

Abstract
The face‐to‐face association of (E)‐1,2‐di(4‐pyridyl)ethylene (bpen) molecules into rectangular motifs stabilized for the first time by chalcogen bonding (ChB) interactions is shown to provide photoreactive systems leading to cyclobutane formation through single‐crystal‐to‐single‐crystal [2+2] photodimerizations. The chelating chalcogen bond donors are based on original aromatic, ortho‐substituted bis(selenocyanato)benzene derivatives 1–3, prepared from ortho‐diboronic acid bis(pinacol) ester precursors and SeO2 and malononitrile in 75–90 % yield. The very short intramolecular Se⋅⋅⋅Se distance in 1–3 (3.22–3.24 Å), a consequence of a strong intramolecular ChB interaction, expands to 3.52–3.54 Å in the chalcogen‐bonded adducts with bpen, a distance (<4 Å) well adapted to the face‐to‐face association of the bpen molecules into the reactive position toward photochemical dimerization.
Keywords: Chalcogens, Crystal Engineering, Cycloaddition, Selenocyanates, Topochemistry
Chalcogen bonding (ChB) interactions are used for the first time to organize photoreactive systems, here (E)‐1,2‐di(4‐pyridyl)ethylene (bpen) molecules, into rectangular motifs favoring cyclobutane formation through single‐crystal‐to‐single‐crystal [2+2] photodimerizations, demonstrating the efficiency and robustness of strong and directional ChB donors such as selenocyanate moieties toward topochemical reactions.

Noncovalent interactions have been used in the last years to direct the solid‐state organization of reactive molecules toward topochemical reactions under external stimuli such as light or heat. [1] This strategy has been applied with success with olefins when using pincer‐like auxiliary molecules or templates to organize them in a face‐to‐face arrangement suitable for [2+2] cycloaddition under UV irradiation. [2] One prototypical example is the association of (E)‐1,2‐di(4‐pyridyl)ethylene (bpen) with rigid ditopic templates able to interact with the pyridinyl nitrogen atoms through hydrogen bonding interactions [3] (Scheme 1) or metal coordination. [4]
Scheme 1.
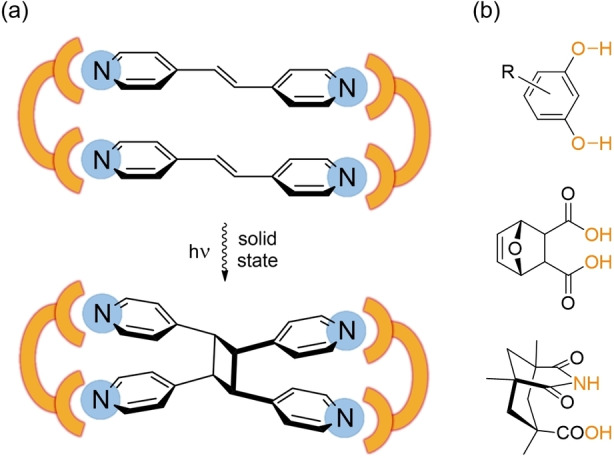
a) Templated photodimerization of bpen in molecular rectangles. b) Examples of ditopic hydrogen bond donors used toward the formation of such molecular rectangles.
Under UV irradiation, the corresponding cyclobutane is formed, rarely in a single‐crystal‐to‐single‐crystal (SCSC) transformation. [5] In many examples, the template is designed as to present two parallel interaction sites to favor the formation of rectangular motifs of 2 : 2 stoichiometry, with the bpen molecules facing each other at a distance short enough (<4 Å) to favor the [2+2] cycloaddition under irradiation. [6] This strategy was recently extended to σ‐hole interactions such as halogen bonding, as shown in Scheme 2. [7] Here the two halogenated (X=I, Br, Cl) aryl groups are either linked together in the proper geometry through covalent bonds or through intermolecular π–π interactions.
Scheme 2.
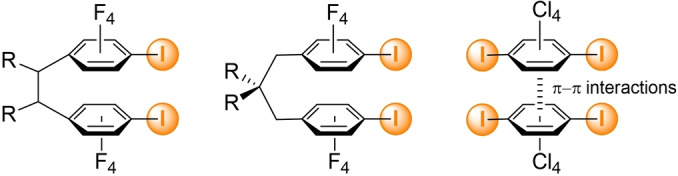
Examples of ditopic halogen bond donors used toward the formation of molecular rectangles with bpen, and exhibiting [2+2] cycloaddition under UV irradiation.
Besides halogen bonding, chalcogen bonding (ChB) has been also the subject of recent investigations for applications in crystal engineering. [8] We can mention first the association of strong ChB donor and acceptor sites on the same molecule, as in 1,2,5‐telluradiazoles, [9] 1,2‐chalcogenazole N‐oxides,[ 10 , 11 ] benzo‐1,3‐chalcogenazoles, [12] leading to the formation of infinite ribbons[ 9 , 13 ] or discrete supramolecular assemblies. [14] Bidentate chelating molecules bearing two activated chalcogen atoms, but able to interact with one single Lewis base, have been also reported in organocatalysis[ 15 , 16 ] or anion recognition[ 17 , 18 ] processes, as in molecules linking two tellurophenes, [19] two 5‐(methylchalcogeno)‐1,2,3‐triazole moieties, [20] two thiophene units, [21] or two selenocyanate groups. [22] Another efficient ChB activation has been reported in chalcogenoacetylenes shown in Scheme 3a‐b but co‐crystal formation with bpen was either unsuccessful or led to structures which appear unreactive under irradiation.[ 23 , 24 ] Considering the efficiency of benzylic selenocyanates (Scheme 3c‐d) to engage in ChB interaction with pyridines, [25] we turned our attention to more rigid derivatives and considered aromatic selenocyanates 1–3 as possible linkers to favor the face‐to‐face association of bpen, with possibly a topochemical [2+2] reactivity under irradiation. We report here the synthesis of original organic bis(selenocyanato)benzene derivatives 1–3 and their ability to form cocrystals with bpen through strong NC−Se⋅⋅⋅Nbpen chalcogen bond interactions. The proper orientation of the photoreactive bpen molecules allows them to enter into efficient [2+2] cycloaddition reactions under UV‐irradiation, while also keeping their single‐crystal quality.
Scheme 3.
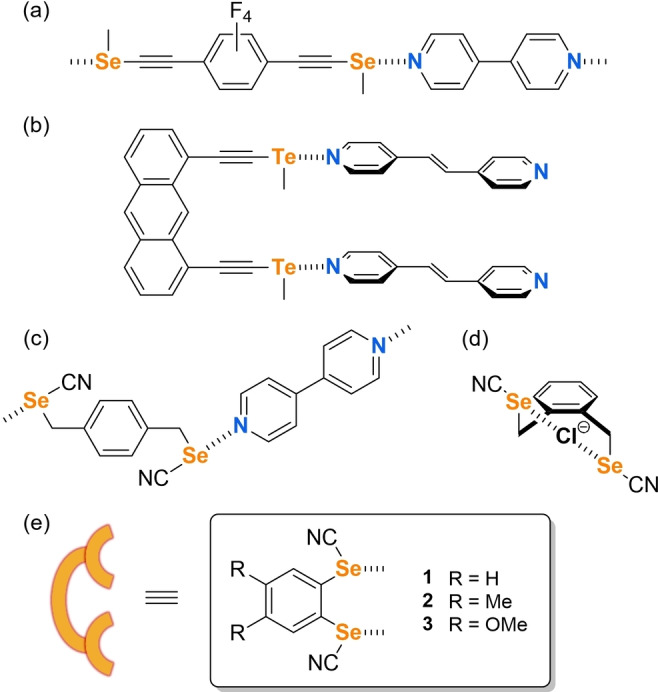
Activation of σ‐hole on selenium atoms and ChB formation with: a), b) alkynyl (chalcogenomethyl), c), d) selenocyanate derivatives and e) targeted ChB donors 1–3.
The preparation of aromatic selenocyanates is usually based on the reaction of KSeCN with either diazonium salts [26] or more electrophilic diaryliodonium salts, [27] both methods suffering from several drawbacks. More recently, the ipso‐functionalization of arylboronic acids has been reported to afford aryl selenocyanates in good yields, using either SeO2 and malononitrile, [28] or Se powder with TMS−CN. [29] Both procedures were developed however only with mono boronic acids, affording the corresponding mono‐selenocyanato derivatives. For the preparation of the desired disubstituted ortho‐bis(selenocyanato)benzene derivatives 1–3, we adapted the first procedure (SeO2/malononitrile) to ortho‐substituted diboronic derivatives. The preparation of the ortho‐diboronic acids normally involves the preparation of intermediate boronic ester through lithiation of ortho‐halogenated aryls, reaction with trimethyl or tri(isopropyl)borane and hydrolysis with HCl. [30] To avoid the isolation and manipulation of the free boronic acids, we considered a modification of reported procedures, performing the selenocyanation reaction directly on boronic esters such as the ortho‐diboronic acid bis(pinacol) esters 4–6 (Scheme 4). Their preparation involves a Miyaura borylation reaction, adapted from the procedure reported earlier [30b] by replacing the iso‐propoxy(pinacol)borane with bis(pinacolato)diborane, while using the same experimental conditions (AcOK in anhydrous DMF at 80 °C with Pd(dppf)Cl2 as catalyst). Under these conditions, the precursors 4–6 were isolated in 50–60 % yields, while the following selenocyanation reaction successfully afforded 1–3 in 75–90 % yields.
Scheme 4.
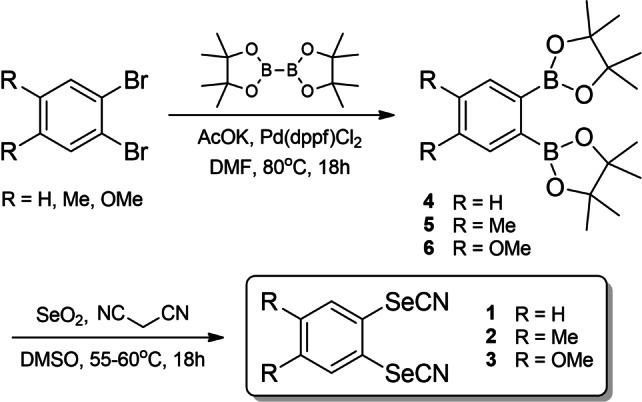
Synthetic route to the ortho‐bis(selenocyanato) derivatives 1–3.
All three compounds were purified by chromatography and recrystallized to afford crystals amenable to single‐crystal X‐ray diffraction. As shown in Figure 1, they all exhibit a recurrent intramolecular chalcogen‐bonded motif, with one NC−Se moiety coplanar with the benzene ring and pointing toward a lone pair of the neighboring Se atom, belonging to the second SeCN moiety, which is essentially perpendicular to the benzene ring. The intramolecular Se⋅⋅⋅Se distances lie in the range 3.22–3.24 Å, notably shorter than the contact distance at 3.80 Å, and corresponding to a reduction ratio (RR) of 3.23 Å/3.80 Å=0.85.
Figure 1.
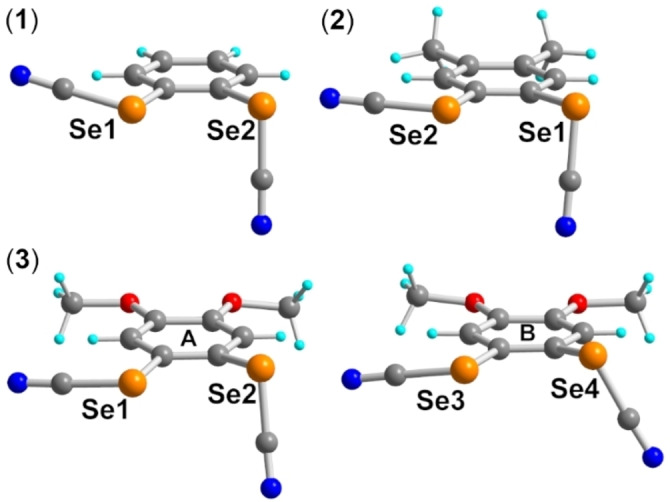
Details of the crystal structures of 1–3. Note the two crystallographically independent molecules (A, B) in 3. Intramolecular Se⋅⋅⋅Se distances amount to 3.245(1) Å in 1, 3.237(1) Å in 2, and 3.216(6) and 3.221(7) Å in 3A and 3B respectively.
In order to rationalize this behavior and the solid‐state organization (see below) of the three compounds, electrostatic potential (ESP) surfaces were calculated for 1–3 (Figure S1, Table S2 in Supporting Information) and shown in Figure 2 for compound 1. On the SeCN moiety perpendicular to the benzene ring, we observe two σ‐holes (in blue), the strongest one (+36.1 kcal mol−1) as anticipated in the prolongation of the NC−Se bond, and a weaker but non‐negligible one (+28.0 kcal mol−1) in the prolongation of the CAr−Se bond, as on the other selenocyanate group (+25.2 kcal mol−1). Moving to the methyl‐ and methoxy‐substituted derivatives 2 and 3, these values are slightly decreased (see Table S2 in Supporting Information), as a consequence of the electron‐releasing nature of the Me and OMe substituents.
Figure 2.
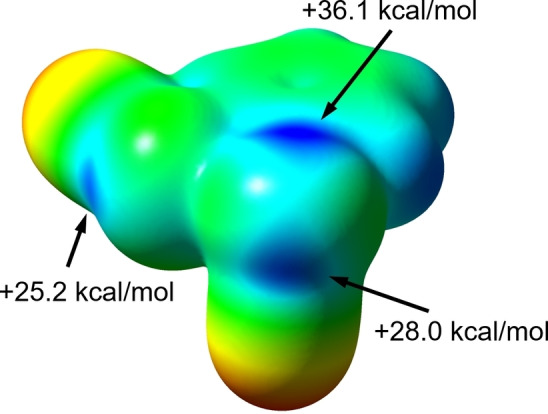
Electrostatic potential (ESP) surface (plotted on the 0.002 e bohr−3 isosurface of the electronic density) for compound 1. Color range from −37.7 (red) to +43.9 (blue) kcal mol−1.
The solid‐state organization of the three compounds (see Figures S2–S4 and Table S3 in Supporting Information) is indeed based essentially on a complex set of chalcogen bonding interactions involving these three “free” σ‐holes on the two Se atoms as ChB donors and, as ChB acceptors, either the selenium atom of neighboring molecules, the nitrogen lone pairs of nitriles, or the oxygen atoms of the methoxy groups in 3. The shortest intermolecular Se⋅⋅⋅N and Se⋅⋅⋅Se distances are associated with a reduction ratio vs. the van der Waals contact distances in the range of 0.85–0.90, comparable with those reported in other organic selenocyanates.[ 31 , 32 ] The main difference to the large set of benzylic selenocyanates reported so far is the notably larger σ‐hole in the prolongation of the CAr−Se bond in 1–3, when compared with that found along ArCH2−Se bonds in benzylic selenocyanates.
Elaboration of co‐crystals of 1–3 with bpen was realized by cooling and/or slow evaporation of equimolar quantities of both components in AcOEt. While 1 and 3 gave co‐crystals with the desired rectangular motifs (see below), cocrystal with compound 2 crystallizes in the P‐1 space group as a water solvate with a 2 : 3 stoichiometry, i.e. with a formulation (2)2(bpen)3⋅H2O, with one 2 and one bpen molecule in general position, and another bpen and water molecule on inversion centers, associated with each other not only by chalcogen bonding but also hydrogen bonding with the water molecule (See Figure S5 and Table S4 in Supporting Information). This compound does not present a favorable relative orientation of the bpen molecules for possible [2+2] cycloaddition and will not be discussed further here.
Co‐crystal of 1 with bpen crystallizes in the triclinic P‐1 space group, with a 4 : 5 stoichiometry, i.e. a formulation (1)4(bpen)5, with two crystallographically independent 1 molecules, two crystallographically independent bpen molecules, and one extra bpen dis molecule disordered on an inversion center. The co‐crystal can be described as an alternation of layers along b, with one layer at y=0 incorporating the bpen dis and molecules 1, and one layer at y= with the other bpen molecules (Figure S6). The solid‐state organization (Figure 3a) is characterized by the association of four bpen molecules into tetrameric units, linked together by short Se⋅⋅⋅Nbpen ChBs (Table 1) and small RR values in the range of 0.75–0.80. The remaining bpen dis molecule is involved in weaker C−H⋅⋅⋅Nbpen hydrogen bonds. We already note that the intramolecular Se⋅⋅⋅Se chalcogen bonds observed in the ChB donors 1–3 alone (see above Figure 1) have been overridden by the stronger ChB with the bpen nitrogen atoms, leading to the formation of the desired rectangular boxes, with face‐to‐face association of bpen molecules within the box and intermolecular C⋅⋅⋅C distances of 3.683(10) and 3.752(10) Å (Figure 3a). Furthermore, two neighboring boxes are related by inversion center and also allow for a face‐to‐face association of bpen molecules, at even shorter intermolecular C⋅⋅⋅C distance of 3.670(9) Å. Both bpen contact lengths are well below the Schmidt rules (<4 Å) for [2+2] cycloaddition. [6]
Figure 3.
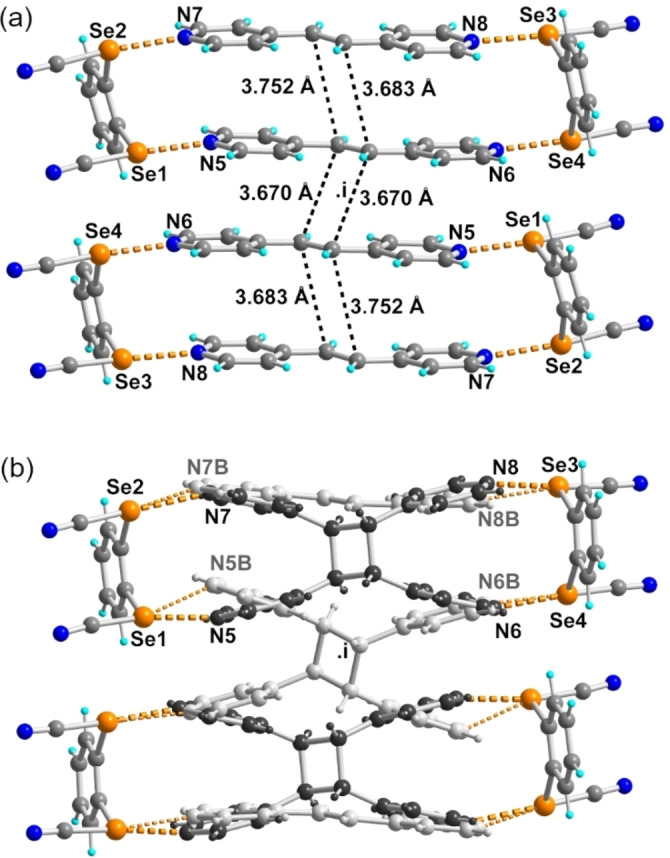
a) Details of the solid‐state association of rectangular, chalcogen‐bonded motifs in (1)4(bpen)5. ChB are indicated by orange dotted lines, and short C⋅⋅⋅C contact between ethylenic moieties by black dotted lines. b) Details of the solid‐state association after UV‐irradiation and formation of the cyclobutane adducts. Occupation parameters for the two components (shown with atoms in black and light grey) are 0.661(6) and 0.339(6).
Table 1.
Structural characteristics of the ChB interactions in (1)4(bpen)5, before and after UV irradiation.
|
ChB |
ChB length [Å] |
RR |
ChB angle [°] |
|---|---|---|---|
|
Before irradiation |
|
|
|
|
NC−Se1⋅⋅⋅N5 |
2.670(3) |
0.77 |
175.83(9) |
|
NC−Se2⋅⋅⋅N7 |
2.754(4) |
0.80 |
173.58(9) |
|
NC−Se3⋅⋅⋅N8 |
2.759(4) |
0.75 |
178.27(9) |
|
NC−Se4⋅⋅⋅N6 |
2.656(4) |
0.77 |
175.42(9) |
|
After UV irradiation, major component | |||
|
NC−Se1⋅⋅⋅N5 |
2.727(11) |
0.79 |
170.52(43) |
|
NC−Se2⋅⋅⋅N7 |
3.017(14) |
0.87 |
179.50(52) |
|
NC−Se3⋅⋅⋅N8 |
2.649(15) |
0.77 |
175.38(50) |
|
NC−Se4⋅⋅⋅N6 |
2.732(12) |
0.79 |
176.38(54) |
|
After UV irradiation, minor component | |||
|
NC−Se1⋅⋅⋅N5B |
2.682(21) |
0.78 |
165.08(57) |
|
NC−Se2⋅⋅⋅N7B |
2.640(25) |
0.77 |
174.57(71) |
|
NC−Se3⋅⋅⋅N8B |
2.850(26) |
0.83 |
172.55(66) |
|
NC−Se4⋅⋅⋅N6B |
2.990(23) |
0.87 |
178.27(63) |
UV‐irradiation of single crystals of (1)4(bpen)5 provided indeed a single‐crystal‐to‐single‐crystal transformation. As shown in Figure 3b, refinement of the crystal structure of the cyclized compound showed, besides the unreacted bpen dis molecule disordered on an inversion center, two components with occupation parameters at 0.661(6) and 0.339(6), corresponding to a 2/3–1/3 statistical distribution expected if the [2+2] reactivity within a rectangular box is the same as the reactivity of inversion‐related bpen molecules between the boxes, which leaves the two outer bpen molecules unreacted. This striking behavior is, to our knowledge, completely new, and can be compared only with another example of tetrameric association of bpen molecules linked through hydrogen bonding by indolocarbazole, but in that case only the inner bpen molecules reacted with each other. [33] It also demonstrates that the ChB interactions provided by this motif of two selenocyanate moieties in ortho position are robust enough to force the face‐to‐face association of the bpen molecules and to stand the severe deformations induced by the photocyclization. Indeed, the small RR values before cyclization (Cf. Table 1) observed in the range 0.75–0.80 (Figure 3a) and characteristic of strong ChB, increase to only 0.77–0.87 in the photo‐cyclized products (Figure 3b).
The success of this approach is further confirmed in the co‐crystal obtained with the dimethoxy‐substituted ChB donor 3 and bpen. It crystallizes indeed in the triclinic system, space group P‐1, with one ChB donor 3 and two bpen molecules, all in general position, corresponding to a 1 : 2 stoichiometry, better described as inversion centered rectangular motifs (3)2(bpen)2, separated from each other by extra bpen molecules (Figure S7).
Within the rectangular motifs (Figure 4a), the bpen molecules are associated in a face‐to‐face manner through ChB with 3 through strong and linear NC−Se⋅⋅⋅Nbpen interactions (Table 2). Contacts between the ethylenic carbon atoms of bpen molecules [3.851(7) Å] are longer than those observed in (1)4(bpen)5 (3.67–3.75 Å), nevertheless shorter than the 4 Å limit. Indeed, UV‐irradiation of single crystals of (3)(bpen)2 led to single‐crystal‐to‐single‐crystal transformation (Figure 4b), with the desired cyclobutane formation while the other bpen molecule interspersed between the rectangles stays unmodified (Figure S8). The ChB interactions (Table 2) are slightly elongated during the photochemical transformation, with RR values evolving from 0.78 to 0.82–0.83.
Figure 4.

a) Details of the solid‐state association within a rectangular chalcogen‐bonded motif in (3)(bpen)2. ChB are indicated by orange dotted lines, and short C⋅⋅⋅C contact between ethylenic moieties by black dotted lines. b) Details of the solid‐state association of the same motif, after UV‐irradiation and formation of the cyclobutane ring.
Table 2.
Structural characteristics of the ChB interactions in (3)(bpen)2, before and after UV irradiation.
|
ChB |
ChB length [Å] |
RR |
ChB angle [°] |
|---|---|---|---|
|
Before irradiation |
|
|
|
|
NC−Se1⋅⋅⋅N4 |
2.679(7) |
0.78 |
178.88(6) |
|
NC−Se2⋅⋅⋅N3 |
2.683(7) |
0.78 |
177.16(6) |
|
After UV irradiation |
|
|
|
|
NC−Se1⋅⋅⋅N3 |
2.863(19) |
0.83 |
176.2(4) |
|
NC−Se2⋅⋅⋅N4 |
2.819(18) |
0.82 |
172.2(3) |
In conclusion, we have unraveled here for the first time the use of chalcogen bond donors such as 1 and 3 to assemble photoreactive bpen molecules into face‐to‐face dimers, allowing for rare single‐crystal‐to‐single‐crystal transformations, with the added originality of the striking reactivity of the tetrameric motif isolated with 1. The structural adaptability of this ortho‐substituted bis(selenocyanato) motif is highlighted by the evolution of the intramolecular Se⋅⋅⋅Se distance, from the short 3.22–3.24 Å values in the molecules alone (cf. Figure 1) due to a strong intramolecular Se⋅⋅⋅Se ChB, to a notably larger distance of 3.52–3.54 Å in the chalcogen‐bonded adducts with bpen, well adapted for the face‐to‐face association of the bpen molecules into reactive position toward photochemical dimerization and cyclobutane formation. Furthermore, as already illustrated by the numerous hydrogen‐bonded systems derived from resorcinol derivatives (Scheme 1b),[ 1 , 2 , 3 , 4 , 7 ] the three different substitution patterns explored here (R=H, Me, OMe) on the 4,5 positions of the aryl ring let us anticipate many other possibilities by varying the nature of these substituents on all four 3‐, 4‐, 5‐ and 6‐positions of the benzene ring. We are now focused on exemplifying this structuring motif to other photo‐ or thermo‐reactive molecules with chalcogen bond acceptor capability, toward the elaboration of novel materials with optical or conducting properties.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Acknowledgements
We thank ANR (Paris) for financial support under grant ANR‐17‐CE07‐0025‐02. This work has been also supported by the Integrated Development Program of the Gdańsk University of Technology program No. POWR.03.05.00‐00.Z044/17 funded by the European Social Fund under the Knowledge Education Development Operational Program.
J. Alfuth, O. Jeannin, M. Fourmigué, Angew. Chem. Int. Ed. 2022, 61, e202206249; Angew. Chem. 2022, 134, e202206249.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. MacGillivray L. R., Papaefstathiou G. S., Friščić T., Hamilton T. D., Bučar D.-K., Chu Q., Varshney D. B., Georgiev I. G., Acc. Chem. Res. 2008, 41, 280–291. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Campillo-Alvarado G., Li C., Swenson D. C., MacGillivray L. R., Cryst. Growth Des. 2019, 19, 2511–2518; [Google Scholar]
- 2b. Bosch E., Kruse S. J., H. R. Krueger, Jr. , Groeneman R. H., Cryst. Growth Des. 2019, 19, 3092–3096. [Google Scholar]
- 3.
- 3a. Bučar D.-K., Sen A., Mariappan S. V. S., MacGillivray L. R., Chem. Commun. 2012, 48, 1790–1792; [DOI] [PubMed] [Google Scholar]
- 3b. Hutchins K. M., Sumrak J. C., MacGillivray L. R., Org. Lett. 2014, 16, 1052–1055; [DOI] [PubMed] [Google Scholar]
- 3c. Ericson D. P., Zurfluh-Cunningham Z. P., Groeneman R. H., Elacqua E., Reinheimer E. W., Noll B. C., MacGillivray L. R., Cryst. Growth Des. 2015, 15, 5744–5748; [Google Scholar]
- 3d. Yelgaonkar S. P., Swenson D. C., MacGillivray L. R., Chem. Sci. 2020, 11, 3569–3573; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Varshney D. B., Gao X., Friscic T., MacGillivray L. R., Angew. Chem. Int. Ed. 2006, 45, 646–650; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 662–666. [Google Scholar]
- 4.
- 4a. Li C., Campillo-Alvarado G., Swenson D. C., MacGillivray L. R., Inorg. Chem. 2019, 58, 12497–12500; [DOI] [PubMed] [Google Scholar]
- 4b. Papaefstathiou G. S., Zhong Z., Gang L., MacGillivray L. R., J. Am. Chem. Soc. 2004, 126, 9158–9159. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Friščić T., MacGillivray L. R., Z. Kristallogr. 2005, 220, 351–363; [Google Scholar]
- 5b. Halasz I., Cryst. Growth Des. 2010, 10, 2817–2823. [Google Scholar]
- 6. Schmidt G. M. J., Pure Appl. Chem. 1971, 27, 647–678. [Google Scholar]
- 7.
- 7a. Kruse S. J., Bosch E., Brown F., Groeneman R. H., Cryst. Growth Des. 2020, 20, 1969–1974; [Google Scholar]
- 7b. Sinnwell M. A., Blad J. N., Thomas L. R., MacGillivray L. R., IUCrJ 2018, 5, 491–496; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7c. Sinnwell M. A., MacGillivray L. R., Angew. Chem. Int. Ed. 2016, 55, 3477–3480; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 3538–3541; [Google Scholar]
- 7d. Caronna T., Liantonio R., Logothetis T. A., Metrangolo P., Pilati T., Resnati G., J. Am. Chem. Soc. 2004, 126, 4500–4501; [DOI] [PubMed] [Google Scholar]
- 7e. Quentin J., Swenson D. C., MacGillivray L. R., Molecules 2020, 25, 907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Scilabra P., Terraneo G., Resnati G., Acc. Chem. Res. 2019, 52, 1313–1324; [DOI] [PubMed] [Google Scholar]
- 8b. Vogel L., Wonner P., Huber S. M., Angew. Chem. Int. Ed. 2019, 58, 1880–1891; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1896–1907. [Google Scholar]
- 9.
- 9a. Cozzolino A. F., Britten J. F., Vargas-Baca I., Cryst. Growth Des. 2006, 6, 181–186; [Google Scholar]
- 9b. Cozzolino A. F., Vargas-Baca I., Mansour S., Mahmoudkhani A. H., J. Am. Chem. Soc. 2005, 127, 3184–3190; [DOI] [PubMed] [Google Scholar]
- 9c. Garrett G. E., Gibson G. L., Straus R. N., Seferos D. S., Taylor M. S., J. Am. Chem. Soc. 2015, 137, 4126–4133. [DOI] [PubMed] [Google Scholar]
- 10. Ho P. C., Szydlowski P., Sinclair J., Elder P. J. W., Kubel J., Gendy C., Lee L. M., Jenkins H., Britten J. F., Morim D. R., Vargas-Baca I., Nat. Commun. 2016, 7, 11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ho P. C., Rafique J., Lee J., Lee L. M., Jenkins H. A., Britten J. F., Braga A. L., Vargas-Baca I., Dalton Trans. 2017, 46, 6570–6579. [DOI] [PubMed] [Google Scholar]
- 12. Kremer A., Fermi A., Biot N., Wouters J., Bonifazi D., Chem. Eur. J. 2016, 22, 5665–5675. [DOI] [PubMed] [Google Scholar]
- 13. Cozzolino A. F., Yang Q., Vargas-Baca I., Cryst. Growth Des. 2010, 10, 4959–4964. [Google Scholar]
- 14. Riwar L.-J., Trapp N., Root K., Zenobi R., Diederich F., Angew. Chem. Int. Ed. 2018, 57, 17259–17264; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 17506–17512. [Google Scholar]
- 15.
- 15a. Wonner P., Vogel L., Dser M., Gomes L., Kniep F., Mallick B., Werz D. B., Huber S. M., Angew. Chem. Int. Ed. 2017, 56, 12009–12012; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12172–12176; [Google Scholar]
- 15b. Wonner P., Vogel L., Kniep F., Huber S. M., Chem. Eur. J. 2017, 23, 16972–16975; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Wonner P., Dreger A., Vogel L., Engelage E., Huber S. M., Angew. Chem. Int. Ed. 2019, 58, 16923–16927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17079–17083; [Google Scholar]
- 15d. Wonner P., Steinke T., Vogel L., Huber S. M., Chem. Eur. J. 2020, 26, 1258–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benz S., Poblador-Bahamonde A. I., Low-Ders N., Matile S., Angew. Chem. Int. Ed. 2018, 57, 5408–5412; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5506–5510. [Google Scholar]
- 17. Taylor M. S., Coord. Chem. Rev. 2020, 413, 213270. [Google Scholar]
- 18. Semenov N. A., Gorbunov D. E., Shakhova M. V., Salnikov G. E., Bagryanskaya I. Y., Korolev V. V., Beckmann J., Gritsan N. P., Zibarev A. V., Chem. Eur. J. 2018, 24, 12983–12991. [DOI] [PubMed] [Google Scholar]
- 19. Garrett G. E., Carrera E. I., Seferos D. S., Taylor M. S., Chem. Commun. 2016, 52, 9881–9884. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. Lim J. Y. C., Marques I., Thompson A. L., Christensen K. E., Félix V., Beer P. D., J. Am. Chem. Soc. 2017, 139, 3122–3133; [DOI] [PubMed] [Google Scholar]
- 20b. Lim J. Y. C., Liew J. Y., Beer P. D., Chem. Eur. J. 2018, 24, 14560–14566; [DOI] [PubMed] [Google Scholar]
- 20c. Borissov A., Marques I., Lim J. Y. C., Félix V., Smith M. D., Beer P. D., J. Am. Chem. Soc. 2019, 141, 4119–4129; [DOI] [PubMed] [Google Scholar]
- 20d. Lim J. Y. C., Marques I., Félix V., Beer P. D., Chem. Commun. 2018, 54, 10851–10854. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Benz S., Macchione M., Verolet Q., Mareda J., Sakai N., Matile S., J. Am. Chem. Soc. 2016, 138, 9093–9096; [DOI] [PubMed] [Google Scholar]
- 21b. Strakova K., Assies L., Goujon A., Piazzolla F., Humeniuk H. V., Matile S., Chem. Rev. 2019, 119, 10977–11005; [DOI] [PubMed] [Google Scholar]
- 21c. Lee L. M., Tsemperouli M., Poblador-Bahamonde A. I., Benz S., Sakai N., Sugihara K., Matile S., J. Am. Chem. Soc. 2019, 141, 810–814. [DOI] [PubMed] [Google Scholar]
- 22. Riel A. M. S., Huynh H.-T., Jeannin O., Berryman O., Fourmigué M., Cryst. Growth Des. 2019, 19, 1418–1425. [Google Scholar]
- 23. Dhaka A., Jeannin O., Jeon I.-R., Aubert E., Espinosa E., Fourmigué M., Angew. Chem. Int. Ed. 2020, 59, 23583–23587; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 23789–23793. [Google Scholar]
- 24. Dhaka A., Jeannin O., Aubert E., Espinosa E., Fourmigué M., Molecules 2021, 26, 4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huynh H.-T., Jeannin O., Fourmigué M., Chem. Commun. 2017, 53, 8467–8469. [DOI] [PubMed] [Google Scholar]
- 26. Nikolaienko P., Rueping M., Chem. Eur. J. 2016, 22, 2620–2623. [DOI] [PubMed] [Google Scholar]
- 27. Guan Y., Townsend S. D., Org. Lett. 2017, 19, 5252–5255. [DOI] [PubMed] [Google Scholar]
- 28. Redon S., Kosso A. R. O., Broggi J., Vanelle P., Synthesis 2019, 51, 3758–3765. [Google Scholar]
- 29. Zhang X., Huang X.-B., Zhou Y.-B., Liu M.-C., Wu H.-Y., Chem. Eur. J. 2021, 27, 944–948. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Durka K., Jarzembska K. N., Kaminski R., Lulinski S., Serwatowski J., Wozniak K., Cryst. Growth Des. 2013, 13, 4181–4185; [Google Scholar]
- 30b. Seven O., Bolte M., Lerner H.-W., Wagner M., Organometallics 2014, 33, 1291–1299. [Google Scholar]
- 31.
- 31a. Jeannin O., Huynh H.-T., Riel A. M. S., Fourmigué M., New J. Chem. 2018, 42, 10502–10509, and references therein; [Google Scholar]
- 31b. Huynh H.-T., Jeannin O., Fourmigué M., New J. Chem. 2021, 45, 76–84. [Google Scholar]
- 32. Kumar V., Leroy C., Bryce D. L., CrystEngComm 2018, 20, 6406–6411. [Google Scholar]
- 33. Stojaković J., Whitis A. M., MacGillivray L. R., Angew. Chem. Int. Ed. 2013, 52, 12127–12130; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12349–12352. [Google Scholar]
- 34.Deposition Numbers 2168861, 2168862, 2168863, 2168864, 2168865, 2168866, 2168867, and 2168868 contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.