 3 | 1 | 27 |
| MRI/CT scan | 1 | 0 | 6 | 1 | 0 | 4 | 2 | 0 | 9 | 2 | 0 | 2 | 0 | 0 | 4 | 6 | 0 | 25 | |
| Adipose substitution | 1 | 1 | 2 | 2 | 6 | ||||||||||||||
| Fibrosis | 0 | 1 | 2 | 2 | 6 | ||||||||||||||
| Muscular biopsy | 0 | 0 | 7 | 2 | 0 | 3 | 3 | 0 | 8 | 1 | 0 | 3 | 0 | 0 | 4 | 6 | 0 | 25 | |
| Fibrosis | 1 | 3 | 1 | 5 | |||||||||||||||
| Adipose infiltration | 2 | 3 | 1 | 6 | |||||||||||||||
| Skin exploration | Skin biopsy | 3 | 0 | 4 | 3 | 0 | 2 | 3 | 0 | 8 | 0 | 0 | 4 | 0 | 0 | 4 | 9 | 0 | 22 |
| Fibrosis | 3 | 3 | 3 | 9 | |||||||||||||||
| Poikiloderma | 3 | 2 | 3 | 8 | |||||||||||||||
| Pancreas exploration | ↓ Pancreatic isoamilase | 0 | 0 | 7 | 2 | 0 | 3 | 1 | 0 | 10 | 1 | 0 | 3 | 1 | 0 | 3 | 5 | 0 | 26 |
| Liver exploration | Liver biopsy | 1 | 0 | 6 | 2 | 0 | 3 | 1 | 0 | 10 | 0 | 0 | 4 | 0 | 0 | 4 | 4 | 0 | 27 |
| Fibrosis | 1 | 2 | 1 | 3 | |||||||||||||||
| Macro‐microvescicular infiltration | 0 | 2 | 1 | 3 | |||||||||||||||
| Bone marrow exploration | Bone marrow biopsy and aspiration | 1 hypocellular BOM | 1 | ||||||||||||||||
| Lung exploration | Restrictive syndrome at PFT | 1 | 0 | 6 | 3 | 0 | 2 | 3 | 2 | 6 | 2 | 1 | 1 | 0 | 0 | 4 | 9 | 3 | 19 |
| ↓ DLCO | 0 | 0 | 7 | 2 | 0 | 3 | 2 | 2 | 7 | 2 | 0 | 2 | 0 | 0 | 4 | 6 | 2 | 23 | |
| Fibrosis at Thoracic CTs | 0 | 1 | 6 | 0 | 1 | 4 | 0 | 3 | 8 | 1 | 1 | 2 | 0 | 0 | 4 | 1 | 6 | 24 | |
| Associated diseases | One asthma, one GER, one VUR | One hypotiroidism | One pancreatic carcinoma | One pancreatic carcinoma | |||||||||||||||
| Sex | 16 M and 15 F | ||||||||||||||||||
Note: Our table retraces and confirms the major signs, symptoms and alterations in radiological, functional and anatomopathological tests, reported by Mercier et al. 2015 table's.
Abbreviations: −, negative; +, positive; DLCO, diffusing lung CO2; GER, gastro‐esophageal reflux; NA, non available; VUR, vescicoureteral reflux.
Liver involvement was described in POIKTMP patients with hepatomegaly, elevated transaminase levels, signs, and symptoms of cholestasis and steatosis. (Goussot et al., 2017; Mercier et al., 2015; Seo et al., 2016)
Hepatomegaly and transaminitis (elevated levels of serum transaminase ALT/AST) are the most common findings among these patients (Dokic et al., 2020; Goussot et al., 2017; Mercier et al., 2015; Seo et al., 2016; Zhang et al., 2019). In the previous literature, there are reports of three patients that underwent liver biopsy. The first two patients were described by Seo et al.: the proband, an eight‐year‐old girl with transaminitis and elevated levels of ALP and GGT, who showed, at liver biopsy performed when she was 2‐year‐old, microvesicular steatosis without symptoms of cholestasis and her older sister (10‐year‐old), with transaminitis, who showed, at two liver biopsies performed when she was 1 and 3‐year‐old macrovesicular and microvesicular steatosis, with a minimal local portal and lobular inflammation. (Seo et al., 2016) The third patient, a 6‐year‐old girl, recently reported by Dokic et al., presented with hepatomegaly and elevated levels of AST/ALT by around the age of 10‐months, with normal levels of ALP and GGT. She underwent several liver biopsies that showed lymphocytic ductulitis and duct loss, with progressive portal inflammation. An increased liver echotexture was observed at liver ultrasound. Due to these findings, the patient started systemic immunosuppressants. She presented with mild portal fibrous extension and hepatic vein thickening since she was 2.5 years old. At the age of 8 years no portal hypertension and standard liver stiffness were observed. (Dokic et al., 2020)
Although not highlighted as a prominent feature of the disease, liver involvement seems to be a significant morbidity cause. We describe a long‐term follow up of a patient with FAM111B mutation who died from liver failure. Furthermore, we present a literature review focusing on the frequency and the impact of hepatic involvement.
2. METHODS
This study aims to describe liver involvement among patients with FAM111B variants reported in previous literature, report the case of patients with severe chronic liver failure and propose a new acronym for the disease caused by FAM111B variants: POIK‐TMPL including POIKiloderma, tendon contractures, myopathy, pulmonary fibrosis/pancreas insufficiency and cancer, liver involvement. All patient data were collected during medical assessment in Bambino Gesù Children Hospital after obtaining informed consent. Data were recorded on a medical database.
3. GENETIC ANALYSIS
Genomic DNA was extracted from peripheral blood by using NucleoSpin tissue, according to the manufacturer's protocol (Macherey‐Nagel, Germany). Whole exome sequencing (WES) was conducted on the proband and his parents by using xGen Exome research panel v1.0 kit (IDT) for exome regions enrichment and Illumina platform for sequencing analysis. Variants were detected by means of the HaplotypeCaller software package of the Genome Analysis Toolkit suite and filtered so that to include only variants covered by at least 20 reads and with mapping quality values exceeding a Phred‐score of 30. Variants were analyzed under presumed autosomal recessive, X‐linked or de novo inheritance models. High‐quality variants were annotated and considered when not reported or having a low‐allele frequency <0.005 in genome AD exomes database and occurring with a frequency <0.01 in our in‐house database. Variants of the proband were filtered to retain all variants predicted to have functional impact (i.e. nonsynonymous variants and changes affecting splice sites) by available bioinformatics tools including PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), Sorting intolerant from tolerant (http://sift.jcvi.org/), Mutation Taster (http://www.mutationtaster.org/), Alamut (http://www.interactive-biosoftware.com/) and Combined annotation dependent depletion (http://cadd.gs.washington.edu/hom).
4. CASE REPORT
We report the case of a 17‐year‐old boy affected by POIKTMP due to a de novo FAM111B: c.1879A > G; (p.Arg627Gly) variant. His karyotype was 46, XY. He presented full expression of dermatological tendon and muscular involvement, but he died due to severe hepatic involvement characterized by hepatic fibrosis. The clinical picture was characterized by: Sparse hair and areas of alopecia, absence of eyelashes and eyebrows, and poikiloderma; severe progressive myopathy and tendon contractures that resulted in wrist ulnar deviation and flexion of hands and feet; muscle wasting and weakness of proximal and distal upper and lower limbs; marked lymphedema of lower limbs.
He was the first of two siblings (he had a healthy brother). His nonconsanguineous and healthy parents, aged 24 years (mother) and 29 years (father) at delivery, originate from a small village of fewer than 10.000 inhabitants in south Italy. He was born at term after an uneventful pregnancy weighing 3 kg (around the 25%). Motor and neurodevelopmental milestones were reportedly normal. At 2 months, he presented convergent strabismus and underwent corrective surgery at 3 years.
From 2 months of life he developed recurrent eczema and photosensitivity with vesicular eruption after sun exposure and later nodular hypochromic/atrophic macules on the face and neck, exuding dermatitis on the neck, wrist, forearm and thigh as well as diffuse telangiectasias. Based on the presence of poikiloderma, erythemato‐squamous plaques on the elbows and knees, and photosensitivity, Rothmund–Thomson syndrome was suspected, but molecular analysis for RECQL4 variants was negative.
A first skin biopsy showed minimal inflammatory infiltrate in the superficial dermis with occasional perivascular melanophages. A second skin biopsy performed at the age of 11, showed ortho‐ and para‐keratotic keratosis of the epithelial lining, which was thinned, and showed hypo‐atrophy of “network ridges.” The upper dermis was characterized by edema, telangiectasia and scattered inflammatory lymphohistiocytic elements with melanophages, a pattern compatible with poikiloderma.
Since the age of four, he presented generalized muscle weakness and wasting that was prominent in distal upper limbs, associated with tendon contractures, leading to ulnar deviation of wrists and finger flexion contractures due to the involvement of the metacarpophalangeal joints. He also had calcaneal tendon contraction with almost absent reflexes. In the following years, he showed a progression in muscular wasting, involving axial and girdle muscles, and a progressive involvement in tendon contractures reduced the ability to extend knees and elbows and led to an inability to walk.
A first muscle biopsy performed at 4 years, showed marked signs of fibrosis with loss of muscle tissue and fatty infiltration. A second muscle biopsy performed at the age of 10, showed increased fibrosis and fatty infiltration with progressive loss of muscle fibers.
Muscular MRI performed at the age of 7 and 11 documented a progressive muscle tissue loss pattern both on thighs and calves with fat replacement as shown by the increased signal intensity in T1‐weighted images.
Electrophysiological examinations excluded peripheral sensory neuropathy, while electromyography showed signs of neurogenic lesion in the right vastus medialis muscle.
Since the age of 5 years old, he additionally manifested progressive lymphedema of the lower legs (Figure 1a–c), and developed recurrent cutaneous infections.
FIGURE 1.
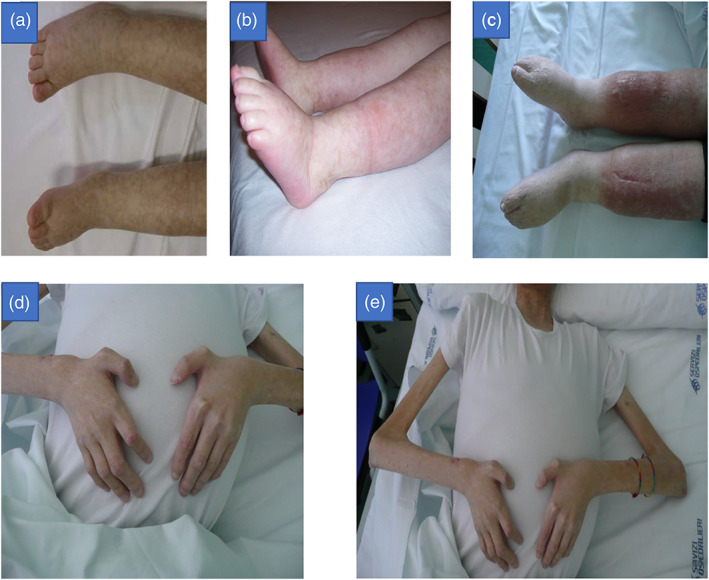
In Figure 1a–c are shown the progression of lymphedema of lower limbs and tendon contractures associated to muscle wasting. In Figure 1d (17‐year‐old) is it possible to observe sclerosis of digits and contractures of upper limbs. In Figure 1e, the patient (17‐year‐old) presented severe muscular wasting, he was cachectic due to the progression of liver disease and showed abdominal bloating due to ascites, secondary to chronic liver failure.
Lung involvement was evident at the age of 16‐year‐old, with a severe restrictive pattern at the spirometry (without diffusion of lung CO2, DLCO) with a forced vital capacity (FVC) of 1.01 (31% of predicted), a forced expiratory volume in the first second (FEV1) of 0.86 (30% of predicted), and a Tiffeneau Index (FEV1/FVC%) of 85.2 (96% of predicted). At nocturnal pulse oximetry, he showed a normal average mean SaO2, associated with significant numbers of desaturation. He reached a maximum height of 146 cm (<3%) and a weight of 39.6 kg (<3%). Bone X‐rays showed severe osteopenia. Cognitive development has always been normal.
Liver involvement appeared in the first year of life with abnormal liver enzymes. An extensive multidisciplinary diagnostic workup ruled out all the main causes of chronic liver disease. Due to persistent hypertransaminasemia (see Table 3), with normal levels of serum creatinine kinase (SCK) liver biopsy was performed showing a vanishing bile ducts syndrome at 2 years of age. A second biopsy was performed 4 years later, revealing the progression of the chronic cholestasis with biliary fibrosis together with micro‐ and macrovesicular steatosis and numerous Kupffer cells with large, leaf‐like, cytoplasm (Figure 2). Around the age of 6‐year‐old, due to marked splenomegaly he started showing signs of hypersplenism, such as thrombocytopenia and leukopenia, in keeping with development of portal hypertension. Primary prophylaxis of variceal bleeding was started with regular upper gastrointestinal endoscopies and band ligation when indicated. Since the age of 13 years, he presented signs of advanced liver dysfunction with coagulopathy, hyperammonemia and severe refractory ascites (Figure 1e) despite intensive medical treatment with albumin infusions and diuretic medications among other chronic liver disease support. During the patient's follow‐up, the regular imaging monitoring of his liver involvement with doppler ultrasound (US) (Figure 3a) and computed tomography (CT) showed the progression of the chronic liver disease from hepatomegaly with increased echogenicity due to steatosis to a final pattern of end stage liver disease (ESLD). The last abdomen CT scan performed at 15 years of age, showed signs of micro and macronodular cirrhotic evolution with portal hypertension (periumbilical/paraesophageal collateral veins, enlarged spleen, and reduced portal flow) (Figure 3b). Liver transplantation was hypothesized but excluded due to the severity of comorbidities.
TABLE 3.
Clinical and molecular data of patients with FAM111B variants and liver involvement
| Mercier et al., 2015 | Seo et al., 2015 | Goussot et al., 2017 | Zhang et al., 2019 | Dokic et al., 2020 | Roversi et al., 2021 (present report) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Characteristics | P1 (13 years) Origin: Italy | P2 (4 years) Origin: France | P3 (5 years) Origin: Ireland | P4 (23 years) Origin: Dominican Republic | P5 (32 years) Origin: Canada | P6 (28 years) Origin: Canada | P7 (8 years) Origin: Canada | P8 (10 years) Origin: Canada | P9 (27 years) origin:France | P10 (14 months) origin: China | P11 (6 years) Origin: Mexican | P12 (17 years) Italian |
| Sex | Female | Female | Female | Male | Male | Female | Female | Female | Female | Female | Female | Male |
| Hepatomegaly | + | − | − | − | n/a | n/a | + | + | − | n/a | + | + |
| Elevated serum ALT/AST | n/a | AST: 63 IU/L, ALT: 56 IU/L | AST:210 IU/L, ALT: 151 IU/L | AST: 100 IU/L, ALT: 132 IU/L | + (data not available) | + (data not available) | AST: 178 IU/L, ALT: 251 IU/L | + (data not available) | + (data not available) | AST: 202 U/L, ALT: 208 U/L | + (data not available) | AST 143 IU/L, ALT 198 IU/L |
| Liver biopsy | n/a | n/a | n/a | n/a | n/a | n/a | 2‐years‐old: Mi | 1 and 3 years‐old: Mi, Ma, F | n/a | n/a | Several biopsies: C, F | 1 and 5 years‐old: Mi, Ma, C, F |
|
Liver ultrasound |
n/a | n/a | n/a | n/a | n/a | n/a | − | n/a | n/a | n/a | US: increased liver echotexture | US: cirrhosis, portal vein dilated |
| Blood test for cholestasis | + (data not available) | ALP: 308 IU/L (<335); GGT: 53 IU/L (<26) | ALP: 772 IU/L (<315); Bil tot: 33 mmol/l (<14) | ALP:129 IU/L (<129); GGT:106 IU/L (<58) | n/a | n/a | ALP 506 IU/L (<380), GGT 407 IU/L (<55) | n/a | n/a | − | − |
ALP 486 U/L, GGT 12 IU/L, Bil Tot 2.09 mg/dL, Bil Dir 1,55 mg/dL |
|
Gene analysis FAM111B mutations |
c.1879A > G (p.Arg627Gly) | c.1883G > A (p.Ser628Asn) | c.1883G > A (p.Ser628Asn) | c.1883G > A (p.Ser628Asn) | c.1261_1263delAAG (p.Lys421del) | c.1261_1263delAAG (p.Lys421del) | c.1261_1263delAAG (p.Lys421del) | c.1261_1263delAAG (p.Lys421del) | c.1884 T > A (p.Ser628Arg) | c.1247 T > C (p.Phe416Ser) | c.1881 C > T, (p.Arg672Ser) | c.1879A > G; (p.Arg627Gly) |
Abbreviations: −, negative; +, positive; C, cholestatis; F, fibrosis; ma, macrovescicular steatosis; Mi, microvescicular steatosis; MRI, magnetic resonance imaging; P, patient; US, ultrasound.
FIGURE 2.
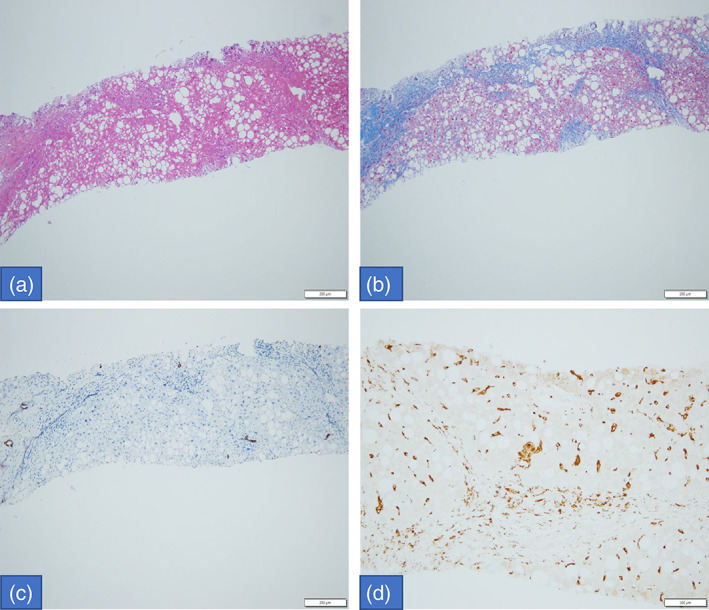
Histologic exam of liver biopsy: macrovesicular steatosis with few portal‐inflammatory cells (HE. 10×); disturbed architecture due to porto‐portal fibrous septa (Masson trichrome stain, 10×); loss of biliary ducts (CK7 IHC, 10×) and increased Kupffer cells, sometimes with large leaf‐like cytoplasm (CD68 IHC, 20×).
FIGURE 3.
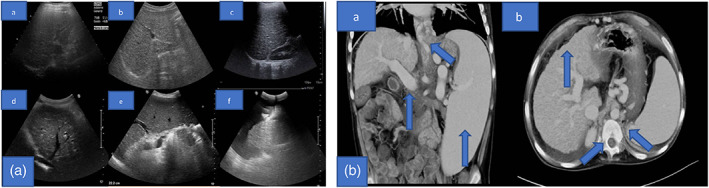
(a) Liver echography in different years: a. Year 2004, b. Year 2007, c. Year 2009, d. Year 2013, e. Year 2015, and f. Year 2016. In this figure it is shown the evolution of liver involvement in our patients in a time span of 12 years, seen on ultrasound scan. He developed a progressive cirrhosis, with a micro‐ and macronodular pattern, predominantly in right liver sections. Portal vein and its intrahepatic branches are dilated. Irregularity in the Glisson's capsule contour. in figure “f” it is possible to observe a massive ascites. The intra and extra‐hepatic biliary tree is not dilated. (b) a, b Abdominal CT: severe reduction in the dimension of the liver, predominantly involving the right sections of the liver, with a clear micro and macronodular pattern, in relation to the cirrhotic involution of the liver. Portal vein and its branches are markedly dilated, with an arterialization of the hepatic parenchyma; activation of the paraumbilical and periesophageal collateral pathways. Extra‐ and intrahepatic biliary tree is in the range of normality. it is possible to observe an important splenomegaly with homogeneous densitometry. Presence of free abdominal fluid. The diaphragmatic crus are bilaterally thickened. Levoscoliosis of the spine.
The rapid progression of liver disease and its related complications brought our patient to death at the age of 17 years.
5. DISCUSSION
POIKTMP is caused by pathogenic variants in the FAM111B gene (MIM#615584) on chromosome 11q12.1. It is inherited in an autosomal dominant manner.
Main features of POIKTMP (POIKiloderma, tendon contractures, myopathy, pulmonary fibrosis) are: (I) congenital poikiloderma on the face and exposed skin, resulting in hypo or hyperpigmentation, telangiectasias and atrophy (Mercier et al., 2015). (II) Tendon contractures of upper limbs and more frequently of distal lower limbs that, in some cases, result in gait abnormalities, requiring surgical tendon lengthening (Mercier et al., 2015). (III) Myopathy, that results in muscle wasting and weakness due to fatty infiltration and focal lymphocytic and macrophage infiltrates revealed on biopsy and adiposis on MRI (Mercier et al., 2013; Mercier et al., 2015). (IV) Pulmonary fibrosis that is a typical manifestation of the second decade of life, ranging from mild involvement with dyspnea to severe interstitial pulmonary fibrosis with restrictive syndrome that, in severe cases, can lead to death (Mercier et al., 2013; Mercier et al., 2015). Other important clinical features reported in the literature are: Pancreatic exocrine insufficiency that can present with diarrhea, fatty stools, liposoluble vitamins deficiency, with insufficient levels of vitamin K that can lead to increased International Normalized Ratio (INR) (Seo et al., 2016).
Malignancies were recently reported in literature among patients with FAM111B variants with a higher risk of developing pancreatic cancer (Goussot et al., 2017; Mercier et al., 2019) and prostate cancer (Akamatsu et al., 2012). Other uncommon associations are growth retardation, cataract, scoliosis and delayed puberty (Mercier et al., 2015).
As described above, liver involvement is another important clinical feature of FAM111B‐related disorder spectrum (Dokic et al., 2020; Mercier et al., 2015; Mercier et al., 2019).
We identified a patient affected by POIKTMP due to the de novo FAM111B deleterious missense variant: c.1879A > G; (p.Arg627Gly). Although this variant has been previously reported in four patients of different ages (ranging from 8‐month to 32‐year‐old), and among them only a 13‐year‐old patient presented hepatomegaly and signs of cholestasis (Mercier et al., 2015), to the best of our knowledge 11 other patients described in the literature showed different degrees of hepatic involvement (Table 3). In this view, it is worthy of mention that their deleterious variants harbor closer to the third predicted active protein site located at FAM111B 650 amino acid residue (Figure 4a) with recurrent single amino acid substitutions at 627–628 positions (six out of six of missense variants retrieved, plus the present report) which may impinge on the efficiency of protein folding in cells.
FIGURE 4.
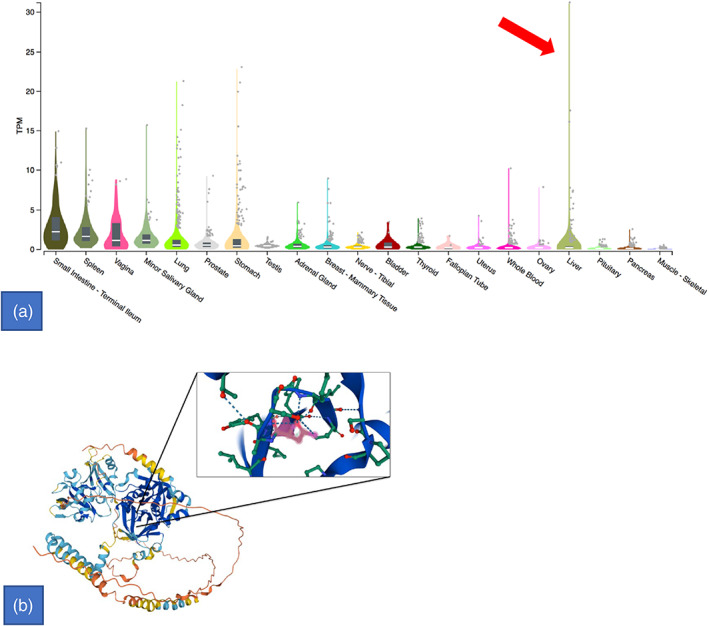
(a) Bulk tissue gene expression for FAM111B. Transcripts per million (TPM) concentration values' violin scale intensity plot (Y‐axis) broken out per all available tissue's transcriptomic analyses (X‐axis) shows outliers values in liver (red arrow). (b) FAM111B structure: tertiary and secondary structure with third predicted active protein site located at 650 amino acid residue showed in the magnification panel. Figure generated by using UniProt (Morgat A. et al., 2015; https://www.uniprot.org). Reference: Morgat, A., Lombardot, T., Coudert, E., Axelsen, K., Neto, T. B., Gehant, S., … & UniProt Consortium. (2020). Enzyme annotation in UniProtKB using Rhea. Bioinformatics
Thus, taking together all the features mentioned above, and also based on the available tissues transcriptome analyses whose FAM111B transcript per million (TPM) values show outliers in liver (Figure 4b), a genotype–phenotype correlation for the liver involvement may be proposed widening the scope of existing of FAM111B‐related disorder's clinical spectrum.
The patient hepatic involvement was severe and progressive, liver enzymes and biopsy were abnormal since the first year of life but clinically silent, while he progressively developed signs and symptoms of liver failure from the age of 13 years.
Although medical literature already reported 11 patients out of 30 with FAM111B variants showing different levels of hepatic dysfunction (Table 3), our patient is the first case described with severe hepatic fibrosis finally causing ESLD who died at the age of 17 years.
Moreover, most of the 11 patients described were young, with a median age of 14.3 years (interquartile range 1.2–32 years).
Interestingly, in a recent case series of 28 patients with FAM111B variants, Chasseuil et al. reported, albeit without giving specific information, a worsening during the patient's lifetime in all extracutaneous manifestations (gastrointestinal, pulmonary and muscular). (Chasseuil et al., 2019).
Due to these significant findings, we suggest emphasizing the gastrointestinal (especially hepatic) aspect in FAM111B proposing to modify the previous acronym of POIKTMP in POIK‐TMPL including POIKiloderma, tendon contractures, myopathy, pulmonary fibrosis/pancreas insufficiency and cancer, liver involvement.
We also speculate on the necessity to rule out liver involvement with first‐line liver function tests clinical and radiological examinations during the first month of life and continue with an appropriate follow up in subsequent years. Finally, liver transplantation should be considered as a possible therapy for ESLD in these patients. However, the important and severe extra‐intestinal complications characteristic of this disease, as observed in the patient we described, may exclude the possibility to perform liver transplantation.
6. CONCLUSIONS
Liver involvement is a key feature in several patients with FAM111B variants described in literature. According to these observations, and to our patient history, we propose the following acronym, POIK‐TMPL: including POIKiloderma, tendon contractures, myopathy, pulmonary fibrosis/pancreas insufficiency and cancer, liver involvement/lymphedema. Moreover, we suggest screening FAM111B patients with an appropriate follow‐up for liver involvement. Further studies are needed to understand this frequent complication better, improve the prognosis and find a possible therapy.
AUTHOR CONTRIBUTIONS
Dr. Marina Macchiaiolo and Filippo M. Panfili and Fabiana Cortellessa conceptualized and designed the article, collected data, drafted the initial manuscript, and reviewed and revised the final manuscript. Dr. Michaela V. Gonfiantini, Paola S. Buonuomo, Davide Vecchio, Paola Francalanci and Lorena Travaglini collected data, supervised data and reviewed and revised the final manuscript. Dr. Andrea Pietrobattista, Maya El Hachem, and Enrico S. Bertini reviewed the manuscript for important intellectual content. Dr. Andrea Bartuli conceptualized and designed the article, collected data, drafted and coordinated and supervised data, reviewed and revised the final manuscript.
FUNDING INFORMATION
No funding was secured for this study. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
CONFLICT OF INTEREST
No disclosure for any prior publication or submission. No conflict of interest or role of any sponsor is present in this work.
ETHICS STATEMENT
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Mauro Paradisi (1949–2020) for the important help in the management and clinical evaluation of our patient.
Macchiaiolo, M. , Panfili, F. M. , Vecchio, D. , Cortellessa, F. , Gonfiantini, M. V. , Buonuomo, P. S. , Pietrobattista, A. , Francalanci, P. , Travaglini, L. , Bertini, E. S. , El Hachem, M. , & Bartuli, A. (2022). Expanding phenotype of FAM111B ‐related disease focusing on liver involvement: Literature review, report of a case with end‐stage liver disease and proposal for a new acronym. American Journal of Medical Genetics Part A, 188A:2920–2931. 10.1002/ajmg.a.62906
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Akamatsu, S. , Takata, R. , Haiman, C. A. , Takahashi, A. , Inoue, T. , Kubo, M. , Furihata, M. , Kamatani, N. , Inazawa, J. , Chen, G. K. , Le Marchand, L. , Kolonel, L. N. , Katoh, T. , Yamano, Y. , Yamakado, M. , Takahashi, H. , Yamada, H. , Egawa, S. , Fujioka, T. , … Nakagawa, H. (2012). Common variants at 11q12, 10q26 and 3p11.2 are associated with prostate cancer susceptibility in Japanese. Nature Genetics, 44(4), 426–429. 10.1038/ng.1104 [DOI] [PubMed] [Google Scholar]
- Chasseuil, E. , McGrath, J. A. , Seo, A. , Balguerie, X. , Bodak, N. , Chasseuil, H. , Denis‐Musquer, M. , Goldenberg, A. , Goussot, R. , Irvine, A. D. , Khumalo, N. P. , King, M. C. , Küry, S. , Lipsker, D. , Mallet, S. , Mayosi, B. M. , Nanda, A. , Puzenat, E. , Salort‐Campana, E. , … Barbarot, S. (2019. Oct). Dermatological manifestations of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis (POIKTMP): A case series of 28 patients. The British Journal of Dermatology, 181(4), 862–864. 10.1111/bjd.17996 [DOI] [PubMed] [Google Scholar]
- Chen, F. , Zheng, L. , Li, Y. , Li, H. , Yao, Z. , & Li, M. (2019). Mutation in FAM111B causes hereditary Fibrosing Poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Acta Dermato‐Venereologica, 99(7), 695–696. 10.2340/00015555-3186 [DOI] [PubMed] [Google Scholar]
- Dokic, Y. , Albahrani, Y. , Phung, T. , Patel, K. , de Guzman, M. , Hertel, P. , & Hunt, R. (2020). Hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis: Hepatic disease in a child with a novel pathogenic variant of FAM111B . JAAD Case Rep., 6(12), 1217–1220. 10.1016/j.jdcr.2020.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goussot, R. , Prasad, M. , Stoetzel, C. , Lenormand, C. , Dollfus, H. , & Lipsker, D. (2017). Expanding phenotype of hereditary fibrosing poikiloderma with tendon contractures, myopathy, and pulmonary fibrosis caused by FAM111B mutations: Report of an additional family raising the question of cancer predisposition and a short review of early‐onset poikiloderma. JAAD Case Rep., 3(2), 143–150. 10.1016/j.jdcr.2017.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khumalo, N. P. , Pillay, K. , Beighton, P. , Wainwright, H. , Walker, B. , Saxe, N. , Mayosi, B. M. , & Bateman, E. D. (2006). Poikiloderma, tendon contracture and pulmonary fibrosis: A new autosomal dominant syndrome? The British Journal of Dermatology, 155(5), 1057–1061. 10.1111/j.1365-2133.2006.07473.x [DOI] [PubMed] [Google Scholar]
- Mercier, S. , Küry, S. , Nahon, S. , Salort‐Campana, E. , Barbarot, S. , & Bézieau, S. (2019). FAM111B mutation is associated with pancreatic cancer predisposition. Pancreas, 48(5), e41–e42. 10.1097/MPA.0000000000001303 [DOI] [PubMed] [Google Scholar]
- Mercier, S. , Küry, S. , Salort‐Campana, E. , Magot, A. , Agbim, U. , Besnard, T. , Bodak, N. , Bou‐Hanna, C. , Bréhéret, F. , Brunelle, P. , Caillon, F. , Chabrol, B. , Cormier‐Daire, V. , David, A. , Eymard, B. , Faivre, L. , Figarella‐Branger, D. , Fleurence, E. , Ganapathi, M. , … Bézieau, S. (2015). Expanding the clinical spectrum of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis due to FAM111B mutations. Orphanet Journal of Rare Diseases, 15(10), 135. 10.1186/s13023-015-0352-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier, S. , Küry, S. , Shaboodien, G. , Houniet, D. T. , Khumalo, N. P. , Bou‐Hanna, C. , Bodak, N. , Cormier‐Daire, V. , David, A. , Faivre, L. , Figarella‐Branger, D. , Gherardi, R. K. , Glen, E. , Hamel, A. , Laboisse, C. , le Caignec, C. , Lindenbaum, P. , Magot, A. , Munnich, A. , … Mayosi, B. M. (2013). Mutations in FAM111B cause hereditary fibrosing poikiloderma with tendon contracture, myopathy, and pulmonary fibrosis. Am J Hum Genet, 93(6), 1100–1107. 10.1016/j.ajhg.2013.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roversi G, Colombo EA, Magnani I, Gervasini C, Maggiore G, Paradisi M, Larizza L. Spontaneous chromosomal instability in peripheral blood lymphocytes from two molecularly confirmed Italian patients with hereditary fibrosis Poikiloderma: Insights into cancer predisposition. Genetics and Molecular Biology 2021;44(3):e20200332. doi: 10.1590/1678-4685-GMB-2020-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, A. , Walsh, T. , Lee, M. K. , Ho, P. A. , Hsu, E. K. , Sidbury, R. , King, M. C. , & Shimamura, A. (2016). FAM111B mutation is associated with inherited exocrine pancreatic dysfunction. Pancreas, 45(6), 858–862. 10.1097/MPA.0000000000000529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi, T. , Nanda, A. , Yang, H. S. , Hsu, C. K. , Lee, J. Y. , Al‐Ajmi, H. , Akiyama, M. , Simpson, M. A. , & McGrath, J. A. (2017. Feb). Syndromic inherited poikiloderma due to a de novo mutation in FAM111B. The British Journal of Dermatology, 176(2), 534–536. 10.1111/bjd.14845 [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Zhang, J. , Chen, F. , Zheng, L. , Li, H. , Liu, M. , Li, M. , & Yao, Z. (2019). Family of hereditary fibrosing poikiloderma with tendon contractures, myopathy and pulmonary fibrosis caused by a novel FAM111B mutation. The Journal of Dermatology, 46(11), 1014–1018. 10.1111/1346-8138.15045 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.